
Solubility properties and spectral characterization of sulfur dioxide in ethylene glycol derivatives†
Abstract
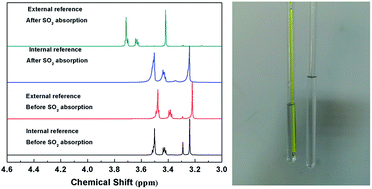
Solubilities of SO2 in ethylene glycol derivatives were determined by dynamic isothermal gas–liquid equilibrium (GLE) experiments, and the thermodynamic parameters of the absorption processes were calculated. The GLE results indicated that the solubilities of SO2 in ethylene glycol derivatives increase in the order: diols < monomethyl ethers < dimethyl ethers, with the enthalpy values ranging from −23.2 to −43.3 kJ mol−1. The regeneration experiment found that the absorption of SO2 in tetraethylene glycol dimethyl ether is reversible, and the solvents can be reused without a significant loss of absorption capacity. The interactions between SO2 and ethylene glycol derivatives were investigated by UV, IR and NMR. In addition, a 1H-NMR spectroscopy technique with external references was used to investigate the physical absorption process of SO2 for the first time, in order to avoid the influence of deuterated solvents. Spectroscopic investigations showed that the interactions between SO2 and ethylene glycol derivatives are based on both the charge-transfer interaction and hydrogen bond. Ethylene glycol derivatives with desirable absorption capacities and excellent regeneration abilities are promising alternatives to conventional sorbents in SO2 separation.
 Please wait while we load your content...
Please wait while we load your content...

