
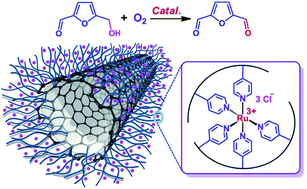
Ruthenium complex immobilized on poly(4-vinylpyridine)-functionalized carbon-nanotube for selective aerobic oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran†
Abstract
Polymer/carbon composite material of poly(4-vinylpyridine)-functionalized carbon-nanotube (PVP/CNT) was prepared by in situ polymerization of 4-vinylpyridine monomer in the presence of CNT suspension. Raman spectra analysis confirmed the almost unchanged graphitized surfaces of CNT moiety in the PVP/CNT after the covalent functionalization of pristine CNT with PVP. Catalyst made of ruthenium complex immobilized on PVP/CNT (Ru-PVP/CNT) was fully characterized by ICP-OES, TG-DTA, FT-IR, Raman, XRD, UV-vis, BET, TEM, XPS and H2-TPR. Moreover, Ru-PVP/CNT shows excellent catalytic performance towards the selective oxidation of biomass-based 5-hydroxymethylfurfural (HMF) to 2,5-diformylfuran (DFF) with molecular oxygen as oxidant. The reaction parameters such as the reaction temperature, reaction time, solvent, catalyst amount, oxidant, and oxygen pressure were systematically investigated for this important biomass-related transformation. Under the optimal condition, a DFF yield of 94% with a full HMF conversion were obtained by using Ru-PVP/CNT in N,N-dimethylformamide (DMF) under 2.0 MPa O2 at 120 °C.
 Please wait while we load your content...
Please wait while we load your content...

