
Functionalization of biodegradable hyperbranched poly(α,β-malic acid) as a nanocarrier platform for anticancer drug delivery†
Abstract
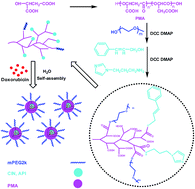
Multiple functionalization of nanoparticles has attracted great interest in drug delivery. In this paper, biodegradable poly(α,β-malic acid) with a hyperbranched architecture was synthesized via the polycondensation of L-malic acid, the functionalized poly(α,β-malic acid) was used as a nanocarrier platform with the immobilization of poly(ethylene glycol) (PEG) for long circulation, cinnamyl alcohol (CIN) for introducing π–π stacking interactions and 1-(3-aminopropyl)imidazole (API) for pH-sensitivity. The conjugates self-assembled into nanoparticles to load anticancer drug doxorubicin (DOX). The morphology, mean size and size distribution, drug release profile and in vitro anticancer activity of DOX loaded nanoparticles were studied. The results showed that the mean size of the nanoparticles was below 200 nm, the drug loading content was higher than 10 wt% and it increased with increasing CIN content because of the π–π stacking interaction between DOX and the carriers. The drug release of the nanoparticles was faster in the medium with pH 6.0 compared to pH 7.4. The nanoparticles exhibited an endosomal escape function to accelerate the release of DOX in cancer cells, which resulted in low IC50s to kill 4T1 breast cancer cells and HepG2 liver cancer cells in vitro.
 Please wait while we load your content...
Please wait while we load your content...

