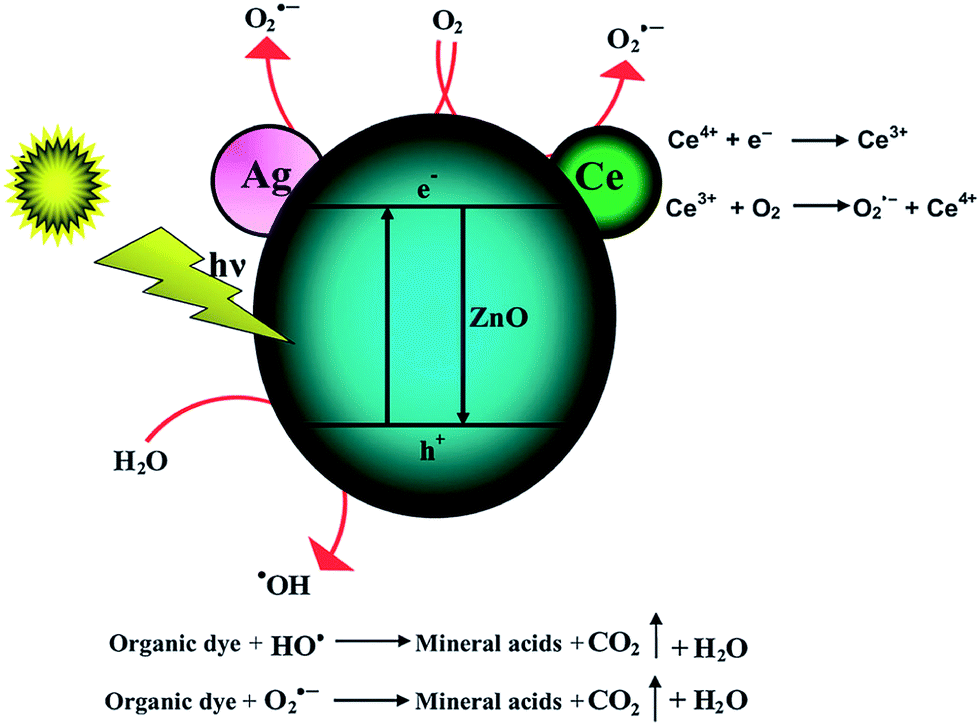
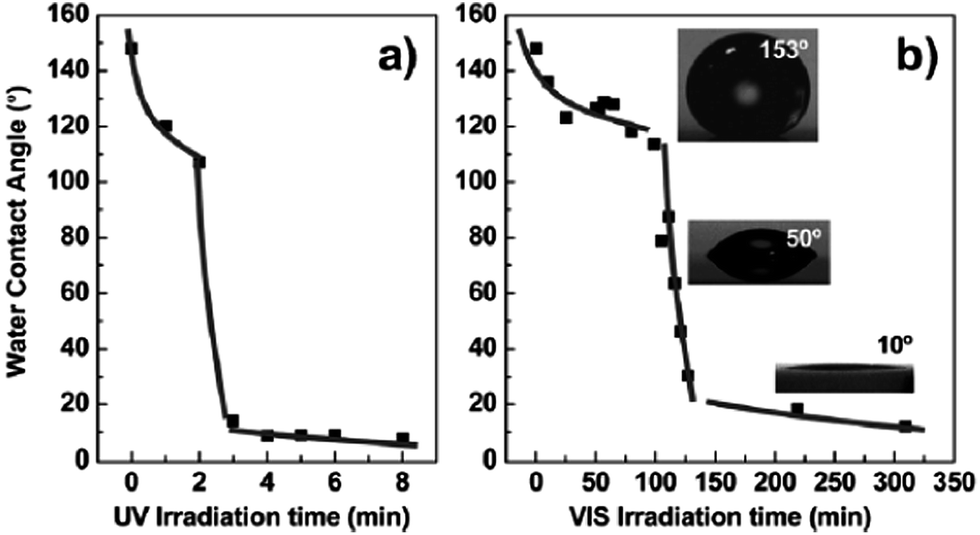
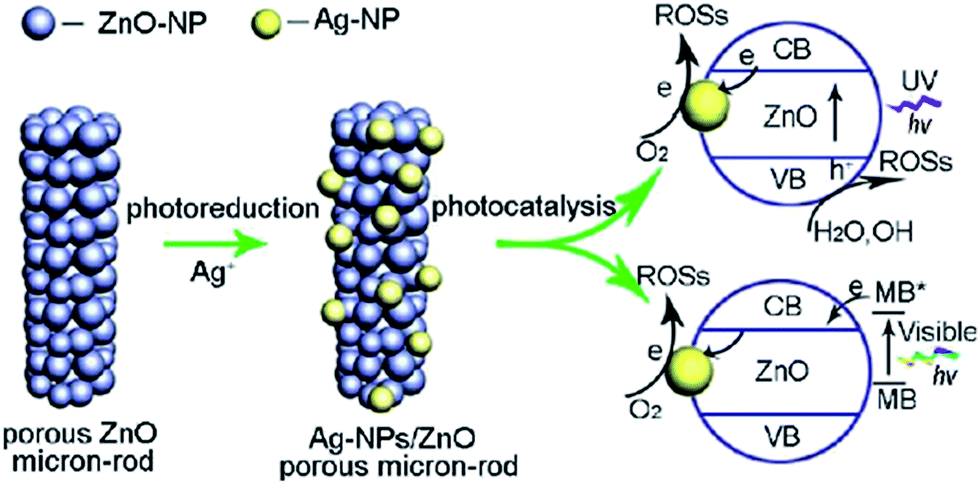
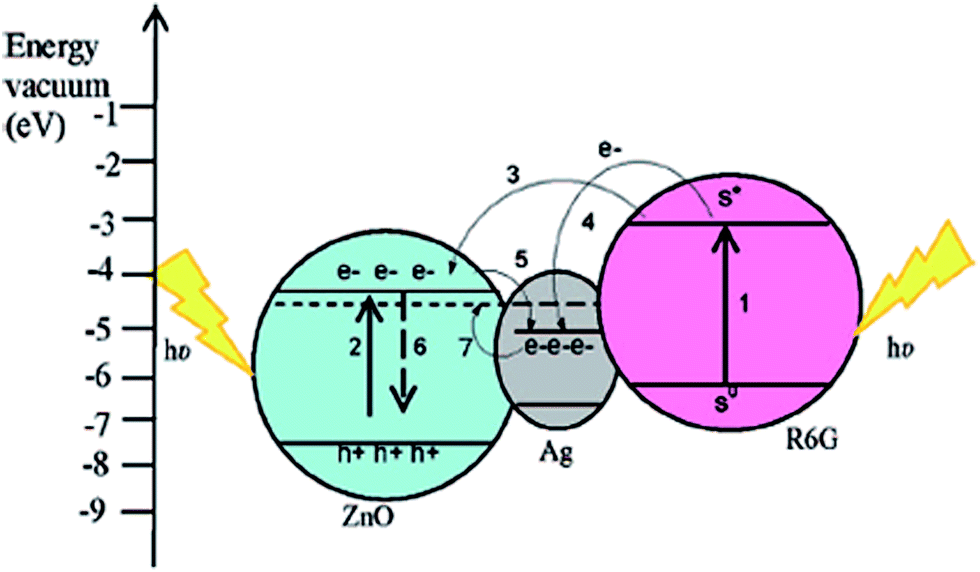
DOI:
10.1039/C4RA13299H
(Review Article)
RSC Adv., 2015,
5, 3306-3351
Zinc oxide based photocatalysis: tailoring surface-bulk structure and related interfacial charge carrier dynamics for better environmental applications
Received
9th September 2014
, Accepted 6th November 2014
First published on 6th November 2014
Abstract
As an alternative to the gold standard TiO2 photocatalyst, the use of zinc oxide (ZnO) as a robust candidate for wastewater treatment is widespread due to its similarity in charge carrier dynamics upon bandgap excitation and the generation of reactive oxygen species in aqueous suspensions with TiO2. However, the large bandgap of ZnO, the massive charge carrier recombination, and the photoinduced corrosion–dissolution at extreme pH conditions, together with the formation of inert Zn(OH)2 during photocatalytic reactions act as barriers for its extensive applicability. To this end, research has been intensified to improve the performance of ZnO by tailoring its surface-bulk structure and by altering its photogenerated charge transfer pathways with an intention to inhibit the surface-bulk charge carrier recombination. For the first time, the several strategies, such as tailoring the intrinsic defects, surface modification with organic compounds, doping with foreign ions, noble metal deposition, heterostructuring with other semiconductors and modification with carbon nanostructures, which have been successfully employed to improve the photoactivity and stability of ZnO are critically reviewed. Such modifications enhance the charge separation and facilitate the generation of reactive oxygenated free radicals, and also the interaction with the pollutant molecules. The synthetic route to obtain hierarchical nanostructured morphologies and study their impact on the photocatalytic performance is explained by considering the morphological influence and the defect-rich chemistry of ZnO. Finally, the crystal facet engineering of polar and non-polar facets and their relevance in photocatalysis is outlined. It is with this intention that the present review directs the further design, tailoring and tuning of the physico-chemical and optoelectronic properties of ZnO for better applications, ranging from photocatalysis to photovoltaics.
 S. Girish Kumar | Dr S. Girish Kumar is a native of Karnataka (Kolar District, Malur Taluk) and obtained his MSc in Physical Chemistry (2005) and PhD (2012) degree in the area of photocatalysis from Bangalore University, Bangalore. He is the recipient of India's most prestigious Dr D. S. Kothari Post Doctoral Fellowship (2012) and works under the supervision of Prof. Rao at the Department of Physics, IISc, Bangalore on CdTe/CdS thin film heterojunction solar cells. The study of phase transition and photoluminescence properties of mixed phase titania and other heterojunctions are his present interests. |
 K. S. R. Koteswara Rao | Dr K. S. R. Koteswara Rao is associate professor at the Department of Physics, Indian Institute of Science, Bangalore, India. He works in the field of semiconductors. His research interests are understanding defects in semiconductor materials and their heterostructures by optical (photoluminescence, optically induced conductivity modulation, etc.) and electrical methods. The growth and study of III–V and II–VI based Binary, Ternary and Quaternary compound semiconductor nano- and micro-structures and their utility for device applications are his current research studies. |
1. Introduction
Clean energy and pollutant-free water/air are the important tasks that we currently face, with a common solution that lies in the design and development of multifunctional nanomaterials for harvesting maximum light energy from solar light. Because environmental pollution has surpassed the threshold of natural purification, an advanced oxidation process seems to be the most effective wastewater treatment methods having high efficiency and low cost. In addition to the photo Fenton process,1–4 semiconductor photocatalysis, which is a ‘green approach’, is at the forefront of fundamental research and consideration for technological applications due to its non-selectivity, low temperature and non-energy intensive approach for complete mineralization of pollutants. The photoinduced charge carrier separation upon the bandgap excitation of semiconductors is vital for redox reactions, followed by charge carrier transfer to solution-phase redox couples, which is essential to accelerate overall photocatalytic reaction rates. Thermodynamically, the redox potential of the VB-hole must be positive to generate hydroxyl radicals, and the CB-electron must be negative to initiate dioxygen reduction.5 As an alternative to illustrious semiconductors, such as TiO2, WO3, Bi2O3, Fe2O3, BiOX (X = Cl, Br and I) and (BiO)2CO3, recently, ZnO is in the spotlight of many research efforts, due to its stupendous benefits, such as low cost and high quantum efficiency, as well as a favourable bandgap and due to its photocatalytic mechanisms.6–15 The admirable attributes of ZnO, such as mechanical-thermal stability, high photosensitivity, low cost, high redox potential, large bandgap offering an excellent driving force to induce redox reactions, non-toxicity, versatility in synthesis with hierarchical morphology, the availability of different precursors (common inorganic salts) and their high solubility in various solvents, ease of crystallization, anisotropic growth, and natural abundance, make it ideal for photocatalysis. The refractive index of ZnO (2.0) is lower than for TiO2 (2.5–2.7), and hence ZnO scarcely scatters light, thereby making it colorless and boosting its transparency. Moreover, ZnO-photocatalyzed reactions perform best in neutral pH conditions, which is an added merit over its competitors. Furthermore, the emission properties of ZnO have made it possible to set up an original catalytic system, which is able to ‘sense and shoot’ the environmental contaminants, thus motivating the further exploration of the properties of ZnO.16
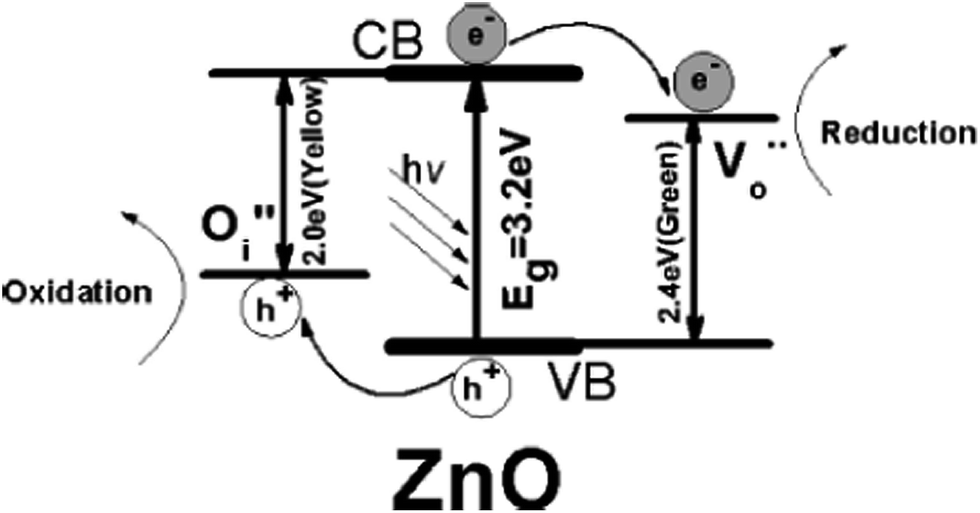
Due to the presence of intrinsic impurities, the electron mobility (200–300 cm2 V−1 s−1) and electron lifetime (>10 s) of ZnO are considerably higher compared to TiO2 (0.1–4.0 cm2 V−1 s−1), which reduces the electrical resistance and promotes the electron transfer efficiency.17,18 Thus, a high concentration of photogenerated charge carriers transfer to the surface, contributing to efficient photocatalysis. In addition, the VB of ZnO is positioned slightly below TiO2 VB, indicating that the hydroxyl radical generated in the former (+3.06 V) has a higher oxidation potential compared to the latter (+2.7 V); moreover, the electron derived from the ZnO CB is believed to be more negative than TiO2 (at pH 0 vs. NHE), whereas the CB edges of both the semiconductors are almost the same at neutral pH conditions (−0.5 V vs. NHE).19,20 The ZnO absorbs a large fraction of UV spectrum and more light quanta, exhibiting a better performance compared to TiO2 for pollutant treatment under light illumination.21–26 The surface band of ZnO is bent upward in air, indicating that the direction of its built-in electric field is from inner to outer, and thus facilitates hole migration to the particle surface, while electrons diffuse to the bulk of the particle.27 Defects like oxygen vacancies, zinc interstitials, oxygen interstitials, and the generation of hydrogen peroxide, superoxide and hydroxyl radicals on the ZnO surface are reported to be responsible for the photocatalytic activity.28–33 Although, different radicals/defects mediate the degradation mechanism depending on the surface-bulk modification of ZnO, it is unambiguously accepted that a low degree of charge carrier recombination is vital to achieve a high photocatalytic efficiency.
ZnO commonly crystallizes in a wurtzite structure (space group P63mc, a = 3.25 Å, c = 5.20 Å) with n-type conductivity (Zn1+σO, σ > 0), with a direct bandgap of 3.37 eV and with a large excitonic binding energy (60 meV), which is even larger than the thermal energy at RT.28–33 The specific physicochemical, optoelectronic and magnetic properties of ZnO stimulates its potential application in various fields, such as photocatalysis, light emitting diodes, solar cells, gas sensors, pyroelectricity, luminescent materials, pigments, UV shielding materials, surface acoustic wave filters, actuators, spin electronics, short-wavelength optoelectronic devices, varistors, antifungal, and piezodielectric nanogenerators.34–39
Despite the versatility, ZnO-based photocatalysis suffers from the following draw backs: (i) ZnO does not absorb the visible portion of the solar spectrum, instead it requires UV light, which is expensive, for bandgap excitation; (ii) rapid recombination of the charge carriers inevitably obstructs the outward diffusion of the charge carriers, and consequently slows down the degradation reactions occurring at the semiconductor–liquid interface; (iii) there are problems associated with the recovery of ZnO powder from the suspension by conventional filtration; (iv) the tendency to aggregate during the catalytic reactions and the susceptibility to corrosion under UV light. The photocorrosion reactions can be represented as follows:40,41
| | |
ZnO + 2h+ + nH2O → Zn(OH)n(2−n)+ + 1/2O2 + nH+
| (1) |
where
n depends on the pH of the solution. The photodissolution of ZnO initially involves hole trapping at the surface, followed by a rapid formation of oxygen molecules and a fast expulsion of Zn
2+ from the surface.
| | |
Osurface2− + h+ → Osurface−
| (2) |
| | |
Osurface− + 3O2− + 3h+ → 2(O–O2−)
| (3) |
| | |
2Zn2+ → 2Zn2+(aqueous)
| (5) |
The overall reaction can be represented as.
| | |
ZnO + 2h+ → Zn2+ + 1/2O2
| (6) |
In addition, the ZnO powder dissolves at strong acidic pH:
| | |
ZnO + 2H+ → Zn2+ + H2O
| (7) |
Under strong alkaline medium, ZnO can undergo dissolution:
| | |
ZnO + H2O + 2OH− → Zn(OH)42−
| (8) |
ZnO passivates to form an inert Zn(OH)2 surface layer upon UV illumination;
| | |
2ZnO + 4H2O + 4h+ → 2Zn(OH)2 + O2 + 4H+
| (9) |
Thus, both strongly acidic and strongly alkaline pH may not favour the photocatalytic process.
To overcome these aforementioned obstacles, research is rapidly progressing to modify the surface-electronic structure of ZnO, largely by altering the ZnO defect chemistry to benefit photocatalysis under ambient conditions. Many insightful review articles are concentrated on the synthesis, properties, growth, defects, and other applications of ZnO.42–47 In contrast, a few seminal review articles associated with the photocatalysis discuss the effects of the initial reaction parameters, such as catalyst dosage, concentration of the dye, solution pH and the presence of electron acceptors, including a brief approach on how to afford the visible light response of ZnO.48–51 Inspired by the advances with interesting and exciting results, the authors have taken up this review on the research progress of ZnO-based photocatalysis to pave the way for its practical application. The interfacial charge carrier transfer dynamics in each strategy correlating to a high activity of modified ZnO are discussed with respect to material properties, such as catalyst dosage, surface charge density, crystallinity, defects (intrinsic and extrinsic), properties of modifiers, and charge carrier generation–separation–recombination dynamics, together with the experimental conditions appropriate for the pollutant structure, pH of the solution, the presence of inorganic electrolytes, and the intensity and wavelength of the excitation source. For the first time, the defect-facet-morphological dependence of ZnO on photocatalytic activity is also highlighted.†
2. Fundamental aspects of ZnO-based photocatalysis
The underlying mechanism of the photocatalysis comprises the bandgap excitation of ZnO with energetic photons, thereby generating an exciton pair with holes in the VB and electrons in the CB. These charge carriers may recombine, which dissipates the input energy as heat. Moreover, they may undergo an interfacial charge transfer process either by trapping at the metastable surface states or by interacting with pre-adsorbed electron donors/acceptors on the catalyst surface or within the surrounding electrical double layer of the charged particles.7,52,53
(a) Surface sensitization and complexation of ZnO
Microscale ZnO decomposes dyes like CV, MB, OG and MO at a faster rate compared to Degussa P25 under UV-visible light, indicating that the photosensitization of ZnO by dyes favours the visible light response with an enhanced charge carrier separation.54 The excited dye molecule transfers electrons to the ZnO CB, whereas the dye itself converts to a cationic radical. The injected electron reacts with dioxygen adsorbed on the ZnO surface to generate a series of active oxygen species, which on subsequent reaction with the dye molecules results in degradation.55,56| | |
Dye + visible light → Dye*
| (10) |
| | |
Dye* + ZnO → Dye˙+ + ZnO(e−)
| (11) |
| |
 | (13) |
| | |
Dye˙+ + reactive oxygenated radicals → products
| (14) |
In addition, an electricity conversion efficiency of 0.23% was obtained for ZnO-based dye sensitized solar cells compared to the Degussa P25 (0.0024%) counterpart. This is an important report to simultaneously realize both dye degradation and the generation of a renewable energy source. The ZnO-sensitized heteroaggregate (CoTPPS + TAPPI) was efficient for the degradation of RhB under visible light compared to ZnO–CoTPPS, ZnO–TAPPI and ZnO.57 The heteroaggregates formed by the intermolecular electrostatic force of attraction between the positively charged TAAPI {tetrakis(4-trimethylaminophyenyl) porphyrin} and the negatively charged CoTPPS {tetrakis(4-sulfonatophenyl) porphyrin cobalt(II)} extends the composite absorption to a wider spectral range compared to the porphyrin monomer. The loading of water soluble porphyrin enhances the hydrophilic character of the ZnO microrods, thereby facilitating their dispersion in the aqueous solution. In addition, the redox potentials of the heteroaggregates align with the energy level of ZnO to promote an electron injection from the excited state of the porphyrin into the ZnO CB and suppress the carrier recombination.57 In the sensitization process, electron transfer and recombination between the sensitizer and ZnO, together with their redox potentials, govern the kinetics of the electron injection.
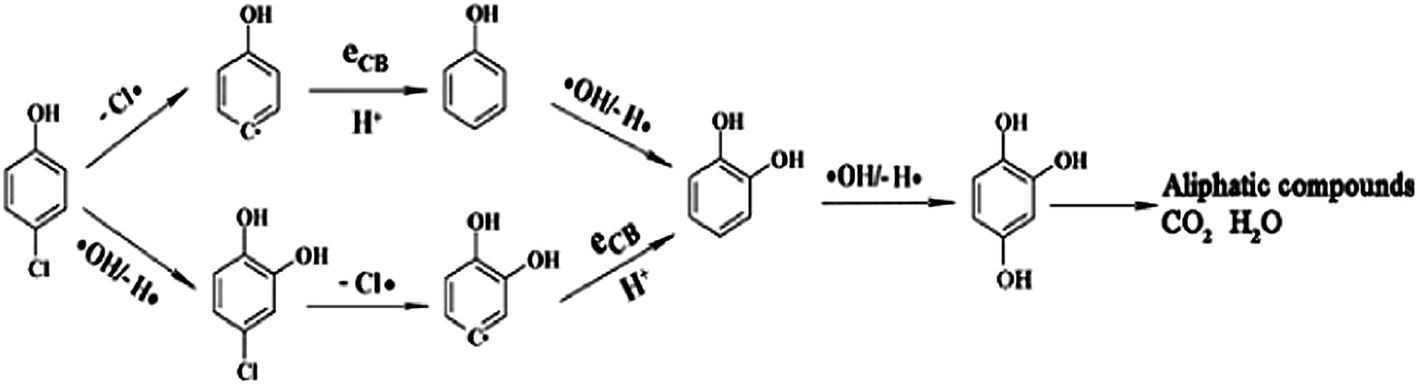
Kamat et al.16 reported a ‘sense and shoot’ approach by monitoring the quenching of the relative emission intensity of ZnO with organic compounds like 4-chlorocatechol, catechol and 4-CP. These phenolic compounds scavenge the photogenerated holes and compete with the charge carrier recombination, which is responsible for the emission properties (Fig. 1). Surprisingly, the original emission was restored following the exposure of the film to deionized water, indicating that the interaction between ZnO and the phenolic compounds was purely physisorption. The varying degree of emission quenching for these compounds arises from the differences in their adsorption and ability to scavenge holes on the ZnO surface. The experimental illustration of the increased emission during photocatalysis confirms that the emission recovery was purely associated with the degradation of the aromatic intermediates in an aqueous solution. In comparison to TiO2, the surface complexation of 4-CP on the ZnO surface via –Zn–O–Ph–Cl linkages extended the photoresponse to the visible range and accelerated the degradation rate of 4-CP.58 The PL studies indicated a slight decrease in the visible emission, suggesting that the surface complexes are mainly formed on the defect sites (oxygen vacancies), which improves the photostability of ZnO. This surface complex is formed via condensation reaction:
| | |
–Zn–OH– + OH–Ph–Cl → –Zn–O–Ph–Cl + H2O
| (15) |
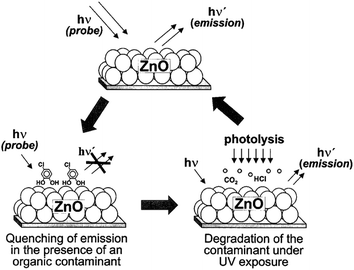 |
| | Fig. 1 Illustration of the ‘sense and shoot’ approach in photocatalysis (reprinted with permission from ref. 16; Copyright 2002 @ American Chemical Society). | |
Unlike the conventional dye sensitization process, the visible light irradiation of the above surface complex directly excites the electron from the ground state of the adsorbate to ZnO CB through the ligand to a metal charge transfer process, provided there exists a significant electronic coupling between the adsorbate orbitals and the Zn ‘d’ orbitals. Similar complexation and degradation pathways for DCP and phenol were noticed under visible light.58 The acetate-capped ZnO crystals (h-ZnO) promoted the degradation of MO and MR at pH 6, but were susceptible to corrosion at extreme pH conditions.59 In contrast, HCA/TBPA-capped ZnO (nh-ZnO) showed a weaker pH dependence for degradation and a high resistance to photocorrosion, which was attributed to its hydrophobic nature. Phosphonic acid strongly bonds to the ZnO surface via a bi/tri-dentate geometry, as opposed to an acetate ligand, which anchors weakly via a unidentate fashion. Thus, the high density of surface sites is expected to be passivated by HCA or TBPA, resulting in a lower activity. In addition, the long alkyl chain can impose a significant barrier towards the adsorption of target molecules. The intermediates formed during the degradation were retarded to a large extent on the nh-ZnO surface compared to the h-ZnO.59 The activity of colloidal ZnO for MO degradation at various calcination temperatures followed an order: 150 > 300 > 500 °C, in correlation with the monodentate-, bidentate-capped and the free acetate group, respectively. With an increase in calcination temperature, the acetate group decomposes, which leads to a larger particle size and a loss in photoactivity.60 The modification of ZnO with Co(II) acetate and trimethylsilanolate inhibited the degradation of 4-NP. This was attributed to the elimination of defect sites (oxygen vacancies) on the ZnO surface, which was crucial for photoactivity.61 Such surface modification with a suitable reagent could significantly improve the stability of ZnO. The integration of organic and inorganic compounds into semiconductor NPs is driving research into an adventurous new set of nanoscale functional architectures suitable for an enormous range of applications.
(b) Effect of crystallite size
The efficiency of ZnO synthesized by the solvothermal route (80 °C for 24 h, followed by calcination at 400 °C for 6 h) using TEAOH and methanol showed a high activity for the degradation of RhB under visible light compared to the samples prepared with ethanol and propanol solvents (Fig. 2).62 The crystallite size of ZnO increased with the length of the carbon chain of the solvent, which can be interpreted by considering the dielectric constant of methanol (32), ethanol (25) and propanol (21). In general, solvents with a low dielectric constant are likely to induce faster and uncontrolled precipitation kinetics, and also lead to the supersaturation of Zn2+ ions due to the lower solubility of zinc salts. This offers the driving force for nucleation and the growth of ZnO NPs, with reduced nucleation time and higher solid particle growth. The smaller crystallite size obtained using the methanol solvent leads to a larger surface area and quantization in the bandgap, which facilitates the easy electron injection from the excited dye to ZnO CB. Upon replacing TMAOH in the synthetic route, the crystallinity appears to be the overriding factor rather than the surface area in governing the photocatalysis. The decolorization of MO for a distinct size of ZnO followed the following order: nanometer (50 nm) > submicron (200 nm) > micron grade (1000 nm).63 This tendency was attributed to the following reasons: (i) the amount of dispersed particles per volume in the reaction solution increases, consequently improving the photon absorption ability; (ii) the increased surface area promotes the adsorption of the dye molecules on the catalyst surface; (iii) the coupling of exciton pairs is suppressed. Surprisingly, ZnO with a particle diameter of 10 nm showed a lower activity compared to its submicron-sized counterpart. Dodd et al.64 also reported that an optimal size of ∼33 nm resulted in an enhanced hydroxyl radical generation, whereas reducing the particle size (∼28 nm) lowered this tendency, as a result of the increased surface recombination.65 Casey et al.66 reported that the photoactivity significantly increased by a factor of 2 to 3 as the mean crystallite size was reduced from 100 to 20 nm. ZnO prepared by the precipitation method from zinc acetate, zinc nitrate and zinc sulfate showed a higher activity for the degradation of RhB compared to the sample obtained through the citrate method.67 In the latter case, a two-step heat treatment at 300 °C (4 h) and 500 °C (or 600 °C, 2 h) was necessary to remove the organic residues and to induce the crystallization of ZnO, whereas the former involved a single step involving calcination (500 °C, 2 h) for the nitrate or acetate precursor and 800 °C for the sulphate precursor. The additional organic residues in the citrate method originated from the formation of citrate complexes. In contrast, the precipitation method produced a crystalline surface devoid of contaminants, which was beneficial for photocatalysis. The activity of ZnO with respect to the precursors obtained via the citrate method followed the following sequence: zinc acetate > zinc nitrate > zinc sulphate, whereas those prepared through the precipitation method showed an almost similar activity. Parameters, such as the surface area, agglomeration and sintering temperature, which varied with the synthesis route and the counter anion of the zinc precursor, did not exhibit any influence on the degradation kinetics.67
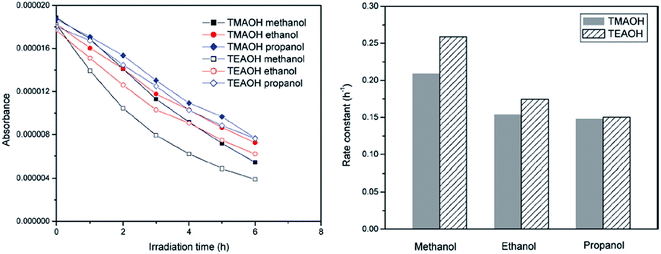 |
| | Fig. 2 Degradation kinetics of RhB (left) and the rate constants (right) with ZnO synthesized with different solvents (reprinted with permission from ref. 62; Copyright 2011 @ American Chemical Society). | |
The photoactivity does not increase monotonically with decrease in the particle size in all the cases, and the relationship between size dependence and the catalytic efficiency is close-knit. A proper size is indispensable to balance the specific surface area, crystallinity and the surface-bulk carrier recombination probability, in order to obtain a better performance.
(c) Influence of reaction pH, electron acceptors and degradation pathways
The photocatalytic activity for the degradation of Acid Brown 14 dye under natural solar light with various catalysts had the following order: ZnO > TiO2 > α-Fe2O3 > ZrO2 > CdS > WO3 > SnO2, which is mainly attributed to the absorption of more light quanta and a large fraction of the solar spectrum by ZnO.68 The photocatalytic oxidation of NO with ZnO–TiO2−xNx produced NO2 gas and HNO3 as the major products, whereas HNO2 produced as a minor compound with acids gets adsorbed on the catalyst surface under UV/visible light.69 The NOx molecule may react with a superoxide radical to form the nitrate anion and finally HNO3, which deactivates the catalyst surface.| | |
NO + 2OH → NO2 + H2O
| (16) |
| | |
NO2 + HO˙ → NO3− + H+
| (17) |
The degradation of phenol was favoured in weakly acidic or neutral pH conditions and was effective under solar light rather than artificial visible light illumination.70 The ZPC of ZnO NPs is in the range of 8–9, and the electrical property changes with the pH value of the dispersion.71 Hence, the surface charge density will be positive in an acidic or weak basic medium and negative under strong alkaline conditions. In a weak acidic solution, the phenol molecules remain undissociated to deliver a strong adsorption of phenol on the ZnO surface, resulting in an efficient degradation. In alkaline conditions, the phenolate intermediates experience an electrostatic repulsion from the ZnO surface, resulting in a poor adsorption of pollutant, and consequently a decline in the degradation rate. It is worth mentioning that commercial ZnO supplied from Merck chemicals was less susceptible to photocorrosion and retained its activity even after recycling for five subsequent runs.70 The degradation and mineralization of SA via the Langmuir–Hinshelwood mechanism was effective in neutral pH, which is attributed to the electrostatic force of attraction between the salicylate anion and the positively charged ZnO. A significant loss in activity was observed only after the fifth run of the recycling test.72 The methyl parathion degradation followed a first-order kinetics on the ZnO and TiO2 surface, with the latter being more effective in complete mineralization.73 The optimum catalyst dosage was found to be 200 mg L−1 and 500 mg L−1 for TiO2 and ZnO, respectively, with the difference attributed to the difference in their characteristics, such as crystal phase, specific surface area, grain size, density of defects, electron–hole recombination kinetics, charge carrier mobility and the surface acid–base properties. At a lower catalyst dosage, the absorption of light controls the photocatalytic process due to the limited catalyst surface, whereas the aggregation and scattering of light would be detrimental at high catalyst loading. The degradation was enhanced with peroxydisulfate as the electron scavenger through the generation of sulfate radical, which is a strong oxidizing agent74
| | |
S2O82− + e− → SO42− + SO4−˙
| (19) |
| | |
SO4−˙ + H2O → SO42− + HO˙ + H+
| (21) |
| | |
SO4−˙ + RH →…→ SO42− + CO2
| (22) |
The toxicity of the treated solution was reduced with TiO2, while the release of Zn2+ ions as a result of the photodissolution increased the toxicity for ZnO. The mineralization studies revealed the absence of phosphate ions due to insoluble Zn3(PO4)2 formation, which was otherwise present with the titania dispersion, indicating the different degradation pathways of these two metal oxides.73 The degradation of Acid Red 14 was improved in the presence of H2O2 at neutral pH under a UV-C source (100–280 nm), while the addition of ethanol suppressed the reaction kinetics, thus confirming the participation of the hydroxyl radical in the degradation mechanism.20 H2O2 served as a better electron acceptor than the dioxygen for trapping CB electrons at a faster rate or by direct photolysis to produce hydroxyl radicals.7a
| | |
H2O2 + O2−˙ → OH˙ + OH− + O2
| (24) |
| | |
H2O2 + e− → OH˙ + OH−
| (25) |
However, a higher dose of H2O2 served as a hydroxyl radical scavenger or a hole scavenger, suppressing the degradation rate. The free radicals may also recombine to form neutral species.
| | |
H2O2 + h+ → O2H˙ + H+
| (26) |
| | |
H2O2 + OH˙ → O2H˙ + H2O
| (27) |
| | |
O2H˙ + OH˙ → H2O + O2
| (28) |
In contrast, the degradation of Acid Red 18 was suppressed with H2O2, but accelerated with (NH4)2S2O8 and KBrO3. This unusual decrease was due to the low adsorption of H2O2 on the ZnO surface.74c The degradation of HD proceeded through the formation of by-products such as thiodiglycol, hemisulfar mustard, divinyl sulfide, 2-chloroethyl vinyl sulfide at the surface of ZnO NPs under dark and visible light irradiation.75 However, HD sulfoxide, HD sulfone, 1,3-dithiane, 2-chloroethanol and CH3CHO, along with hydrolysis and elimination products, were formed under sunlight and UV-A light illumination. In the former case, the degradation through elimination and surface complexation reactions played a dominant role, whereas in the latter case, photocatalytic reactions involving C–S bond cleavage, an oxidation of carbon and sulphur atoms, were observed.75 The ZnO exhibited a superior visible light activity for the degradation of Basic Blue 11 compared to TiO2.76 The rapid degradation rate under alkaline conditions (pH 10) was attributed to the better adsorption of the dye on the ZnO surface and the generation of more hydroxyl radicals in the reaction system. Interestingly, the degradation was considerably better at pH 3 compared to neutral conditions. The positively charged ZnO surface promotes the migration of electrons from interior of nanocrystals to the surface and prevents carrier recombination. The degradation mechanism was followed by both N-dealkylation and oxidation pathways (pH 9). In another study, the degradation of EV followed oxidative degradation (cleavage of the chromophore) at acidic pH and through N-de-ethylation, which lead to an N-de-ethylated EV species, along with their N-hydroxyethylated intermediates under an alkaline medium. The former proceeded via the formation of a carbon centred radical, whereas the latter was through the generation of a nitrogen centred radical.77
(d) Influence of excitation source and radical scavenger
The photocatalytic degradation of metamitron herbicide under a mixed UV-A (315–400 nm) and UV-B (280–315 nm) light source was efficient at acidic pH (2.1–4), but was inhibited with anions like carbonate and sulphate, whereas chloride showed a weak influence.78 The surface sites available at the ZnO–metamitron interface for adsorption and electron transfer between the catalyst and substrate were blocked by the anions, which are very resistant towards oxidation. These deposited anions deactivate the catalyst surface towards the targeted pollutant and also scavenge the hydroxyl radicals in the solution. The generated carbonate or sulphate radicals, although they behave as oxidants, have lower oxidation potential compared to hydroxyl radicals.74,78| | |
OH˙ + CO32− → OH− + CO3−˙
| (29) |
| | |
OH˙ + HCO3− → H2O + CO3−˙
| (30) |
| | |
OH˙ + Cl− → Cl˙ + OH−
| (33) |
| | |
SO42− + OH˙ → SO4−˙ + OH−
| (35) |
The ZnO effectively oxidized the iodide ion under the influence of the increased air flow-rate and decreased water content in the reaction medium.79 The high water content promotes an indirect recombination via the trapping of the CB electrons by the hydroxyl radical. The hole reacts with the adsorbed iodide ion to form an iodine atom and further reacts with iodide ions to produce I2−, which then undergoes a disproportionate reaction to form tri-iodide and iodide ions.
The coupling of the photo Electro Fenton process with the immobilized ZnO was effective for decolorising Basic Yellow 28 compared to the individual process at pH 3 using CNT-poly tetrafluoroethylene cathode and a Pt sheet anode in the presence of Na2SO4 electrolyte.80 This is a consequence of the enhanced generation of hydroxyl radicals from both electrocatalysis and the photo Fenton process.
| | |
At anode: H2O → 1/2O2 + 2H+ + 2e−
| (39) |
| | |
At cathode: O2 + 2H+ + 2e− → H2O2
| (40) |
| | |
Fe2+ + H2O2 → Fe3+ + HO˙ + H2O
| (41) |
The excitation source UV-C was found to be more powerful for photoelectrochemical degradation compared to UV-B and UV-A, which is due to the production of hydroxyl radical, which arises from the photolysis of H2O2. The initial step of the mechanism of degradation involves the hydroxylation of the dye, leading to 1,2,3,3-tetramethyl indoline and 4-methoxy benzenamine, followed by a subsequent attack of hydroxyl radical, which leads to the formation of simple aliphatic acids.80
(e) Defect-mediated photocatalysis
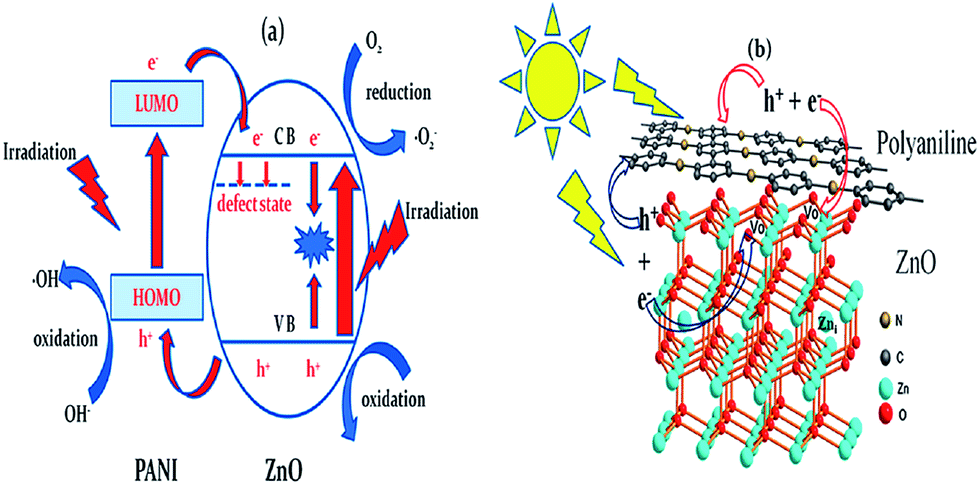
The use of nanoscopic ZnO embedded in Nafion membranes resulted in a faster degradation of RhB compared to ZnO powder.81 Initially, a blank Nafion was soaked overnight in an aqueous zinc nitrate solution to facilitate Zn2+ ion exchange. Then, the Zn2+–Nafion was soaked in C2H5OH (ZNE) or NaOH (ZNA) to introduce hydroxyl anions. The Zn(OH)2 formed is unstable in hydrophilic cavities (reverse micelles), which either dehydrolyzes it to give ZnO or it reacts with a hydroxyl anion to form the growth units of [Zn(OH)4]2−, followed by its polymerization, resulting in a ZnO nuclei. ZNE showed a superior activity and an excellent photostability compared to ZNA, although both the samples had comparable particle size, crystal structure, morphology and surface area. The PL evidenced a large concentration of oxygen defects, i.e. oxygen vacancy/interstitial oxygen with ZNE, as a result of the rapid crystal growth. Under UV excitation, the electrons are trapped by oxygen vacancies, whereas the holes are captured at interstitial oxygen, thereby restraining the recombination (Fig. 3). Ethanol swelled the membrane film and made the entrance of hydroxide ions easy and uniform. This modified ZnO did not lose its activity even after recycling ten times. The concentration of Zn2+ ions in the solution after the photoreaction was ∼0.0148 mg L−1 for ZnO–Nafion film, whereas it was ∼0.2318 mg L−1 with the commercial ZnO.82 These Nafion membranes form an excellent support for semiconductor nanocrystals because of the improved chemical stability, exceptional mechanical strength and high optical quality. The advantages of the Nafion-templating approach to synthesize metal oxide NPs are as follows: (i) it provides a stable matrix to prevent the agglomeration and corrosion of embedded NPs; (ii) NPs embedded are easy to operate and can be recycled for catalytic purpose; (iii) the Nafion membrane has a small absorbance in the UV-visible region and their hydrophilic cavities and the channels possess a strong polarity and excellent ion-exchange properties.83–85 These features enhance the adsorptive capacity of the materials, leading to the enrichment of pollutants on the catalyst surface. The hybrid effect between the 1.0% monomolecular-layers PANI dispersed on the ZnO surface inhibited photocorossion and also improved the activity for MB degradation under UV/visible light.86 The degradation was quenched with EDTA (a hole scavenger) for the ZnO system, and with EDTA or TBA (a hydroxyl radical scavenger) for the ZnO–PANI system. This indicates that the holes were the dominant oxidising agent in the former case, whereas both holes and the hydroxyl radical contribute to photocatalysis in the latter under UV light. In contrast, hydroxyl radicals predominately participated in the degradation of MB for the composite under visible light. Under UV light, VB holes transfer to the HOMO of PANI, and then migrate to the catalyst surface, thereby directly oxidizing the contaminants. Under visible light, PANI absorbs the incident photons, and the excited electron is transferred from its LUMO to the ZnO CB.86 The coating of PANI via the cold-plasma treatment technique intentionally Introduces defects (i.e. oxygen vacancies and interstitial zinc) and enhances the activity of PANI–ZnO for MO and 4-CP degradation.87 In addition, PANI effectively stabilized these defects on the surface of ZnO even after prolonged UV illumination. The surface oxygen vacancy traps the electron from ZnO CB and the LUMO of PANI to suppress the charge recombination process (Fig. 4). In addition, the increased donor density due to the presence of Zni ( and
and  ) and Vo (
) and Vo ( and
and  ) improves the charge transport and shifts the Fermi level towards the CB, which facilitates charge separation at the semiconductor–electrolyte interface and ultimately supplements the photocatalytic efficiency. The PANI behaves like a p-type semiconductor, which is an excellent hole transporting material and has become a better choice for preparing organic–inorganic hybrid photocatalysts. The oxygen deficient ZnO obtained from the calcination of ZnO2 was found to be efficient for the decomposition of DCP under visible light.88 The oxygen defects are introduced during the decomposition of O22− and were tailored by heat treatment at various temperatures. At high calcination temperatures (800 °C, 2 h), the pale yellow colour of oxygen-deficient ZnO was transformed to a white color. However, annealing under an argon atmosphere preserves the visible light absorption edge, as the oxygen vacancies are retained under an oxygen free condition. The impurity states associated with these high oxygen vacancies become more delocalized and overlap with the VB edge, and this raises the position of VB, making the ZnO more efficient for visible light absorption. The photocatalytic activity of oxygen-deficient ZnO gradually decreased with increases in the calcination temperature from 400 to 800 °C, which is in agreement with the concentration of oxygen vacancies. However, very low oxygen vacancies did not exert any impact on the optical properties. In contrast, oxygen deficiency was not observed for ZnO prepared by the calcination of Zn(OH)2.88 Thus, the electronic and optical properties of ZnO are strongly dependent on the nature and concentration of the defects generated during the crystallization process. For ZnO-based photocatalysis, excitation at high energy wavelength (254 nm) triggers both photolysis and photocatalysis;89 however, low energy (340 nm) induces only photocatalysis, due to its close association with the bandgap absorption. This indicates an increase in the surface or near surface reactions with the latter excitation source, and that the former operates by a MVK type mechanism, in which the oxygen from the crystal lattice is removed and used in the oxidation reaction. On the basis of a series of experiments, Ali et al.90 reported that a different degradation mechanism of MB operated for different ZnO films at different excitation wavelengths, with competition between the two distinctive mechanisms: conventional redox radicals and lattice oxygen driven oxidation. Surface photocatalyzed radical formation was prominent for highly aligned and more crystalline morphologies of ZnO with plentiful oxygen. However, the lattice oxygen mediated photodegradation was significant for less aligned and more amorphous morphologies with more defects. The high energy associated with 254 nm allows the activation energy barrier to overcome for lattice oxygen abstraction; furthermore, a high number of lattice defects lower the overall activation energy. However, the MVK type mechanism deactivates the catalyst surface, thereby inhibiting the redox reactions.90 Despite the debate concerning the exact role of defects in photocatalysis, it is generally accepted that the surface defects (states) act as shallow trapping sites, whereas the localized bulk defects promote the recombination process. The defects in mediated photocatalysis are vital for realizing the photocatalysis pathways, as these defects not only modulate the photoactivity, but are also inherent in pristine metal oxides.
) improves the charge transport and shifts the Fermi level towards the CB, which facilitates charge separation at the semiconductor–electrolyte interface and ultimately supplements the photocatalytic efficiency. The PANI behaves like a p-type semiconductor, which is an excellent hole transporting material and has become a better choice for preparing organic–inorganic hybrid photocatalysts. The oxygen deficient ZnO obtained from the calcination of ZnO2 was found to be efficient for the decomposition of DCP under visible light.88 The oxygen defects are introduced during the decomposition of O22− and were tailored by heat treatment at various temperatures. At high calcination temperatures (800 °C, 2 h), the pale yellow colour of oxygen-deficient ZnO was transformed to a white color. However, annealing under an argon atmosphere preserves the visible light absorption edge, as the oxygen vacancies are retained under an oxygen free condition. The impurity states associated with these high oxygen vacancies become more delocalized and overlap with the VB edge, and this raises the position of VB, making the ZnO more efficient for visible light absorption. The photocatalytic activity of oxygen-deficient ZnO gradually decreased with increases in the calcination temperature from 400 to 800 °C, which is in agreement with the concentration of oxygen vacancies. However, very low oxygen vacancies did not exert any impact on the optical properties. In contrast, oxygen deficiency was not observed for ZnO prepared by the calcination of Zn(OH)2.88 Thus, the electronic and optical properties of ZnO are strongly dependent on the nature and concentration of the defects generated during the crystallization process. For ZnO-based photocatalysis, excitation at high energy wavelength (254 nm) triggers both photolysis and photocatalysis;89 however, low energy (340 nm) induces only photocatalysis, due to its close association with the bandgap absorption. This indicates an increase in the surface or near surface reactions with the latter excitation source, and that the former operates by a MVK type mechanism, in which the oxygen from the crystal lattice is removed and used in the oxidation reaction. On the basis of a series of experiments, Ali et al.90 reported that a different degradation mechanism of MB operated for different ZnO films at different excitation wavelengths, with competition between the two distinctive mechanisms: conventional redox radicals and lattice oxygen driven oxidation. Surface photocatalyzed radical formation was prominent for highly aligned and more crystalline morphologies of ZnO with plentiful oxygen. However, the lattice oxygen mediated photodegradation was significant for less aligned and more amorphous morphologies with more defects. The high energy associated with 254 nm allows the activation energy barrier to overcome for lattice oxygen abstraction; furthermore, a high number of lattice defects lower the overall activation energy. However, the MVK type mechanism deactivates the catalyst surface, thereby inhibiting the redox reactions.90 Despite the debate concerning the exact role of defects in photocatalysis, it is generally accepted that the surface defects (states) act as shallow trapping sites, whereas the localized bulk defects promote the recombination process. The defects in mediated photocatalysis are vital for realizing the photocatalysis pathways, as these defects not only modulate the photoactivity, but are also inherent in pristine metal oxides.
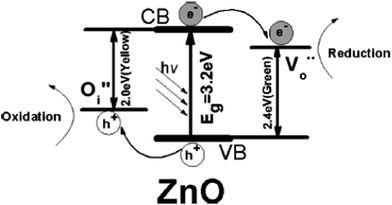 |
| | Fig. 3 Band structure and charge transfer pathways of ZnO nanocrystal with oxygen defects (reprinted with permission from ref. 81; Copyright 2009 @ American Chemical Society). | |
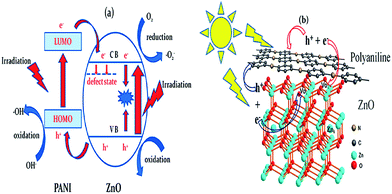 |
| | Fig. 4 (a) Proposed photocatalytic mechanism; (b) atomic level illustration of ZnO–PANI hybrids (reprinted with permission from ref. 87; Copyright 2014 @ American Chemical Society). | |
On the basis of the above results, it can be surmised that heterogeneous photocatalysis is a delicate function of catalyst dosage, particle size, surface acid–base properties, defect density, surface anchored groups, photocatalyst stability, substrate concentration and their redox levels, pollutants' molecular structures, the presence of electron scavengers other than oxygen, the presence of inorganic electrolytes, the formation of active free radicals, solution pH, the affinity of pollutants and intermediates to react with free radicals, degradation pathways, intensity and the wavelength of the excitation source.
3. Morphological dependence of ZnO on the photocatalytic activity
Further improvements in the photoactivity of ZnO can be realized by providing a suitable geometric structure for effective carrier transfer pathways. ZnO has the richest morphologies, which are very complex and diversified and can be easily manipulated with a desirable structure, as well as allow for rational tailoring of the surface to volume ratios. The capability to control the particle morphology and understanding the surface signatures of ZnO governed by the particle size and shape may be vital for constructing nanoscale electronic devices. The growth of metal oxides in an aqueous solution is influenced by variable parameters such as temperature, precursor chemistry, chelating agents, solvents, precursor concentration, mineralizers, inorganic electrolytes, templates and the pH of the solution.91–94 Zn(II) exists as Zn2+, Zn(OH)2, Zn(OH)3− and Zn(OH)42− in aqueous solution, with their concentration ratio depending on the pH of the reaction medium. In general, a relatively large quantity of Zn(OH)42− under alkaline conditions will act as seeds for the nucleation of ZnO growth units.
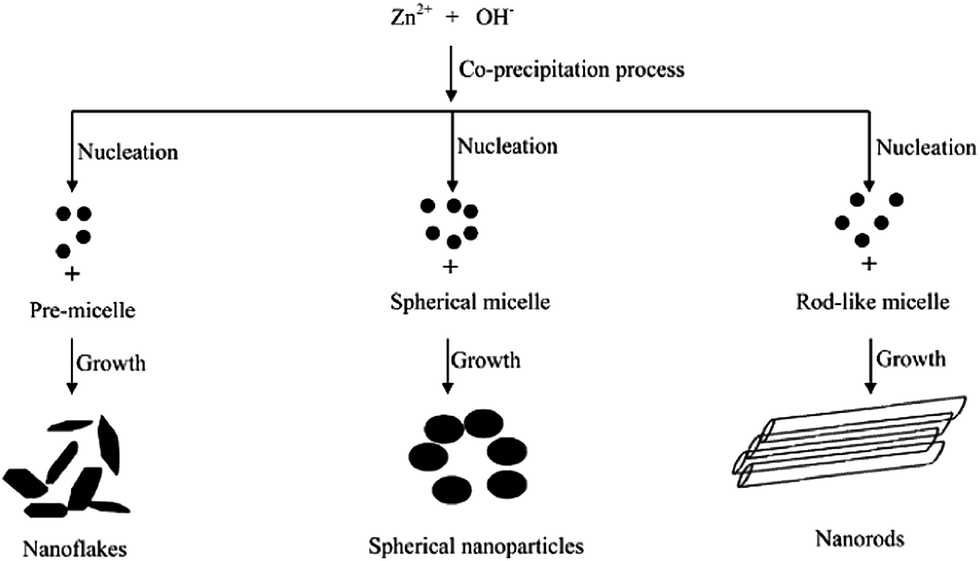
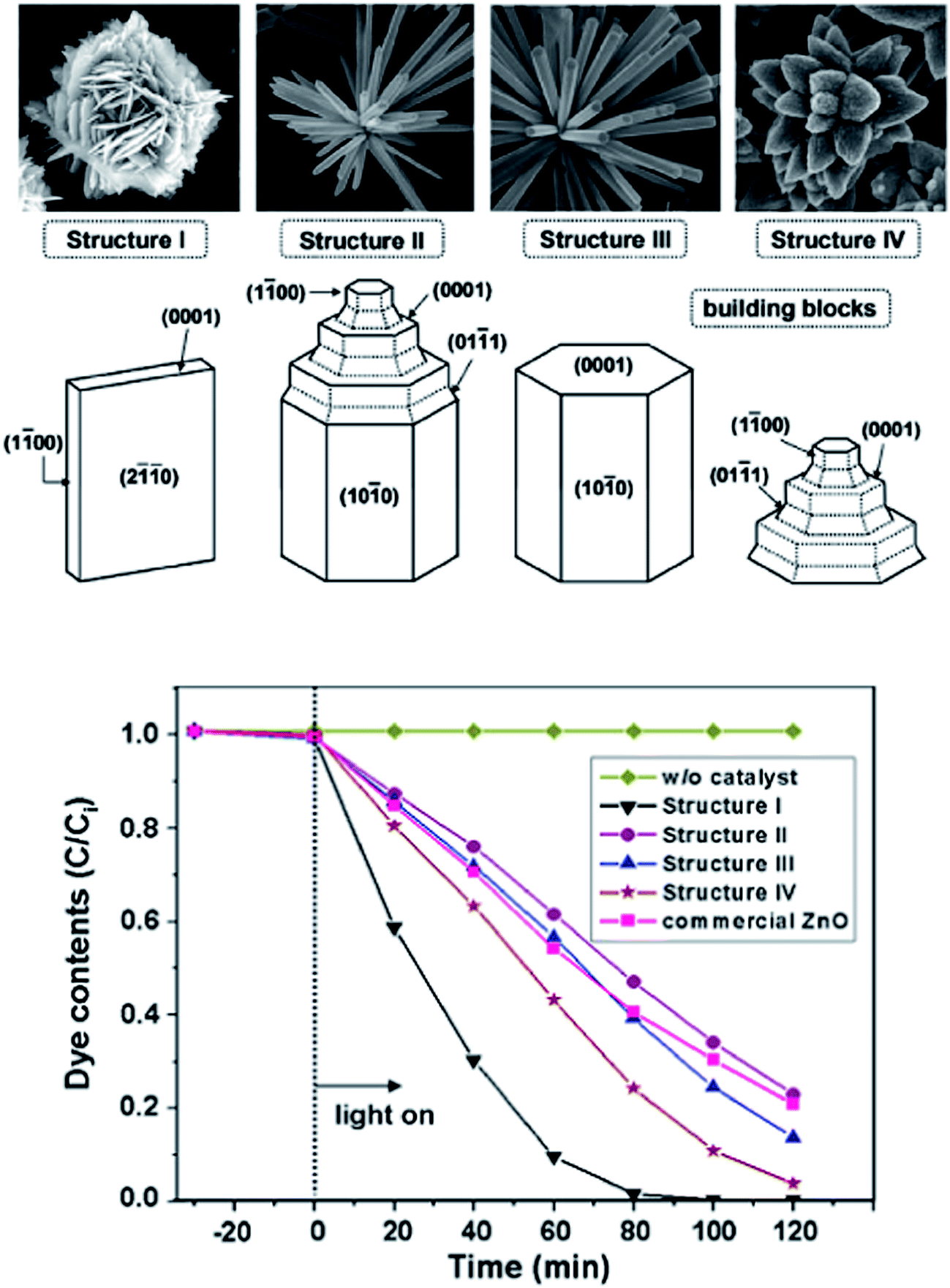
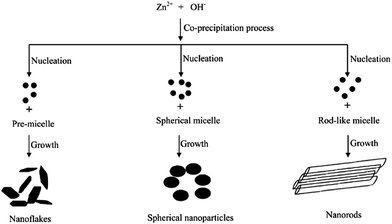
The photocatalytic activity of ZnO towards resorcinol degradation follows the order: spherical shape > rod-like > flake-like. The activity of the sperhical shape was due to the formation of a non-faceted morphology comprising a high surface area, small crystalline size distribution, and a high concentration of electron (oxygen vacancies) and hole traps (oxygen interstitials).95 The appreciable activity of the rod-like morphology was attributed to the presence of a zinc-terminated {0001} and an oxygen terminated {000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) } polar face with a high surface energy. However, the formation of smooth {1101} and {1010} facets without a high surface energy for the flake-like morphology resulted in poor activity. These morphologies could be tuned by changing the capping agent, i.e. the Triton X-100 concentration from PMC to CMC during the synthesis step (Fig. 5).96 In the case of PMC (2.1 × 10−4 mol L−1), a high yield of ZnO nanoflakes was obtained. During the particle formation, monomers (the capping agent) adsorb onto the preferred crystallographic planes and alter the growth kinetics. Thus, growth along all the preferred directions will be retarded to produce a nanoflake-like morphology during the nucleation stage. The nanoflakes are aggregated due to their nanoscale forces. The spherical micelles are formed at their first CMC (3.2 × 10−4 mol L−1), leading to the formation of spherical NPs. Due to the smaller dimension of the spherical-shaped granular particles, the polar fields generated in each particle are weaker.97 Consequently, a lower propensity of agglomeration between the single particles leads to the formation of an unagglomerated assembly of NPs. The second CMC (1.3 × 10−3 mol L−1) indicates the structural transition from a spherical micelle to rod-like ones to facilitate the formation of the NRs. The collective behaviour of the van der Waals forces and the electrostatic interaction support the self-aggregation of the ZnO NRs.98
} polar face with a high surface energy. However, the formation of smooth {1101} and {1010} facets without a high surface energy for the flake-like morphology resulted in poor activity. These morphologies could be tuned by changing the capping agent, i.e. the Triton X-100 concentration from PMC to CMC during the synthesis step (Fig. 5).96 In the case of PMC (2.1 × 10−4 mol L−1), a high yield of ZnO nanoflakes was obtained. During the particle formation, monomers (the capping agent) adsorb onto the preferred crystallographic planes and alter the growth kinetics. Thus, growth along all the preferred directions will be retarded to produce a nanoflake-like morphology during the nucleation stage. The nanoflakes are aggregated due to their nanoscale forces. The spherical micelles are formed at their first CMC (3.2 × 10−4 mol L−1), leading to the formation of spherical NPs. Due to the smaller dimension of the spherical-shaped granular particles, the polar fields generated in each particle are weaker.97 Consequently, a lower propensity of agglomeration between the single particles leads to the formation of an unagglomerated assembly of NPs. The second CMC (1.3 × 10−3 mol L−1) indicates the structural transition from a spherical micelle to rod-like ones to facilitate the formation of the NRs. The collective behaviour of the van der Waals forces and the electrostatic interaction support the self-aggregation of the ZnO NRs.98
 |
| | Fig. 5 Mechanism of formation of ZnO nanostructures with different morphologies (reprinted with permission from ref. 95; Copyright 2012 @ American Chemical Society). | |
Flower-like ZnO thin film (160 nm thick) deposited on the FTO substrate via spray pyrolysis (400 °C) was photoelectrochemically active for the degradation of organic pollutants, such as toluene, SA and 4-CP under natural solar light due to the large content of surface oxygen vacancies and the high surface area.99 Mesoporous ZnO prepared by a solution combustion route using oxalic acid as the fuel had higher activity for the degradation of OG dye compared to ZnO prepared with other fuels such as citric acid, dextrose, glycine, oxalyl dihydrazide, and urea.100 The furnace temperature was varied between 350–450 °C depending on the number of carbon atoms in fuel and also on the nature of groups. The equivalence ratio (Φ), defined as the absolute value of the ratio of oxidizer valency to reducer valency, was <1 for citric acid and dextrose, and greater than unity for glycine, oxalic acid and urea, equals to unity for oxalyl dihydrazide fuel. ZnO with a low crystallite size and a high strain was obtained for Φ < 1, similarly due to the smoldering combustion, which arises at a combustion temperature of <650 °C.101 This results in an improper crystal growth favouring the formation of small crystals. For Φ ≥ 1, the maximum amount of heat will be produced, helping to render proper crystal growth. The morphology of ZnO was agglomerated, irregular, spherical, cylindrical, and flower-like for citric acid, dextrose, glycine, oxalyl dihydrazide, oxalic acid and urea, respectively. The surface area of the catalyst had no correlation with its activity in this study.
The nanostar ZnO assembled from NRs obtained under hydrothermal conditions (160 °C, 2 h) with gelatin under aqueous ammonia was active for MO degradation. The surface and capacious interspaces of the nanostars provided more opportunity for the diffusion and mass transportation of MO and hydroxyl radicals during the photocatalytic reactions.102 However, NSs, microrods, microsheets and hexagonal prisms were observed using NaOH, ethanediamine, urea and HMT, respectively. The optimum amount of gelatin to obtain nanostars was found to be 0.02 g and high content (0.036 g) favored the formation of peanut-like structures. The carboxylic groups of gelatin bind to metallic ions via coordination or electrostatic interactions to form complexes such as gelatin–Zn(NH3)42+ and gelatin–Zn(OH)42−. The gelatin biomolecules are mainly dispersed as random coils in the solution and behave as soft biotemplates to tailor the shape of ZnO NPs under the reaction conditions. The ZnO nuclei generated from the above complexes grow in free space and aggregate through the orientation and alignment to decrease the free energy, forming NRs in the initial stages. Then, the gelatin confines the growth of ZnO NRs and transforms them into star-like morphologies via the Ostwald ripening process.102 The ZnO NFs (78 nm) were active for the degradation of RhB, Amido Black 10B and Acid Fuchsin compared to the ZnO NPs (30 nm) under visible light, although the latter had a smaller particle size.103 The porous structure of the nanofibrous mats improved the contacting surface areas between the catalyst and the dye molecules, whereas an aggregation of NPs in solution lowered its efficiency. The ZnO TPs showed a high efficiency for MO, Acid Orange 7, MB and R6G degradation compared to the NPs, although a lower concentration of hydroxyl radicals and a reactive oxygen radical were observed for the TPs structure.104 Due to the low concentration of non-radiative defects, PL decay was the longest for the TPs, whereas a rapid decay of luminescence was observed for the small size NPs, attributed to the non-radiative quenching at the surface defects/impurities. In another study, the abundant surface states and the high surface-to-volume ratio of the ZnO TPs contributed to them exhibiting high activity for RhB degradation.105 The EDS confirmed the oxygen deficiencies in the TPs structure (atomic ratio of Zn and O is 51.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48.8), which then serve as reactive electron capture centres. Both PL and photosensitivity measurements revealed a high density of surface states, which would be beneficial for photocatalytic and device fabrication.105 The photoactivity of ZnO TPs surpassed the irregular-shaped ZnO NPs towards MO degradation, which is attributed to their 3D branched morphology, which resists aggregation during photocatalytic reactions.63 From a geometrical point of view, the TPs nanostructures, which assembled into the 3D networks may act as ideal candidates for harvesting the maximum number of photons from a light source. In the reaction solution, the TPs provide all the contactable surfaces, leading to a spatial stearic effect against the dense agglomeration, owing to their unique structure with four arms growing from one centre and multiple pathways for diffusion of the reactants and the products.
:48.8), which then serve as reactive electron capture centres. Both PL and photosensitivity measurements revealed a high density of surface states, which would be beneficial for photocatalytic and device fabrication.105 The photoactivity of ZnO TPs surpassed the irregular-shaped ZnO NPs towards MO degradation, which is attributed to their 3D branched morphology, which resists aggregation during photocatalytic reactions.63 From a geometrical point of view, the TPs nanostructures, which assembled into the 3D networks may act as ideal candidates for harvesting the maximum number of photons from a light source. In the reaction solution, the TPs provide all the contactable surfaces, leading to a spatial stearic effect against the dense agglomeration, owing to their unique structure with four arms growing from one centre and multiple pathways for diffusion of the reactants and the products.
Highly oriented ZnO NRA synthesized on zinc foil exhibited the best activity for 4-CP degradation compared to a ZnO NR grown on a titanium substrate. A cross-sectional SEM observation revealed that the rods with typical lengths of about 500 nm were well separated from each other but densely aligned, and that a preferential growth occurs along the c-axis perpendicular to the substrate. In contrast, the hexagonal rods were packed in a disordered manner for ZnO grown on Ti-foil. In this study, the zinc foil was used both as a zinc ion source and as a substrate for the direct growth of NRA.106 The unique surface features of well-aligned NRs and an excess appearance of polar-{0001} facets in the NRA serve as the most active sites for photocatalysis.107 In addition, the NRA exhibited remarkable activity with little loss even after a fifth run without any change in morphology. Enhanced interfacial charge transfer can be achieved in such a 1D material because the delocalization of the carriers is increased owing to their free movement throughout the length of the crystal with minor resistance. Thus, the occurrence of surface trapping states is reduced and a more efficient charge separation is ensured.108
Porous ZnO nanopyramids with a greater density of basic sites exhibited a high rate of adsorption and degradation for acidic dyes, such as Fluorescein, Acid Green, Acid Blue and the Acid Black, compared to ZnO nanopyramids without pores and to ZnO mesoporous ellipsoids, but remained insensitive for the adsorption of basic dyes (RhB and Basic Red).109 Thus, their integration in adsorption and photocatalysis facilitates the porous nanopyramids to contact and react with more dyes per unit time. This ZnO had unique structural features, such as high specific surface area (127.7 m2 g−1), uniform nanopores (4.7 nm) and large pore volume, and was assembled from 4.7 nm ZnO nanocrystal building blocks with an oriented attachment without any template assistance.109 Meso-macroporous ZnO NSs prepared by the one-step polyol refluxing process (198 °C, 4 h) rapidly degraded MB compared to ZnO dense NSs without any porous structure and/or ZnO nanospheres.110 The EG (polyol) in the synthesis step played a crucial role in the formation of NSs; (i) it has a relatively high dielectric constant and most of the metal species are soluble, providing a suitable growth conditions for NSs; (ii) it lowers the hydrolysis rate of the metal oxides; (iii) it also serves as a reducing agent.111,112 In contrast, the presence of nitrilotriacetic acid and PVP in the reaction system results in dense NSs and nanospheres, respectively.110 The photoactivity of ZnO hollow spheres for the degradation of RhB under hydrothermal conditions (180 °C, 24 h) of glucose–ZnCl2 mixtures increased with increase in the molar ratio of glucose to zinc ions up to 15, which is attributed to the combined effects of multiple factors, such as the porous structure, enhancement in the carrier redox potential and the formation of a bimodal meso-macroporous structure.113 The formation of hollow spheres proceeded through three steps: (i) dehydration of glucose with a subsequent carbonization, resulting in the formation of carbon spheres with hydrophilic functional groups such as –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –OH; (ii) entrapment of Zn2+ ions into the hydrophilic shell via coordination/electrostatic interactions; (iii) removal of carbon spheres, densification and cross-linking of incorporating metal cations in the layer via heat treatment, which leads to the hollow sphere formation. In addition, the hollow spheres were stable against photocorrosion at neutral pH, and could be easily separated from the slurry system by simple filtration or sedimentation, due to their large weight, weak Brownian motion and good mobility. These hollow spheres possess an unusual hierarchical nanoporous structure, which allows more efficient transport for the reactant molecules to reach the active sites on the framework walls by enhancing the photocatalytic efficiency. Moreover, hollow spheres allow multiple reflections of the UV-visible light within the interior cavity, which facilitates more efficient usage of a light source.114 This activity was reduced after grinding the hollow spheres, suggesting that solvent entrapment and sequestration within the enclosed spheres were beneficial for the photocatalysis. The porous structure of the ZnO plates (sintered at 700 °C, 1 h) was efficient for Reactive Orange 16 and the degradation of Reactive Red 180 dye.115 The ZnO plates retained their photoactivity after the recycling tests when placed under darkness.116 The illuminated surface became less negatively charged during the photocatalytic process, when positive holes are drawn to the illuminated surface by the layer of space charge. Conversely, the crystal attains a negative charge when the catalyst is placed under darkness. In order to balance the charge, surface diffusion take place and restrains the hole trapping rate under UV illumination, thus retaining the activity instead of deactivating it. Compared to the powder form, the plate structure significantly reduces the cost and time required for the catalyst removal from the suspension, thereby allowing better recycling and reuse of the plates.115 The rate constants for the degradation of MO were 1.03, 1.73 and 1.96 h−1 for ZnO solid nanospheres, hollow nanospheres and yolk–shell nanospheres, respectively, which were in exact (linear) correlation with the intensity of the visible light emission in the PL spectra.117 The evolution of morphology for the zinc citrate microspheres with aging time had the following pathway: solid microspheres to hollow microspheres via yolk–shell microspheres, according to the Ostwald ripening mechanism associated with the progressive redistribution of matter from the cores to the shells of the microspheres, as the cores have a higher energy due to their larger curvatures compared to the outer shells.118 Thus, the inner cores gradually dissolve into shells and are consumed and shrink with an increase in aging time. Simultaneously, zinc citrate redeposits on the outer shell, which increases the shell thickness, leading to yolk–shell microspheres. Thus, the growth of the shell and shrinkage of the cores continues with aging time. The core eventually disappears after aging for 12 h, leaving behind a spherical hollow sphere (Fig. 6). This ripening process is a classic phenomenon in particle growth, wherein the growth of larger particles takes place at the expense of smaller particles due to the higher solubility of the latter.118 Novel ZnO composed of a core (pyramid)–shell (nanosheet) composite with open and porous nanostructural surface layers exhibited excellent activity and durability for the degradation of MO against NPs, NSs and nanoneedles.119 Moreover, the higher redox potential of size-quantized NSs (∼10 nm) standing on the micro-sized conical-shaped particles promotes electron transfer from the conduction band of a nanosheet with a high electric potential to those of a core-part micropyramid with a low electric potential.120 The vertical and net-like or grid-like arrangement of the NSs, as well as the conical shape of the ZnO micro/nanostructures, successfully avoid aggregation and preserve its large specific surface area. On the basis of the experimental observations, it was proposed that the initial step involves the formation of ZnO hexagonal pyramid-like nanocrystals followed by the build-up of a NSs network on the facets of pre-formed ZnO microcrystals. This two-step growth mode was favoured only for a certain release rate of Zn2+ ions (in this study, Zn foil was used as the zinc source and the ratio of Vdistilled water/Vehtylene diamine = 3:37 to 1:7) because a high growth rate leads to a single morphology (i.e. one-step growth) and a low growth rate results in quasi-equilibrium growth, leading to vertically arranged NRAs on a Zn foil. In addition, replacing the Zn foil with zinc nitrate results in irregular shaped particles.119 The high performance of ZnO NT arrays compared to ZnO NRA for NOx degradation was attributed to the unique surface features of its well-aligned structure. The NTs differ from NRs from the prospect of having a hollow cavity structure.69 Noticeably, the NT arrays with outer and inner surfaces have a relatively large surface area (∼twice) compared to the NRA with the same length and the diameter. The peculiar tubular structure provides more interfaces for NO decomposition, thereby increasing the activity of the NT arrays. In addition, incident light is preferably trapped in the NT arrays and reduplicatively gets absorbed, both inside and outside of the tubes, thus facilitating easy carrier generation.69
O and –OH; (ii) entrapment of Zn2+ ions into the hydrophilic shell via coordination/electrostatic interactions; (iii) removal of carbon spheres, densification and cross-linking of incorporating metal cations in the layer via heat treatment, which leads to the hollow sphere formation. In addition, the hollow spheres were stable against photocorrosion at neutral pH, and could be easily separated from the slurry system by simple filtration or sedimentation, due to their large weight, weak Brownian motion and good mobility. These hollow spheres possess an unusual hierarchical nanoporous structure, which allows more efficient transport for the reactant molecules to reach the active sites on the framework walls by enhancing the photocatalytic efficiency. Moreover, hollow spheres allow multiple reflections of the UV-visible light within the interior cavity, which facilitates more efficient usage of a light source.114 This activity was reduced after grinding the hollow spheres, suggesting that solvent entrapment and sequestration within the enclosed spheres were beneficial for the photocatalysis. The porous structure of the ZnO plates (sintered at 700 °C, 1 h) was efficient for Reactive Orange 16 and the degradation of Reactive Red 180 dye.115 The ZnO plates retained their photoactivity after the recycling tests when placed under darkness.116 The illuminated surface became less negatively charged during the photocatalytic process, when positive holes are drawn to the illuminated surface by the layer of space charge. Conversely, the crystal attains a negative charge when the catalyst is placed under darkness. In order to balance the charge, surface diffusion take place and restrains the hole trapping rate under UV illumination, thus retaining the activity instead of deactivating it. Compared to the powder form, the plate structure significantly reduces the cost and time required for the catalyst removal from the suspension, thereby allowing better recycling and reuse of the plates.115 The rate constants for the degradation of MO were 1.03, 1.73 and 1.96 h−1 for ZnO solid nanospheres, hollow nanospheres and yolk–shell nanospheres, respectively, which were in exact (linear) correlation with the intensity of the visible light emission in the PL spectra.117 The evolution of morphology for the zinc citrate microspheres with aging time had the following pathway: solid microspheres to hollow microspheres via yolk–shell microspheres, according to the Ostwald ripening mechanism associated with the progressive redistribution of matter from the cores to the shells of the microspheres, as the cores have a higher energy due to their larger curvatures compared to the outer shells.118 Thus, the inner cores gradually dissolve into shells and are consumed and shrink with an increase in aging time. Simultaneously, zinc citrate redeposits on the outer shell, which increases the shell thickness, leading to yolk–shell microspheres. Thus, the growth of the shell and shrinkage of the cores continues with aging time. The core eventually disappears after aging for 12 h, leaving behind a spherical hollow sphere (Fig. 6). This ripening process is a classic phenomenon in particle growth, wherein the growth of larger particles takes place at the expense of smaller particles due to the higher solubility of the latter.118 Novel ZnO composed of a core (pyramid)–shell (nanosheet) composite with open and porous nanostructural surface layers exhibited excellent activity and durability for the degradation of MO against NPs, NSs and nanoneedles.119 Moreover, the higher redox potential of size-quantized NSs (∼10 nm) standing on the micro-sized conical-shaped particles promotes electron transfer from the conduction band of a nanosheet with a high electric potential to those of a core-part micropyramid with a low electric potential.120 The vertical and net-like or grid-like arrangement of the NSs, as well as the conical shape of the ZnO micro/nanostructures, successfully avoid aggregation and preserve its large specific surface area. On the basis of the experimental observations, it was proposed that the initial step involves the formation of ZnO hexagonal pyramid-like nanocrystals followed by the build-up of a NSs network on the facets of pre-formed ZnO microcrystals. This two-step growth mode was favoured only for a certain release rate of Zn2+ ions (in this study, Zn foil was used as the zinc source and the ratio of Vdistilled water/Vehtylene diamine = 3:37 to 1:7) because a high growth rate leads to a single morphology (i.e. one-step growth) and a low growth rate results in quasi-equilibrium growth, leading to vertically arranged NRAs on a Zn foil. In addition, replacing the Zn foil with zinc nitrate results in irregular shaped particles.119 The high performance of ZnO NT arrays compared to ZnO NRA for NOx degradation was attributed to the unique surface features of its well-aligned structure. The NTs differ from NRs from the prospect of having a hollow cavity structure.69 Noticeably, the NT arrays with outer and inner surfaces have a relatively large surface area (∼twice) compared to the NRA with the same length and the diameter. The peculiar tubular structure provides more interfaces for NO decomposition, thereby increasing the activity of the NT arrays. In addition, incident light is preferably trapped in the NT arrays and reduplicatively gets absorbed, both inside and outside of the tubes, thus facilitating easy carrier generation.69
 |
| | Fig. 6 Morphology evolution for zinc citrate microspheres with aging time (reprinted with permission from ref. 117; Copyright 2012 @ Royal Society of Chemistry). | |
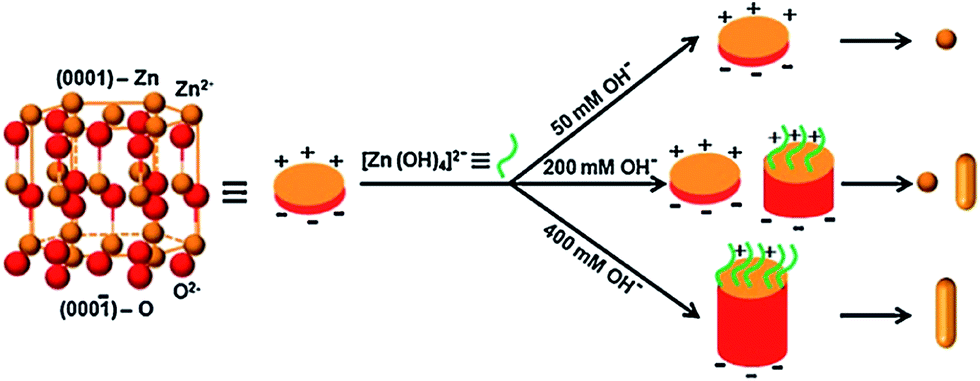
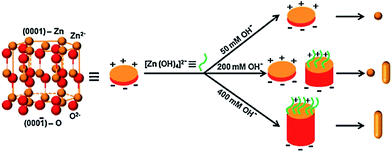
The spherical particles of the ZnO increased its activity for the degradation of MB compared to NRs and a mixed morphology of particles and rods, which was attributed to the presence of a large amount of oxygen vacancies, which trap CB electrons temporarily and reduce the surface recombination.121 Furthermore, the oxygen vacancies favour the adsorption of oxygen to capture CB electrons, thereby simultaneously producing oxygenated free radicals. These morphologies were fine-tuned by varying the concentration of hydroxyl anion from 50 to 400 mM NaOH through a soft chemical approach (Fig. 7). At low concentration, spherical NPs (∼8 nm) were formed due to the uniform crystal growth as the concentration of hydroxyl anions was very low to favour anisotropic growth. While at higher concentrations of hydroxyl anions, ZnO growth along the {0001} surface was blocked by the binding of [Zn(OH)4]2−, which changed the surface free energy, thereby favouring the growth of a {000} surface to form NRs (length 30–40 nm). However, at an intermediate concentration of 200 mM, both uniform, as well as anisotropic, crystal growth occurs, leading to the formation of both NPs and NRs. The chemical reaction for ZnO nanostructure formation is as follows;
| | |
Zn2+ + 2OH− → Zn(OH)2
| (43) |
| | |
Zn(OH)2 + 2OH− → [Zn(OH)4]2−
| (45) |
| | |
[Zn(OH)4]2− → ZnO + H2O + OH−
| (46) |
 |
| | Fig. 7 Mechanism of formation for different ZnO nanostructures under the influence of hydroxyl anions (reprinted with permission from ref. 121; Copyright 2011 @ Elsevier). | |
The Zn(OH)2 is unstable and directly hydrolyses to ZnO nuclei or reacts with a hydroxyl anion to form a growth unit of [Zn(OH)4]2−, followed by polymerization to form a ZnO nucleus. The abundant concentration of hydroxyl anion favours the formation of a [Zn(OH)4]2− intermediate, which is likely to control the surface morphology of ZnO.121 Thin films of ZnO microrods with an hexagonal shape of submicron-size diameters were active for the degradation of MB compared to ZnS, although the latter was more striking for MB adsorption.122 The morphology of ZnO remained the same after the photocatalytic reaction, whereas the surface of a ZnS microrod was occupied by dye molecules with partial damage in some areas. However, both ZnS and ZnO lost their activity after three cycles in subsequent runs.
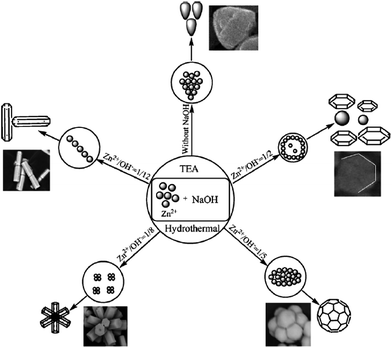
The high performance of sheet-like ZnO for the degradation of MO compared to flower-like and sphere-like ZnO was attributed to a large quantization in the bandgap, as the mean thickness of the sheets was comparable to the Bohr radius of ZnO.123 This enables the materials to harvest the maximum number of photons from the excitation source, producing surplus charge carriers and thereby enhancing the activity.124 The photodegradation was faster than mineralization, indicating the high resistance of the accumulated by-products to react with free radicals. Two steps were followed to obtain such a morphology: (i) the formation of ZnO spheres as seeds on a quartz substrate; (ii) the hydrothermal growth of ZnO in an aqueous solution by the reaction of zinc nitrate and HMT, with the latter providing a controlled supply of hydroxide ions. The HMT also reacts with water to produce NH3, which subsequently forms NH4+ ions. Thus, the two tetrahedral complexes [Zn(NH3)4]2+ and [Zn(OH)4]2− served as precursors for ZnO nucleation. Flower-like NSs were epitaxially formed from the seeds after the hydrothermal reaction (150 °C, 4 h), which transforms them to sheet-like ZnO in 6 h without the agglomeration of the ZnO seeds.123 The combined effects of surface roughness, crystallinity, and an appropriate film thickness of porous ZnO obtained by chemical bath deposition from a methanolic solution of zinc acetate (0.05 mol L−1) and calcined at 500 °C (1 h) make it active for the degradation of MO compared to those calcined at 300 °C and 800 °C, having different precursor concentrations.125 At a low precursor content (0.01–0.05 mol L−1), the morphology of porous ZnO film was nest-like assemblies, while globular aggregates were observed at 0.1 mol L−1. The porous structure originates from the formation of a flake-like layered zinc acetate, which stands on the alumina substrate with its edge pointing to the surface normal direction and forms a highly porous 2D nest-like structure. The nest-like porous structure also provides sufficient surface roughness, which is advantageous for the adsorption of pollutants during the photocatalytic reactions. The globular agglomerates easily depart from the film during the photocatalysis because of the low adhesion with the substrates, and also the effective surface area absorbing the photons from the excitation source is drastically reduced, which lowers the activity.125 The degradation of MB with respect to a ZnO morphology followed the following order: flower > oblate > hexagonal sphere > hexagonal biprism > nut-like > NRs, indicating that the different morphologies display large differences in their activity.126 The high performance of flower-like ZnO was attributed to the greater exposure of its polar surface, and hence more hydroxyl radicals could be formed to participate in redox reactions.127 These morphologies were obtained by varying the Zn2+:NaOH and Zn2+:TEA ratio under hydrothermal conditions (140 °C, 12 h). The NRs were observed without any addition of TEA, whereas a nut-like structure was formed in the absence of NaOH, revealing that the concentration of hydroxyl ions was indispensible for obtaining such diverse morphologies (Fig. 8). Initially, TEA will hydrolyze to produce the hydroxyl ions (eqn (47)), resulting in Zn(OH)2, which further transforms into ZnO after calcination (eqn (43)–(46)).
| | |
TEA + H2O ↔ [TEA]+ + OH−
| (47) |
 |
| | Fig. 8 Mechanism of formation of ZnO microstructures (reprinted with permission from ref. 126; Copyright 2012 @ Elsevier). | |
With increases in the content of NaOH, an excess of hydroxyl anions favours the formation of [Zn(OH)4]2−, which transforms to ZnO nuclei at a concentration above its critical solubility.91–93 These zinc hydroxyl complexes preferentially adsorb on the ZnO nuclei, which lifts the growth along the direction of the c-axis and leads to a hexagonal biprism shape. When [Zn(OH)4]2− ions are relatively insufficient, TEA molecules will occupy the dominant position and serve as the structure-directing and assembling agent, resulting in nut-like, hexagonal sphere-like, oblate and flower-like structures.126 The photoactivity towards the degradation of phenol with respect to various solvents (morphology) followed the following sequence: THF (cauliflower-like) > decane (truncated hexagonal conical) > water (tubular and a rod-like) > toluene (hourglass-like) > ethanol (NRs) > acetone (spherical shape).128 The ZnO synthesized in water were less homogeneous compared to those prepared in organic solvents, as the zinc precursor was better dispersed in the latter. The solvents like THF, ethanol and acetone have high vapor pressure, which limits the growth of ZnO nuclei to favour the NPs nucleation. The aggregation of these NPs and the formation of clusters reduce the overall surface area and the surface energy. In contrast, larger ZnO crystals with faceted morphologies were obtained for relatively low saturated vapour pressure solvents, such as decane, toluene, and water.
These varied nanostructured morphologies encourage charge carrier separation, in addition to enhancing the surface reactions, which ultimately results in the rapid decomposition of organic pollutants. The morphological stability even after recycling many times indicates the resistance towards agglomeration and a low degree of by-products accumulation on the catalyst surface during the degradation reactions. The hierarchical structure, combining the features of nanoscaled building blocks, not only show unique properties different from those of the bulk, but also provide more opportunity for the surface photochemical reaction to realize the morphology dependent surface reactivity.129 Therefore, it is desirable to develop a facile protocol for the morphology controlled ZnO with low cost precursors and without special equipment, harsh experimental conditions or toxic reagents. Despite having comprehensive studies relating to the effects of the synthesis parameters on the formation of ZnO, the literature is not quite convincing in establishing relationships among the synthesis parameters, morphological properties and the photocatalytic activity. Understanding such interdependences will provide an insight into the origin of the chemistry behind the photocatalysis, which should advance our capability to utilize ZnO nanocrystals.
4. Photocatalytic activity of metal ion-doped ZnO
The doping of metal oxides (TiO2 or ZnO) with foreign ions/impurities initially changes the coordination environment of host metal ions in the lattice and also modifies the electronic band structure via introducing localized electronic energy levels within the bandgap states. The former alters the pristine defect structure, and thus affects the mobility of charge carriers, whereas the latter enables a more efficient manipulation of incident photons. The dopant impurity level situated either above the VB or below the CB momentarily traps the photogenerated charge carriers, thus affecting their redox pathways.130
(a) Doping with alkali metal ions
Li (10%)–ZnO was more active for the degradation of 4-NP under visible light compared to Na–ZnO and K–ZnO, primarily due to the electron trapping ability of Li+ ions, the small crystallite size and high surface roughness.131 The bandgap widening and efficient charge carrier transfer process were identified as the factors responsible for the sunlight-driven activity of Mg (0.1%)–ZnO for the degradation of MB.132 The increase in optical bandgap energy by Mg2+ doping was attributed to the Moss–Burstein effect, which was caused by electrons generated from the oxygen vacancies.133 The substitution of Zn2+ by Mg2+ intensifies the oxygen vacancies and the electron concentration due to the differences in electronegativity and the ionic radius between the host (Zn) and the guest (Mg). This increase in carrier density lifts the Fermi level into the CB of a degenerate semiconductor, resulting in the bandgap widening. Cubic MgO crystallized at a higher Mg2+ doping (0.2%) served as the recombination centres.132
(b) Doping with transition metal ions
The enhancement in the defect concentration of ZnO by Zr4+ (1.5 wt%) doping resulted in a faster degradation of resorcinol.95 The small crystallite size decreases the diffusion path length for the movement of charge carriers from the bulk to the surface, and the size quantization of the bandgap enhances the redox potential of the charge carriers. However, a complete mineralization was observed only with a 365 nm excitation source rather than at 254 nm. The UV light photolysis indicated that the intermediates formed during the course of degradation significantly absorb light energy at 254 nm, thus shunting the photons away from the ZnO surface. Because resorcinol does not absorb at 365 nm, the complete absorption of light by Zr–ZnO facilitates the generation of reactive free radicals in the solution, which accounts for the complete mineralization. The doping of Ta5+ into ZnO by a modified Pechini-type method using water soluble peroxo–citrato tantalum complexes as a Ta5+ source was positive for the degradation of MB under visible light, which is attributed to a competitive trade-off between the crystallinity, surface hydroxyl groups and the specific surface area.134 It was predicted that the crystallinity should dominate over the other two factors in enhancing the efficiency. The activity at different annealing temperatures of Ta (1 mol%)–ZnO had the following order: 700 > 800 > 900 > 600 > 500 °C. As the annealing temperature increases, the crystallinity is improved with the loss of surface hydroxyl groups and surface area. The degradation rate increases by two fold with a change in the pH of the dye solution from 5 to 8 but declined at higher pH values. The high degree of hydroxylation combined with the efficient adsorption between cationic MB and the surface negatively charged Ta–ZnO contributes to the overall efficiency at pH 8. Although the adsorption of MB was drastically improved at pH > 8, the density of the hydroxyl groups decreased and the rupture of hydroxylation on the catalyst surface suppressed the activity. The degradation rate with respect to the dopant content followed the following order: 1 > 1.5 > 0.5 > 2.0 > 3.0 mol%.135 The particle size increased, resulting in the loss of surface area with increase in dopant concentration (>1 mol %) and an orthorhombic ZnTa2O6 was formed at a very high content of Ta5+ (4 mol%). The substitution of Ta5+ into the host Zn2+ resulted in the formation of defects, such as zinc and oxygen vacancies, which form an intermediate electronic level within the bandgap, enabling the visible light absorption and also restraining the carriers from recombination.| |
 | (48) |
| |
 | (49) |
| |
 | (50) |
The oxygen vacancy forms a deep donor at ∼1.0 eV below the bottom of the CB, and the zinc vacancy defect is relatively shallow at ∼0.4 eV above the top of the VB.136 The doping changed the morphology of ZnO from NRs to NPs with a spheroidal shape. The catalyst still retained its activity even after storage for 90 days in air, attesting to its excellent stability due to its high crystallinity.
The defect-free Cr (∼3 at.%)–ZnO obtained via the solvothermal treatment (120 °C, 12 h) in ethanolic solution under an alkaline medium showed visible light activity towards the degradation of MO.137 The visible light response originated from the ‘sp–d’ exchange interactions between the CB electrons and localized ‘d’ electrons of the Cr3+ ions, which substitute for the Zn2+ ions.138 The ‘s–d’ and ‘p–d’ exchange interactions lead to a negative and a positive correction to the CB and VB edges, respectively, resulting in a red shift in the bandgap absorption.139 Mn (1%)–ZnO obtained by the wet chemical precipitation technique was detrimental to the degradation of MB under UV light, but was beneficial under visible light.140 The contribution from the donor states (oxygen vacancies and interstitial zinc) and acceptor states (zinc vacancies and interstitial oxygen) enhanced the optical absorption in the visible region, and thus the photoactivity.141 The Mn–ZnO creates electron–hole pairs at the tail states of CB and VB, respectively, under visible light. The excited electron transfers to the adsorbed MB molecule on the particle surface and disrupts its conjugated system, leading to its complete decomposition. The VB holes react with water or hydroxide ions to generate hydroxyl radicals, which further degrade the pollutant molecules.140 In another study, both Mn (1%)–ZnO and ZnO obtained by a conventional method (i.e. a slow crystallization process) showed a low activity for the degradation of MB under visible light against the defect rich (i.e. zinc interstitials and oxygen vacancies) ZnO, which was crystallized under microwave conditions.142 Heating of the precursor solution occurs locally due to the molecular rotation of the medium, arising from their interaction with the electromagnetic fields of incoming microwave radiation. This assists in a quick energy supply for the reaction, thus resulting in a faster nucleation and growth of nanocrystallites.143 The rapid inclusion of zinc and oxygen atoms into the crystal lattice creates interstitial defects, which then induce the mid-gap states, which serve as intermediate steps for the electrons and holes during the photoexcitation process. This means that instead of absorbing one UV-photon, an electron arrives at the CB by utilizing multiple photons from visible light. In the case of a conventional heat treatment to induce crystallization, heat passes by convection from the walls of a container towards the centre and takes a long time before establishing thermal equilibrium between the precursor and its surroundings. Thus, the thermal decomposition of the precursor contents and the subsequent formation/crystallization of ZnO or Mn–ZnO occur very slowly, resulting in the formation of nearly defect-free nanocrystals. The doping of Mn2+ (2 mol%) was positive, while Co2+ (1–5 mol%) incorporation was detrimental for the generation of hydroxyl radicals, wherein, these doped catalysts were prepared by a three-stage process, consisting of a high energy mechanical milling, heat treatment and washing.144 It was proposed that Co2+ doping served as recombination centers, while the presence of a secondary phase like Mn3O4 and Mn3−xZnxO4 favoured the charge separation in Mn–ZnO. Casey et al.66 and Barick et al.145 suggested that the dopants such as Fe2+, Ni2+, Co2+, and Mn2+ were not beneficial to enhance the photoactivity of ZnO. Interestingly, Co–ZnO with bicrystalline wurtzite–zinc blende phases showed high activity for the degradation of MB and phenol under visible light.146 The cobalt doping induced a red shift in the visible light absorption and homojunction between the mixed phases, together with increased surface oxygen vacancies, thus promoting the charge carrier generation–separation pathways. The undoped ZnO had a wurtzite structure and that doping of Co2+ led to the formation of CoO having a zinc blende crystal structure, and which served as substrate for the growth of ZnO in the zinc blende phase. This is a very rare report on the activity of mixed phase ZnO and further research should focus on tuning the ratios of the mixed phases and understanding the charge carrier transfer pathways.
The doping of Ni2+ (1–15 wt%) into ZnO thin film via a combined sol–gel and dip coating method was detrimental for the degradation of MG under UV/visible light, despite Ni2+ doping reduced the crystallite size of the catalyst.147 The Ni2+ served as a p-type dopant in ZnO and acts as a recombination centre.148 The decrease in the effective charge carrier concentration lowers the band bending near the crystallite surface, thus suppressing the driving force for the charge carrier separation. However, the photostability of both the films were excellent, as revealed by SEM images, in which the ganglia-like hills and wrinkles morphology of the films were not changed even after recycling in three subsequent runs. In contrast, an increase in the doping concentration of Ni from 0% to 5% into ZnO NRs increased the degradation of RhB, as the dopant induced a distortion of the local electric field and the charge carriers were then trapped around the dopant. At a higher dopant concentration (∼10%), the bandgap of a doped catalyst decreases, thus reducing the energy of excited electrons and then the excess dopant promotes the recombination process.149 However, the doping of Pd2+ into ZnO decreased the surface area and improved the activity for the degradation of MO.150 The Pd2+ doping introduced a new electronic energy level within the bandgap states, which served as a shallow trap for charge carriers. The charge carriers were efficiently separated with an increase in the Pd2+ content from 2% to 3%, followed by a sudden decrease at high concentration. At this optimum dopant concentration, the surface barrier becomes higher and the space charge region becomes narrower. The electron–hole pairs within the region are efficiently separated by a large electric field traversing the barrier, which promotes the charge carrier generation–separation–transfer pathways. For higher dopant concentrations, the space charge region becomes very narrow, and as the penetration depth of light into ZnO greatly exceeds the space charge layer, this facilitates carrier recombination.151
The sequence in activity of Cu–ZnO with different dopant contents for the degradation of MO was in correlation with the FS technique, namely, the lower the intensity, the higher the activity.152 Because the FS emission arises from the recombination of excited charge carriers, a lower FS intensity indicates a reduced recombination. Thus, Cu–ZnO with 0.5 wt% of Cu2+ was beneficial, while a higher dopant level was found to hinder the activity. The doped Cu2+ traps the electron and detraps it to adsorbed oxygen to produce superoxide radicals, whereas the surface hydroxyl groups trap the holes to form hydroxyl radicals. The optimum heat treatment for this catalyst was found to be 350 °C (3 h).152 The Cu-doped Zn–ZnO composite modified by a surface carbon species exhibited visible light activity for the degradation of Reactive Brilliant Blue KN-R dye.153 In the preparation method, CO2 dissolved in solution was converted to carbon, which then adhered onto the composite surface. The doping of copper promotes the crystal growth of ZnO, inhibits the phase transfer of metallic Zn to ZnO and enhances absorption in the visible region. The composite with a 2.0 at.% Cu loading under hydrothermal treatment (180 °C, 2 h) was reported to have a high activity, which was attributed to: (i) the substituted Cu2+ serving as a deep acceptor along with the oxygen vacancy, which substantially improves the electron transport properties;154 (ii) the metallic zinc clusters acting as electron sinks, due to the Schottky barrier formation at Zn–ZnO interface, in which the bulk Zn is surrounded by a ZnO shell; (iii) the surface carbonaceous species having a high propensity for adsorbing the reactants, which facilitates the transfer of reactants to the active sites, and thus improves the surface chemical reactions.155 The formation of (Cu,Zn)7(SO4)2(OH)10·3H2O impurity at a higher copper doping level (4–6 at.%) did not influenced the activity. The surface doping of copper (Cu1+δO) on ZnO enhanced the reaction rate for the decomposition of OG and MB under UV/visible light. The optimum concentration of copper was found to be 1.5 and 0.5 mol% for the catalysts prepared by the aqueous method and the solid state reaction, respectively.156
(c) Doping with rare earth ions
Nd3+(1%)–ZnO nanoneedles synthesized via an ultrasonic-assisted method were active for MB degradation, as Nd3+ served as an electron scavenger to inhibit the recombination.157 The doping of Ln3+ (Nd3+-4 wt%, Sm3+-8 wt% and La3+-4 wt%) into the ZnO matrix by the polymer pyrolysis method was promising for the 4-NP degradation, which is attributed to the change in the structural properties, such as the surface area and the crystallite size.158 A suitable loading of the dopant is compulsory for producing a significant potential difference between the surface and the centre of the particles to separate the photoinduced electron–hole pairs, as an excess of Ln2O3 covering the ZnO surface may facilitate the carrier recombination. The secondary phase of La2O3 nucleated at ≥3 mol% doping of La3+ into the ZnO matrix via the precipitation route, whereas it was observed at 10 mol% La3+ in the ball milling method, indicating a high degree of dopant solubility at room temperature in the latter.159 The doping of La3+ (1 mol%) into the ZnO matrix was constructive in the former and detrimental in the latter case for the degradation of MB, which is mainly attributed to an increase or decrease in the oxygen vacancies after doping, respectively. The relative photonic efficiency of La (0.8 wt%)–ZnO for TCP and MCP degradation was relatively higher compared to ZnO under the excitation wavelengths of 254 nm and 365 nm.160,161 This was attributed to the combined factors of large surface area, quantized bandgap and high density of oxygen vacancies, which collectively lead to an efficient adsorption of pollutants on the catalyst surface, a high redox potential for charge carriers and an enhanced production of hydroxyl radicals, respectively. The charge carrier separation was effective with the 254 nm excitation source, as it excites the electron with high kinetic energy, and hence they escape from the recombination channels. The degradation rate reached a maximum at pH 10 for MCP, but declined thereafter. Because MCP is not protonated at pH > 10, the electrostatic repulsion between the surface charges on the adsorbent and adsorbate leads to an inefficient adsorption. The low reaction kinetics at acidic pH was due to the overcoverage of MCP on the ZnO surface, which prevents the catalyst from photon absorption.161 The Ce4+ doping changed the morphology of ZnO from nanoplates to nanospheres and enhanced its activity for the degradation of MB (pH 5.4–9). In this study, a simple refluxing method (90 °C) was used to incorporate Ce4+ into the ZnO lattice. A Ce4+ content of 0.05 mol%, refluxing time of 9 h, catalyst dosage of 0.15 g and a calcination temperature of 400 °C (2 h) were found to be optimum conditions to achieve high performance.162 The photoactivity of Ce–Ag–ZnO towards the decomposition of Napthol Blue Black dye increased under solar light up to pH 9, but declined thereafter, exactly correlating to dye adsorption on the catalyst surface at different pH conditions.163 The synergistic effect between silver and Ce4+ (i.e. Lewis acid) to trap a CB electron with its subsequent transfer to the adsorbed oxygen was responsible for such a high activity (Fig. 9).
 |
| | Fig. 9 Charge transfer pathways in Ce–Ag–ZnO (reprinted with permission from ref. 163; Copyright 2012 @ Royal Society of Chemistry). | |
The doping of foreign ions into ZnO appears to be an alternative approach to modify the physical (i.e. crystallite size and surface area), electronic (i.e. bandgap energy and defect concentration) and morphological features and make them suitable for photocatalytic applications. However, the literature reveals widespread disagreement related to the abovementioned properties as follows: (i) a shift in bandgap towards the visible region is observed with transition metal ion doping, whereas quantization in the bandgap is noticed upon doping with alkali metal ion and rare earth ion doping; (ii) the change in crystallite size does not correlate with the bandgap energy; (iii) the morphological changes after the doping are unclear, as they may also originate from the counter-anion of the precursor; (iv) the change in the defect density distribution of defect-free or defect-rich sites is almost impossible to predict owing to its dependence on the experimental conditions; (v) the dopants can either inhibit or promote the crystal growth; and (vi) the precipitation of impure dopant oxides may change the chemical composition of ZnO itself. The corrosion–dissolution of ZnO at extreme pH conditions leaches the substituted metal ion from the matrix, thus creating toxicity. These issues must be taken into consideration for designing the doped ZnO nanostructure in near future.
5. Doping with nitrogen (N–ZnO or ZnO1−xNx)
The cation doping usually produces additional defects or impurities in ZnO, which may act as recombination centers. In contrast, doping with non-metals with small radii are promising candidates, because they easily substitute for lattice oxygen sites of ZnO or can occupy interstitial sites. Due to its larger solubility in metal oxides and its comparable size with oxygen, nitrogen is widely used as a dopant to modify the electronic structures of metal oxides, as only small formation energy is required for the substitution process. In addition, the low electronegativity and low ionization energy of nitrogen compared to oxygen reduces the electron density around the doped nitrogen in the pristine oxide.164
(a) Photoactivity of N–ZnO
Highly crystalline N–ZnO obtained via a vacuum atmosphere method (380 °C, 2 h) using melamine as the nitrogen source was active for the degradation of MO under simulated daylight illumination.165 The melamine transformed into isocyanic acid upon heating (120 °C), which then reacted with residual oxygen or water in the vacuum tube chamber to form ammonia, which subsequently mixed with ZnO to form N–ZnO.| | |
xNH2− + ZnO ↔ ZnO1−xNx + xH2O
| (52) |
In addition, melamine combines with oxygen and a small amount of N2 was also generated, further promoting the nitrogen doping into ZnO via diffusion of the nitrogen atoms. Interestingly, unreacted isocyanic acid was transformed to melamine during the cooling process, which could then be collected for reuse.165 The surface doping of nitrogen on the ZnO TPs showed visible light activity for the degradation of bis-phenol A.166 It was proposed that the nitrogen forms isolated N 2p states above the VBM, which intensifies the absorption in the visible region. The electron–hole pairs generated between the N 2p states and the CB of Zn 3d levels are separated through trapping from surface adsorbed oxygen and the hydroxyl groups, respectively. The N–ZnO prepared by the decomposition of zinc nitrate in air (350 °C, 3 h) showed a high performance for the reduction of Cr(VI) and oxidation of MO under visible light.167 However, the activity of N–ZnO was lower for the oxidation of MO and better for the reduction of Cr(VI) compared to pure ZnO under UV light. Based on first principle calculations, it was proposed that nitrogen doping into ZnO will cause the appearance of a redundant carrier, i.e. a hole near the top of VB. The mutual exclusion effect between the holes will enable the carriers to form a narrow-deep acceptor level in the energy gap and induce a conductivity conversion of N–ZnO from the n-type to the p-type, which was also supported by iodometric titrations.168 Thus, photoformed holes move towards the interior of the p-type semiconductor under illumination, while electrons migrate to the surface to favour the reduction process. In another study, N–ZnO obtained using zinc nitrate under solvothermal conditions (260 °C, 20 min, followed by calcination at 400 °C, 2 h), showed an appreciable performance towards the degradation of RhB.169 The increased oxygen defects, improved absorption ability of the incident photons and extended carrier lifetime were responsible for the high photocatalytic activity. In the synthesis step, zinc nitrate coordinates to oleylamine and forms zinc–oleylamine complex (130 °C), followed by a pyrolysis to form N–ZnO at high temperatures. The oleylamine serves as a structure directing agent, influencing the nucleation and aggregation process of the NPs, which are ultimately assembled to microspheres with a mesoporous structure. Liu and co-workers170 suggested that substitutional nitrogen doping in semi-crystallized ZnO NPs was effective for the degradation of RhB under visible light. In contrast, such successful doping failed in highly crystallized ZnO, during the calcination in gaseous ammonia atmosphere (400 °C, 30 min). The partially disordered structure and defects in the surface layers of semicrystallized ZnO serve as reactive sites (i.e. energetic) for the substitution of nitrogen during the heating process. In contrast, the long-range ordered surface structure of crystallized ZnO requires a high energy for the substitution of nitrogen into oxygen and could not be accomplished at 400 °C. The N–ZnO bundle-like NPs obtained from the ZnOHF precursor demonstrated exceptional visible light induced water oxidation without an applied bias, and the activity was further enhanced by loading IrO2 co-catalyst.171 The ZnOHF decomposed to ZnO and HF during heat treatment in a NH3 gas flow (973 K). The ZnO intermediate was subsequently doped with nitrogen, while HF reacts with ammonia gas to form a NH4F flake-like by-product. However, this N–ZnO was less active for the degradation of MB under visible light. As the mechanism for water oxidation and pollutant degradation are entirely different, it was suggested that the surface of N–ZnO possesses energetic sites for oxygen evolution, but lacks reactive sites for pollutant degradation. The ZnO is partially reduced in a highly reductive atmosphere and the resulting Zn is vaporized due to its low melting point at high calcination temperature (1073 K).171 The doping of nitrogen as No acceptor states is detrimental for the degradation of MO degradation.172 The PL studies indicate that N-related defects increase with the increase in nitrogen content and are accompanied by a rapid recombination process. The heavily doped (No) acceptor states provide a large number of internal holes, which may hinder the formation of photogenerated holes.
(b) Coupling of N–ZnO with other semiconductors
The glass–ZnO–ZnO1−xNx and glass–ZnO1−xNx–ZnO heterojunction was positive for the degradation of humic acid, indicating a synergism between ZnO and ZnO1−xNx.173 However, the activity was found to depend on the mode of the light illuminating the composite: (i) the former was active when light was illuminated from the film to the glass side, as holes accumulate on the ZnO1−xNx layer to produce more hydroxyl radicals, which then react with pollutants. While few carriers will be generated in the outer ZnO layer and then the inability of humic acid to diffuse into the ZnO1−xNx layer in the latter case results in a low efficiency; (ii) the glass–ZnO1−xNx–ZnO was beneficial when light was illuminated from the glass to the film side, which is attributed to the efficient absorption of visible light by the ZnO1−xNx layer. In the case of glass–ZnO–ZnO1−xNx, most of the incident light will be lost via diffuse reflection in the ZnO layer, and also, the outer ZnO1−xNx may poorly absorb the incident photons.
The nitrogen doping into ZnO via spray pyrolysis at 600 °C improves the activity for the decomposition of CH3CHO under visible light.174 The nitrogen atom exists in two chemical forms: one was site-N (i.e. nitrogen substituting the oxygen atoms of ZnO), while the second was residual nitrogen originating from the precursors. A further improvement in the performance of N–ZnO was achieved by coupling with WO3 (or V2O5) under UV/visible light, while the efficiency was hampered in the case of Fe2O3.175–177 The nitrogen content in the catalyst increased monotonically with the increase in WO3 (or V2O5) added, while it was insensitive to the addition of Fe2O3. The highest activity was observed at 0.7 wt% Fe and 5.8 wt% for W series powders at the spray pyrolysis temperatures of 800 °C and 1000 °C, respectively. Under visible light, the nitrogen dopant forms an energy state above the VB of ZnO, serving as a “stepping stone” for electron transfer from VB to the impurity level and from the impurity level to the ZnO CB. Then, the electron transfers to WO3 (or V2O5) CB, while the hole migrates in the opposite direction. Tennakone et al.178 reported that W(VI) in WO3 could easily be reduced to W(V) by excited electrons and may be reoxidized to W(VI) by detrapping them to an adsorbed oxygen. In contrast, the CB and VB of Fe2O3 sandwiches between the ZnO band edges, and thus both the electron and hole transfer in the same direction, resulting in the recombination. The high activity of V2O5–ZnO1−xNx compared to WO3–ZnO1−xNx is probably due to the larger difference in the CB edge between V2O5 and ZnO, which facilitates a thermodynamically faster electron transfer process. A typical feature of sprayed catalysts is the homogeneous distribution of ingredients throughout the particle surface, as they are derived from a homogeneous solution.174–177 The fabrication of hybrid SnO2 NFs decorated with surface bound N–ZnO showed a high performance for the degradation of RhB under UV/visible light.179 According to the band energy diagram, photoelectrons generated in the CB of SnO2 and the holes in ZnO VB can be separated at the heterojunction by a favourable bias between the two sides. In addition, the lower resistance caused by the formation of oxygen vacancies upon nitrogen doping indicates an easy carrier transport and enhancement in the VB of ZnO.
Compared to the broad literature on N–TiO2, N–ZnO has seldom been investigated, mostly due to the inconvenience associated with realizing the nitrogen doping process by the commonly developed synthesis routes. The undesired transformation of substituted nitrogen to molecular nitrogen at elevated temperature impairs the understanding of doping process itself. However, low-temperature doping quite often leads to poorly crystallized samples that cannot always guarantee high activity. At the atomic level, the homogeneous distribution of nitrogen dopant throughout the matrix is essential for narrowing the bandgap of the semiconductor, while surface doping often leads to localized states, which may not be very effective for a visible light response. The efficiency of N–ZnO reported to date is not satisfactory, and thus leaves scope for further investigation.
6. Deposition with noble metals
Metal oxide–metal heterojunctions represent a novel class of advanced functional materials that have attracted tremendous interest, owing to their promising applications in various fields.180 The deposition of metal NPs on a semiconductor surface is gainful, whereby the particle size could be independently controlled and a very narrow size distribution could be obtained. It is assumed that the metal NPs modify the reactive sites and the deposited noble metal itself serves as a catalyst to decompose the pollutants.181 The metal islands capture the photoinduced charge carriers and intensify the light absorption in semiconductor oxides, with both effects facilitating the redox reactions and amplifying the photocatalytic efficiency.180 The double layer charging around the metal NPs facilitates the storage of electrons within them.182
6.1 Photocatalytic activity of Ag-deposited ZnO (Ag–ZnO)
The Ag (1.0 wt%)–ZnO obtained by the deposition–precipitation method showed a high activity for the degradation of MO due to the maximum distribution of Ag and Ag–ZnO interface with a low concentration of surface oxygen defects. However, a sample obtained by coprecipitation did not disperse well in dye solution, resulting in a very low UV light utilization efficiency.183 The Ag NPs distributed along the hollow interior of the ZnO NRs, supported over Si (100) wafers, switch from superhydrophobic with a WCA > 150° to superhydrophillic having a WCA < 10°, and also have an inflexion point at ∼110° under UV/visible light.184 The visible light was less effective in inducing superhydrophilicity, as a WCA < 10° was reached after an irradiation of 150 min, while it was otherwise attained in 8 min under UV illumination (Fig. 10). The surface recovered its superhydrophobic character after the samples were stored in the dark, with a full recovery after five days. It was proposed that the ZnO close to the Ag NPs behaved as a superhydrophillic surface because of the hydroxyl enrichment under the light illuminating conditions. The Ag NPs (1.66 wt%)–ZnO microrods with an interface between the {100} plane of ZnO (0.28 nm) and the {101} plane of Ag (0.235 nm) showed a strong performance for the degradation of MB under UV/visible light, and furthermore their efficiency was maintained even after five consecutive runs.185 The high crystallinities of ZnO (20 nm) and Ag (20–50 nm) are responsible for the high performance, while a low concentration of surface defects in the micro/nano-configured structure improve the photostability. These structures possess an overall dimension in micrometer with nanosized units. The assembled nanosized units are stabilized to ensure an improved structural stability, which reduces the aggregation. The Ag islands on the ZnO surface act as electron wells to inhibit the recombination and the plasmatic effect of Ag NPs promote the photocatalytic activity (Fig. 11). Dimer type Ag–ZnO NRs were synthesized by the solvothermal method (160 °C, 24 h); wherein, Ag metals were embedded in the ZnO NR via Zn–O⋯Ag bond formation. The PL spectra revealed that the oxygen vacancy defect density increased to upto 5 at.% of Ag, but decreased thereafter, exactly correlating to the observed efficiency of Ag–ZnO for the degradation of MO.186 The formation of oxygen vacancies was due to the intermolecular dehydrolysis of the precursors to form a ZnO–Ag2O heterostructure, followed by treatment with ethanol under solvothermal conditions.| |
 | (53) |
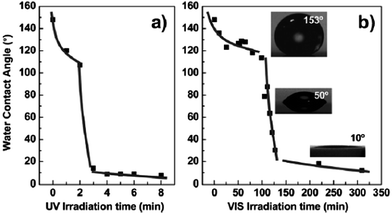 |
| | Fig. 10 Switching the Ag–ZnO surface from superhydrophobic to superhydrophilic under UV (a) and visible light (b) illumination (reprinted with permission from ref. 184; Copyright 2012 @ Royal Society of Chemistry). | |
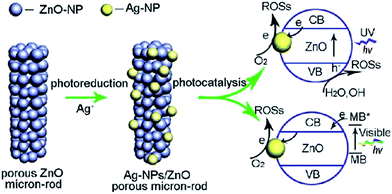 |
| | Fig. 11 Illustration of the process of the degradation of MB using Ag–ZnO microrods under UV and solar light irradiation (reprinted with permission from ref. 185; Copyright 2012 @ American Chemical Society). | |
Surprisingly, the concentration of ZnO after Ag deposition did not have any effect on the degradation rate. This composite had an excellent stability because of the surface-to-volume ratio of ZnO and the close contact between the Ag and ZnO. According to the proposed mechanism, an electron transfer from ZnO to Ag or to the oxygen defects and finally to the solution effectively suppress the recombination process.186 Highly ordered ZnO NRA fabricated via a wet chemical route and photodeposited with Ag (0.05 M AgNO3) was shown to be promising for the degradation of MB, and without a loss in activity even after five consecutive runs.187 The ZnO microspheres modified with Ag NPs, prepared by a “one pot” strategy in an EG medium performed better for the degradation of RhB.188 The EG served as a reaction medium for the formation of ZnO via a forced hydrolysis and also reduced Ag+ to Ag metal. The efficiency with respect to the Ag content had the following sequence: 2.5 > 1.25 > 0.625 > 5.0 mol%. Thus, the deposition of Ag clusters with an appropriate amount on ZnO surface captures and transfers the CB electrons to adsorbed species. The Ag NPs were found to aggregate and/or the size of the Ag particles increased at a high Ag content, thus promoting the recombination process. In addition, the large particle size of the composite was easily settled from the reaction system within 10 min even without centrifugation.188 The deposition of 0.2 wt% Ag around the oxygen vacancy sites on the ZnO surface reduced the defects, and improved the photostability and the efficiency for CV degradation without any change in the crystal structure, even after recycling many times.189 The CB electrons trapped by Ag react with chemisorbed/physisorbed/dissolved oxygen in the dye solution to form a reactive oxygen species. The suppression of the degradation kinetics under the argon atmosphere revealed that the active oxygen formed by the reaction of the CB electron and the oxygen chemisorbed on Ag was the potential oxidant responsible for the dye degradation. This suggests that the metallic Ag promotes the reaction between chemisorbed oxygen and CB electrons to promote a charge separation, and hence the photostability. Although PL measurements revealed the lowest intensity for 6.7% Ag–ZnO, the photoelectrochemical measurements indicated a reduced photocurrent, as the holes get trapped by a large number of negatively charged Ag particles, which is detrimental to photocatalytic reactions.190 This process of hole capture proceeds through a non-radiative pathway, which cannot be detected by PL measurements. The hydrothermal synthesis (120 °C, 6 h) of Ag (6.2%)–ZnO in the presence of HMT resulted in a faster degradation of RhB.191 During the composite preparation, HMT was transformed to NH3 and HCHO, which served as a precipitating agent for the formation of ZnO and as a reducing agent for Ag+ to metallic silver. Pure ZnO exhibits a pillar and bundle-like morphology, while the addition of silver resulted in a rod-like structure. In contrast, an increase in HMT concentration resulted in a pillar-like morphology, as it prevents the influence of silver on the equilibrium between Zn2+ and NH3. The UV emission of ZnO was enhanced after depositing with Ag due to an increase in the concentration of the excitons, as a result of the interface between the Ag islands and ZnO.191
Ag (3 mol%)–ZnO prepared by a non-aqueous sol–gel process exhibited a higher activity compared to Degussa P25 and unmodified ZnO for the degradation of R6G under sunlight.192 The segregation of Ag around the ZnO grain boundaries (when calcined at 400 °C, 2 h) inhibits the grain growth and provides more surface active sites, which enables the charge carriers to react with the surface adsorbed molecules to form active free radicals. The Ag modification was detrimental at high temperature (800–1000 °C), as Ag promoted the densification and grain growth of ZnO by forming high surface energy silver islands, which reduces the surface active sites for pollutant adsorption, as well as for photon absorption. The degradation of R6G on Ag–ZnO surface takes place by multiple charge transfer pathways under visible light (Fig. 12). The high reduction potential of R6G was found to be a vital factor that dominates the electron transfer process and thereby defines the degradation pathway.192 The optimum Ag content towards hydrogen evolution from a water–methanol mixture was found to be 1.6 and 8.3 mol% under UV-vis and solar light, respectively.193 The better dispersion of the Ag NPs and the large exposure of the ZnO surface facilitate efficient light absorption in the former case, while the formation of a Ag2O–ZnO heterojunction additionally promotes the activity in the latter case.194 The electron transferred from ZnO CB to Ag NPs effectively reduces the H+ to H2 on its surface, while the holes oxidize the alcohol.
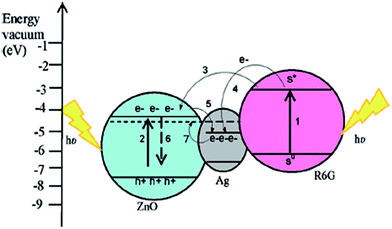 |
| | Fig. 12 Electron transfer events of the Ag–ZnO system in the presence of R6G: (1) excitation of R6G from the ground singlet to an excited singlet; (2) bandgap excitation of ZnO to generate an electron–hole pair; (3) a sensitized electron transfer from the dye to ZnO CB; (4) sensitized electron migration from the dye or Ag islands; (5) electron transfer from the ZnO CB to Ag; (6) charge carrier recombination; (7) shifting of the Fermi level of silver (reprinted with permission from ref. 192; Copyright 2008 @ American Chemical Society). | |
The worm-like Ag (2.8 at.%)–ZnO core–shell composite with the single crystal Ag NWs serving as its core, on which a dense coating of ZnO particles grow as the shell, exhibits the maximum activity for RhB degradation.195 The synthesis route involves the dispersion of single crystal Ag NWs into a mixed zinc acetate and TEA aqueous solution. The ZnO particle seeds are then partially deposited onto the surface of the Ag NWs to form nucleation sites for the subsequent growth. During the early stages of ultrasonication, ZnO crystal nuclei are formed on the Ag lateral surface by the reduction reaction of zinc acetate. The initial particle seeds so formed continually aggregate and crystallize over the course of the reaction time, leading to an increased diameter of ZnO shell. Finally, a dense and complex worm-like heterostructure is formed.195 Gu et al.196 reported a high activity of dendrite-like Ag (8 at.%)–ZnO, wherein the ZnO NRs were grown hetero-epitaxially on the Ag NWs of the {111} and {100} lateral surfaces. This heterostructure formation involved two steps: (i) single crystal Ag NWs with a truncated-rhombic cross-section are dispersed in zinc nitrate and HMT solution. The Ag NWs have four {111} and {100} lateral surfaces, on which the ZnO NRs were grown; (ii) the nucleus of crystalline ZnO is formed on the Ag lateral surfaces by the thermal decomposition of Zn2+ amino complex, followed by a preferential growth along the 〈0001〉 direction during the continuous chemical reaction. Thus, the ZnO NRs grow perpendicular to the length direction of Ag NWs, in order to minimize the interaction or competition among themselves. Finally, ultrathin ZNWs or NTs also grow from ZnO NRs.196
6.2 Photocatalytic activity of M–ZnO (M = Pd, Pt and Au)
The photocatalytic performance of ZnO for the degradation of gas phase n-C7H16 and phenol in the liquid phase could be improved after depositing Pd (0.5 wt%) on the ZnO.27,197 The lattice oxygen on the ZnO surface was reduced after Pd deposition, accompanied by an enhancement in the adsorbed oxygen content as well as surface hydroxyl groups. The electron transfer from CB of ZnO to the deposited metal and the adsorbed oxygen depends on the size and distribution of metal on the semiconductor surface. If the sizes of metal NPs are too large, its Fermi level will be lower than that of adsorbed oxygen and the electrons cannot move uphill. In contrast, the Fermi level of very small size metal NPs lies above the CB of ZnO itself, due to the size quantized effect, thus disabling the electron transfer from ZnO CB to metal NPs. Thus, an optimal particle size is essential for the metal NPs, to ensure that the Fermi energy level lies between ZnO CB and the adsorbed oxygen, to facilitate unidirectional electron transfer from ZnO CB to the metal NPs, and then to the adsorbed oxygen.198
Pt–ZnO porous nanocages were fabricated through the incorporation of Pt into ZnO porous shells by an ultrasonic irradiation-assisted two-step etching of Zn–ZnO core–shell NP colloids.199 The diameter of the Pt clusters was tuned from 2.5 to 1.1 nm with a change in ultrasonic power from 160 to 400 W, and the size distribution became narrower. The first step in H2PtCl6 etching induces core hollowing, and then Pt incorporates through the diffusion–redox–deposition process. The second step in C4H6O6 etching induces the nanoshell opening, due to the amphoteric nature of ZnO. These nanocages were active for the degradation of MO, which is attributed to their outstanding features: (i) abundant nanoscale Schottky barriers, formed because of their small size and high density of embedded Pt clusters, which endow these nanocages with a strong interface to separate the charge carriers; (ii) a hollow and porous structure, which increases their specific surface area and the active sites for redox reactions.199
Au–ZnO hybrid with a hexagonal pyramid-like morphology was active for the degradation of RhB.200 These nanopyramids possess a narrow size distribution, comprising Au NPs at the tip and hexagonal ZnO nanopyramids as the tail. In the presence of oleylamine and dodecanol as the solvent and capping agent, respectively, ZnO nanopyramids epitaxially grow over the presynthesized Au seeds, in which Au decorates at the basal surface of hexagonal ZnO nanopyramids ({0001} polar plane) as a result of the lower interfacial energy at the polar–metal interface.201 In another report, a multifunctional hybrid structure of Au NPs and a 3D hierarchical flower-like ZnO comprising a large specific surface area and a small Au NPs was positive for the degradation of RhB.202 The morphology of the Au–ZnO was very stable, and its efficiency was retained even after three runs, underlining its extraordinary stability and reuse in photocatalysis. The unique features of this composite are as follows: (i) interspaces in its flower-like nanostructures allow facile diffusion and the mass transportation of RhB molecules and hydroxyl radicals during the photochemical reactions; (ii) the strong electronic interaction between the plasmonic Au NPs and ZnO result in an efficient charge transfer process; (iii) the Au NPs on the ZnO surface lower the work function203 and the adsorbed oxygen at the interface between Au and the ZnO, thereby facilitating an easy electron transfer to oxygen, which produces reactive oxygenated species to enhance the degradation reaction. The Au (25.5%)–ZnO NRs had higher activity for the degradation of RhB and 4-CP over ZnO NRs.204 In this study, gold was decorated on ZnO NRs via galvanic displacement reactions at room temperature with the aid of Cu foil and CoCl2 solution. The dynamic force for a galvanic displacement reaction stems from the difference in the half-cell potentials between the metal ions to be reduced and the substrate to be oxidized. For the deposition of metal NPs, the half-cell potential of a reduced species must be higher than that of an oxidized substrate. Consequently, Au NPs with a narrow size distribution can be directly deposited onto a Cu foil without any additional reducing or capping agents. CoCl2 improves the hindrance towards homogeneous nucleation, while an autocatalytic growth of particles rapidly depletes the gold monomer concentration in the solution, with both the effects collectively favouring a heterogeneous nucleation. The mechanism of the degradation of phenol involves the following steps; (i) scission of the ring C–Cl bond to give a short-lived hydroxyphenyl radical, which rapidly combines with a proton to form phenol, followed by o-hydroxylation, which results in catechol; (ii) the 4-chlorocatechol resulting from the hydroxylation of 4-CP undergoes p-dechlorination and subsequently reacts with the protons in the presence of a CB electron to form catechol; (iii) oxidative hydroxylation of the catechol and phenol gives hydroxyhydroquinone and hydroquinone. Finally, the phenolic rings are opened to form aliphatic compounds, which on continuous oxidation lead to mineralization (Fig. 13).204 The thiolate-protected Au NPs (2 wt%) decorated on ZnO exhibited an enhanced activity without requiring any thermal activation for both oxidative (R6G degradation) and reductive (thionine degradation) pathways under visible light,205 which was in contrast to TiO2 with thiolate-protected gold NPs.206 The presence of ligands on the Au NPs imposed a large kinetics barrier for an electron transfer from TiO2 to the gold NPs, and thus the catalytic activity was accelerated only after removing the ligand after thermal treatment. In contrast, the thermal treatment of Au–ZnO did not significantly improve the activity, indicating that electron transfer through a linker ligand to the Au NPs occurs at a relatively short time-scale. The strong electrostatic force of attraction between a negatively charged carboxylate anion of a glutathione molecule and a positively charged ZnO surface under neutral pH conditions resulted in Au–ZnO without the need of any additives. The monodispersed Au NPs surface avoids both direct- and trap-related charge carrier recombination channels by extracting electrons from the photoexcited ZnO and supressing recombination within the metal oxide.205 The photocatalytic activity increased with an increase in gold size from 1.1 to 2.8 nm, which is in accordance with the electron transfer rate from ZnO to Au NPs, which followed the size-dependent capacitance model. Thus, both the photocatalytic and electron transfer rates could be controlled by varying the size of the mediating gold capacitors.207
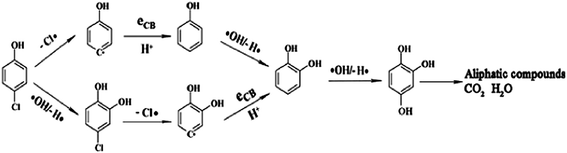 |
| | Fig. 13 Photodegradation pathways of 4-CP (reprinted with permission from ref. 204; Copyright 2009 @ American Chemical Society). | |
The Au (3 wt%)–ZnO obtained by the flame spray pyrolysis was effective for the degradation of MB, whereas Pt (1–3 wt%)–ZnO exhibited a poor efficiency, although PL measurements revealed the low degree of carrier recombination for both the composites.208 The deposition of Pt renders an ohmic contact, which neither induces the Fermi level shift towards more negative potentials nor stores the photogenerated electrons, but rather it discharges them into the electrolyte.209,210 In contrast, the Schottky contact between the Au and ZnO facilitates efficient charge separation and induces a Fermi level equilibration between the Au and ZnO. The metallic gold has an unusual capacity to store electrons without facilitating their discharge into solution, and thus the photogenerated electrons get distributed between the semiconductor and the metal layer. However, such an ohmic contact promotes the efficiency of certain photocatalytic reactions.211 The photoactivity of Au–ZnO NRs for the degradation of MO was found to depend on the diameter and the densities of the Au NPs on the ZnO surface. For instance, Au deposited on ZnO NRs using 10−4 to 10−5 M HAuCl4 with photoirradiation for 30 min exhibited a higher activity compared to other reaction conditions (10−3 or 10−4 M HAuCl4, 10 min).212 The maximum activity was observed for Au NPs < 15 nm, but then the activity declined for 30 nm. It was suggested that the Au NPs with a smaller diameter and a high density on the surface of ZnO NRs induce a greater reduction in the built-in potential barrier, due to a large shift in the Fermi level with CB.182,210 At higher metal loadings, the intensity of the incident light for the ZnO absorption will be reduced by the scattering, due to the larger Au NPs on the tips. In addition, the formation of a Schottky barrier at the junction of ZnO NRs and Au NPs inhibits the electron injection from the semiconductor to metal.209,213 The charge distribution between UV-irradiated ZnO and gold NPs results in the Fermi level equilibration. Additionally, the transfer of electrons to the metal nanocore, followed by equilibration continues until the Fermi level reaches close to the CB edge of ZnO, and then each injected electron within a gold NP shifts the Fermi level by 0.1 V, which obviously increases the photopotential of the nanocomposite electrode.210,214 The different optimum content of metal NPs on the ZnO surface for maximising the photoactivity, as reported by several research groups, is due to the varied morphologies of the composite, the crystallinity of the metal or ZnO, the charge density distribution and the density of the interface contacts.
The multiple roles of the deposited metal, such as altering (i.e. increasing or decreasing) the surface defect concentration, increasing the chemisorbed oxygen and hydroxyl group density, and improving the charge carrier separation and visible light response, together with changes in the morphology, make it an ideal candidate for modifying the electronic properties of ZnO. The size of the deposited metal, the crystallinity of metal oxide, the segregation of the metal on the surface defective site of ZnO and the dispersivity of the metal–ZnO composite in the reaction solution are the decisive factors dictating the photocatalysis. The disadvantage of metal aggregation to clusters and the relatively low coverage on the metal oxide surface at high and low loadings, respectively, affect the photocatalytic properties of the composite materials.196,215 The decoration of noble metals onto varied morphologies of ZnO under a wide variety of preparative methods attests to its structural stability, which is required for photocatalysis. More importantly, this composite easily settles down and retains its efficiency even after many times of recycling and without an agglomeration of particulates. Rather than surface deposition, further research should focus on metal cores or metals embedded within metal oxides for practical applications. However, the exact role of a deposited metal in such an enhancement is yet to be realized in a nanoscale regime. The combination of well defined metal NPs with ZnO opens the avenue to develop tunable nanoscale materials, and whose reactivity can be controlled by varying their intrinsic and extrinsic properties.
7. Coupled semiconductors
The electronic-coupling of semiconductors with different bandgaps to form heterojunctions has become a key focus of research because of their unique properties that arise from interfacial interactions at the nanoscale, which are not encountered in individual nanomaterials.216 A solid interfacial structure is an underlying factor in establishing the charge transfer behavior between different semiconductors in their heterostructure, and thus their photocatalytic activities. It is widely accepted that an extended carrier lifetime and an enhancement in the interfacial charge transfer to the targeted substrates can be attained by such heterostructures, i.e. through the formation of a heterojunction interface or an intrinsically staggered band offset.217 This improvement is related to the interface electric field built into junctions, originating from the band bending and the vectorial transfer of electrons and holes in the composite. Although pure ZnO has a defect-rich chemistry, intense interest is devoted to hybridize it with other nanophase materials, as this is advantageous to overcome some intrinsic defects of ZnO and to impart it with new physico-chemical and optoelectronic properties, in order to extend its applications in various diverse fields.
7.1 Coupling of ZnO with metal oxides
(a) Photocatalytic activity of ZnO–TiO2. Photocurrent generation and the degradation of MB was enhanced for the ZnO NRs–TiO2 composite (Zn/Ti molar ratio of 2:1) under UV-vis illumination, as the large surface area enabled maximum light harvesting ability and improved the electron transport properties.218 The deposition of titania NPs on the ZnO NR passivates the surface recombination centers and act as a radial energy barrier that can repel electrons from the surface of the ZnO NRs. Alternatively, electron transport within a single crystalline ZnO NR is considerably faster and the surface fields within each NR reinforces the charge separation because of the longer diffusion length for the charge carriers.219 The preserved crystallinity of TiO2 NPs and ZnO NRs permit an efficient electron injection and transport within the composites. The Mott–Schottky plot revealed a large negative shift of CB and a high donor density, which is attributed to the high concentration of surface states.218 The dispersion of titania on ZnO reduces the defect density and inhibits photocorrosion.54 Anatase TiO2 immobilized on single crystalline TPs, such as ZnO, enhance the degradation of phenol under a Hg lamp without a cut-off filter, although its activity is lower compared to Degussa P25 under excitation wavelengths of >340 nm. The close match between the {101} crystal plane of anatase with the ‘a’ and ‘b’ values of hexagonal ZnO, facilitates an efficient heterojunction via an epitaxial growth mechanism.220 The highly crystalline ZnO–TiO2 prepared by the direct mixing of sol and calcination (500 °C, 2 h) was active for the degradation of MO.221 The activity exhibited a linear tendency of rate constant against the value of Ti/(Ti + Zn), excluding at 75% because of the poor crystallization. This fact was confirmed by the increment in the degradation rate with increases in the calcination time to 5–10 h. The derived linear fit followed the relation:| |
 | (54) |
Alternatively, treating the above composite with a 7–28% NH3 atmosphere (24 h, room temperature) and calcination at 500–700 °C for 2 h also improved the crystallization and promoted the anatase to rutile phase transition, consequently enhancing the photoactivity.222 The dominant hexagonal ZnTiO3 was observed at 700 °C for the NH3-treated composite, while both the cubic and hexagonal ZnTiO3 phases were formed for the untreated composite. However, a poorly crystallized composite prepared by the in situ mixing of sol followed by calcination showed very low activity.221
Macro-mesoporous thick films of ZnO–TiO2 with homogeneously distributed open porosity have shown promise for the degradation of MO.223 The macroporous channels improve the light harvesting capacity by enhancing the light transfer paths for photon energy distribution onto the mesoporous network. Such a structure-in-structure arrangement favours a molecular transport control and prevents catalyst deactivation by the inert deposits. Thus, the macro-/meso-porous structures offer readily accessible pore-walls for molecules and also diminish the pressure drop over a monolithic material.223 However, the composite activity was lower than for pure TiO2, indicating that the presence of ZnO was found to hamper the composite activity.224 It was proposed that the TiO2 particles were surrounded by ZnO film, and an optical excitation results in electron accumulation in the ZnO film, while holes are confined to the particles, which reduces the number of sites available for degradation.223 The sulfated ZnO–TiO2 obtained via the modified sol–gel route and calcination (550 °C, 4 h) using citric acid as a complex reagent with zinc sulfate showed a superior performance for the degradation of MO compared to the composite prepared with zinc chloride and zinc nitrate precursors, indicating the role of the sulfate group in achieving a high activity.225 In addition, the sulfate group restrained the anatase phase even at a high calcination temperature in the composite. The sulfated ZnO–TiO2 showed a superacid behaviour, which favours the transfer of CB electrons to the surface, resulting in an efficient charge carrier separation. The activity was reduced after washing with water due to the removal of the sulfate groups and also on calcination at 700 °C (4 h), because of the decomposition of the sulfate group, the increase in the rutile content and the formation of impure ZnTiO3.225
The self-supporting ZnO–TiO2 fabricated by the tape casting and lamination process followed by calcination (600 °C, 3 h) exhibited a high activity for the degradation of Remazol Brilliant Red F3B. At this temperature, the interdiffusion between ZnO and TiO2 greatly prevents the formation of binary compound and coarsening of the particles that favour an efficient photocatalysis.226 The formation of less active phases, such as Zn2TiO4 and Zn2Ti3O8, reduce the activity of the composite at high calcination temperatures (650–700 °C). The tricomponent ZnO–TiO2–SnO2 prepared by the sol–gel method using a Sn(Zn)/Ti molar ratio of 0.05 and calcined at 500 °C (3 h) was more beneficial for the degradation of MO compared to a composite obtained by a solid state reaction under UV/visible light.227 The XPS revealed the formation of Ti3+ species, indicating the presence of many oxygen vacancies in the sol–gel composite. The TiO2 has a CB edge sandwiched between the ZnO and SnO2, giving a “staggered” type-II heterojunction at the composite interface. Under light illumination, an electron transfers from ZnO to TiO2 to SnO2, whereas a hole migrates in the opposite direction to favour charge separation (Fig. 14). The loss of crystallinity and rutile formation at high Sn(Zn)/Ti molar ratios lower the activity. In another study, ZnO–TiO2–SnO2 (4:1:1) synthesized via a solid state reaction (calcined at 500 °C, 2 h) was remarkably active for the degradation of MO compared to TiO2 and SnO2, but had a lower activity compared to ZnO.228 This situation was due to the presence of a highly active ZnO in association with a less active SnO2 in the tricomponent system. The activity decreased with an increase in the calcination temperature (≥700 °C), due to the nucleation of photoinactive inverse spinel Zn2TiO4 and Zn2SnO4 polymorphs, and it was completely inert after thermal treatment at 1300 °C (42 h) because all the oxides reacted to form solid solution Zn2Ti0.5Sn0.5O4.228 The photocatalytic activity of the ZnO NT arrays-TiO2−xNy and ZnO hollow sphere-TiO2 towards NOx decomposition decreased with increase in the emission wavelength of the excitation source.69,229 The vectorial electron transfer takes place from ZnO CB to TiO2−xNy (or TiO2) CB, whereas the hole transfers in the opposite direction to facilitate charge separation. In addition, an additional energy level created by Ti5O9 binds the excited electrons and also enables the formation of exciton by light with lower energy.229
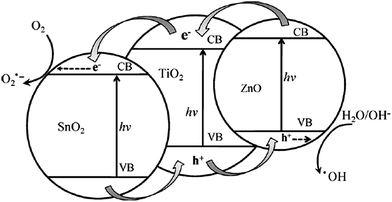 |
| | Fig. 14 Energy band structure and electron–hole separation process of a tri-component SnO2–TiO2–ZnO system (reprinted with permission from ref. 227; Copyright 2012 @ Elsevier). | |
The advantage of ZnO–TiO2 stems from the high absorbance of ZnO and the better stability of TiO2. The three binary compounds formed in this system are Zn2TiO4 (cubic), ZnTiO3 (cubic or hexagonal) and Zn2Ti3O8 (cubic),230 and any of these solid compound can nucleate via a solid state reaction, depending on the annealing environment, and the crystal structure of ZnO and TiO2, together with their relative contents. As a consequence of the structural resemblance, Zn2Ti3O8 and ZnTiO3 were formed with anatase and rutile, respectively. Both ZnTiO3 and Zn2Ti3O8 are stable phases below 900 °C, whereas Zn2TiO4 is most stable above this temperature.226 In fact, Zn2Ti3O8 is known to be a defect form of an inverse spinel structure of Zn2TiO4 because of the missing Ti ions in the lattice.
(b) Photocatalytic activity of ZnO–SnO2. ZnO–SnO2 NFs synthesized by a combined sol–gel and electrospinning technique have shown a high activity for the degradation of RhB compared to the ZnO NFs and SnO2 NFs, due to their arrangement of a stable heterojunction and a large specific surface area.231 The average grain size of ZnO (42 nm) in the composite is larger compared to pure ZnO (30 nm), whereas the average grain size of SnO2 (13 nm) in a coupled system is smaller than pure SnO2 (31 nm). These electrospun–NF composites could be reclaimed easily by sedimentation without any loss of activity, due to their 1D nanostructural properties. A mesoporous-network-structured semicrystalline SnO2-high crystalline ZnO was active for the degradation of MO.232 This was due to the presence of more surface reaction sites, which favour the adsorption and transportation of the dye molecules. The TEM image revealed a multipod framework for the composite and that each NP is attached to several other NPs, indicating the formation of a stable heterojunction. In another study, the degradation of MB using mesoporous ZnO–SnO2 collapsed in an acidic medium but improved in basic conditions, which was rationalized on the basis of the surface charge density of SnO2 (PZC 4) and ZnO. The adsorption of cationic dye is favoured on negatively charged metal oxide surfaces under an alkaline medium.233 A high roughness of ZnO–SnO2 thin films is beneficial for the degradation of MB upon calcination (500 °C, 1 h) with a Zn:Sn atomic ratio of 80:20. In addition, this composite film was very resistant to acid etching with respect to photostability.234 The maximum photocatalytic activity of ZnO–SnO2 prepared by a co-precipitation method for the degradation of MO was observed at 33.3 mol% of SnO2 at an annealing temperature of 500 °C (10 h).235 The grain sizes of ZnO and SnO2 were smaller in the composite compared to their respective pure phases, indicating the mutual interference of SnO2 and ZnO on the particle growth dynamics. The activity was reduced at a high temperature treatment (≥700 °C), as ZnO reacts with SnO2 (4.8–95.2 mol%) to form a less photoactive Zn2SnO4, which was otherwise observed at 1000 °C in a solid state reaction method.236 The uniform coating and tight decoration of SnO2 NPs on ZnO NRs resulted in a high performance for the degradation of RhB. The PL investigation revealed a decreased UV emission for the composite compared to the bare ZnO NRs, which reflects the spatial charge separation from the type-II band alignment.237 An extremely high adhesion of SnO2 NPs on ZnO NRs was observed even after ultrasonicating the composite in ethanolic medium. This composite was achieved by a two-step thermal evaporation approach: (i) ZnO NRs were obtained through the thermal evaporation of zinc powder; (ii) SnO2 was coated on the pre-fabricated ZnO NRs from the thermal disproportionation of SnO powder.| |
 | (55) |
| |
 | (56) |
Hydrogen generation was observed from dye sensitized SnO2 (10–15 nm) attached to the large ZnO (∼600 nm) particles under visible light in the presence of a hole scavenger and a Pt co-catalyst.238 On the basis of the series of modifications, it was found that the premixing of ZnO and SnO2 prior to dying and the presence of a dye on SnO2 and Pt islands on ZnO favour hydrogen evolution. A photon absorbed by a dye molecule on the surface of a SnO2, could inject an electron across a few SnO2 crystallites without losing energy to reach ZnO CB at a distance of 120 nm from the ZnO surface, suggesting the involvement of hot electrons with ballistic transport. Then, the electrons on ZnO CB were captured by Pt islands to initiate water reduction.
| | |
2H2O + 2e− → 2OH− + H2
| (57) |
The bandgap, work function and electron affinity of ZnO are 3.37 eV, 5.2 eV and 4.3 eV, whereas those of SnO2 are 3.5 eV, 4.9 eV and 4.5 eV, respectively. The VB of ZnO is positioned between the CB and VB of SnO2 and the CB of ZnO is positioned above the VB and CB of SnO2. Accordingly, a ZnO–SnO2 heterojunction involves the vectorial transfer of an electron from ZnO to SnO2 and hole transfer in the reverse direction. Hence, this heterojunction not only serves as a source of carriers but also acts as a sink for electrons and holes. The XPS revealed ΔEv = 0.7 ± 0.05 eV and ΔEc = 0.2 ± 0.05 eV for this heterojunction.233 The surface acid–base properties, low-cost and cheap availability of SnO2 and ZnO, combined with the compatibility of some specific crystallographic planes to form a heterojunction, would be very useful for the further design of this bicomponent system.239
(c) Photocatalytic activity of ZnO coupled with other metal oxides. ZnO–CuO prepared by the coordination–oxidation–homogeneous coprecipitation method exhibited a better activity for MO degradation under UV-vis light.240 The composite was prepared by using a metallic copper as the Cu2+ source, NH3 as the coordination agent, air as the oxidizer and ammonium bicarbonate as the precipitating agent, along with ZnO. The change in the activity of the composite was in agreement with the analysis from particle growth dynamics. The activation energy for the growth dynamics in low (≤450 °C) and high temperature (≥450 °C) regions are 14.94 kJ mol−1 and 59.84 kJ mol−1, respectively. At low temperatures, crystal particles with a small size, an incomplete crystal lattice, and many vacancies are obtained leading to a low crystal grain growth activation energy, which contributes to the high photocatalytic activity, while grain growth will be rapid, resulting in the loss of surface area and surface activity at high temperatures. Thus, the vacancies will be reduced, and hence the crystal grain growth activation energy increases. In addition, ZnO–CuO was also effective for the treatment of real textile waste water containing MO and MB dyes under visible light.241ZnO–ZnO2 with small ZnO2 granules fused at the surface of a prismatic ZnO exhibited significant activity for MO decomposition, due to the presence of either structural or energetic heterogeneity within the interfacial region of the composite.242 As-obtained ZnO2 from the aqueous reaction of zinc sulfate and H2O2 under an alkaline medium was less active, due to its poor crystalline nature. The hydrothermal treatment (120–180 °C, 2 h) of ZnO2 gradually transformed it to ZnO, and a high photoactivity was noticed in the range of 140–150 °C, while pure ZnO obtained at 180 °C showed poor performance (Fig. 15). The difference in the electronic band structure between ZnO and ZnO2 (4.12 eV) results in a potential gradient at the interface, and the interfacial structural defects serve as a trap site for excitons, thereby suppressing recombination.242
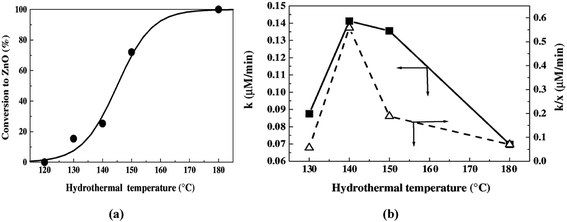 |
| | Fig. 15 (a) Phase transformation from ZnO2 to ZnO; (b) rate constant (k) and normalized rate constant (k/x) vs. hydrothermal temperature (x is the ZnO2-to-ZnO conversion) for the degradation of MO (reprinted with permission from ref. 242; Copyright 2005 @ Elsevier). | |
The ball milling of n-ZnO in water doped with p-CaFe2O4 exhibited better activity for MB degradation, which was attributed to the stable integrated micro p–n heterojunction.243 The activity increased with the increase in the content of p-CaFe2O4 by up to 1.0 wt% and decreased thereafter as the absorption of light and the generation of electron–hole pairs were lowered, together with a decrease in the space charge region on the particle surface. When the ball milling times were 3, 6, 12 and 24 h, the degradation efficiencies were 50.8, 73.4, 78.7 and 75.6%, respectively. With increases in the ball milling time, the specific surface area and the number of active sites per unit weight of the catalyst increases, thus enhancing the activity. Beyond this optimum time, it is proposed that the fresh surface formed by the high energy ball milling possesses a robust surface energy, which favours agglomeration, with a loss in surface area. In the process of ball milling, the crystal lattices of CaFe2O4 and ZnO are vulnerable to severe plastic deformation that induces stress and strain. This creates a crystal lattice distortion, and hence many defects are formed within the particles, and which have a high surface energy. Under this situation, the activation energy for the diffusion of elements decreases to a large extent, which allows the facile atomic or ionic diffusion among the elements at room temperature. When the activity of a powder system is sufficiently high, the collision between the balls and the grains of powder produce a rise in the interface temperature, which induces a coupling reaction between the semiconductors.243,244 ZnO–ZnFe2O4 was ineffective for MG degradation for all the contents of ZnFe2O4, both in the thin film and in the powder form.245 Interestingly, the ZnO–ZnFe2O4 core–shell nanocable was active for RhB degradation under visible light. The nanocable arrays configuration are associated with a large surface-to-volume ratio, leading to an optical path increase of incident light in the structure, which suppresses reflection, thus enhancing the light absorption. In this composite, the unidirectional electron transfer takes place from the excited dye to ZnFe2O4 CB to ZnO CB, while holes are left in the VBM of ZnFe2O4. The degradation of RhB with a composite involved both the de-ethylation and destruction of the chromophore, while only the latter mechanism was observed with methanol.246 ZnO–Ag3PO4 at an optimum content of 3 wt% Ag3PO4 prepared by the ball milling was active for RhB degradation under visible light.247 As evidenced by the XPS studies, the ΔEv and ΔEc for this heterostructure were found to be 0.3 eV and 1.05 eV ± 0.05 eV, respectively. The reaction rate was suppressed with isopropanol compared to benzoquinone, indicating that the hydroxyl radicals were the dominant oxidative species in the degradation mechanism. During the photocatalytic process, the holes residing on ZnO VB induce oxidation reactions, while electrons accumulated on Ag3PO4 CB trigger hydrogen peroxide generation. It was proposed that the CB and VB levels of Ag3PO4 were positioned at 0.45 eV and 2.9 eV (vs. NHE), respectively.248 The bottom of CB was mainly composed of hybridized Ag 5s, 5p and a small quantity of P 3s orbitals, while the top of VB constituted hybridized Ag 4d and O 2p orbitals.247
ZnO–AZO thin film with a AZO thickness of 30 nm increased the degradation efficiency of RhB and MB, while it was lowered for 80–200 nm.249 This was ascribed to IEF differences induced by the dissimilarity in carrier concentration between AZO (1.5 × 1018 cm−3) and ZnO single crystals (8.8 × 1016 cm−3). The difference in the Fermi levels of AZO and ZnO single crystals gave rise to a built-in potential of 0.07 eV, an IEF with a maximum intensity (Em) of 5.2 × 104 V cm−1 and a width of 27 nm. When ZnO is excited by UV light, most of the charge carriers are produced in a thin, effective absorption layer, which is about a 100 nm below the surface.250 Thus, IEF lies completely within the light absorption layer and the charge carriers are efficiently separated by a 30 nm AZO film thickness. When the thickness is further increased (80–100 nm), part of the IEF will be located in the effective absorption layer and thus, the separation efficiency will be near the edge of light absorption layer. Under these circumstances, only a few electron–hole pairs will be generated in the thickness zone of IEF, because of the exponential attenuation of UV light (Fig. 16). In fact, AZO thin film with a higher thickness (200 nm) had a lower efficiency compared to ZnO itself, as the charge carriers were completely generated in AZO, which had more recombination centres than ZnO single crystals.249 The incorporation of In2O3 to ZNWs resulted in a visible light response and glucose oxidation via a photoelectrochemical approach at a potential bias of 0.8 V.251 Linear sweep voltammetry and EIS studies revealed the generation of larger number of charge carriers with a charge transfer rate constant of 3.23 s−1 and a very low charge transport resistance of 19083 Ω for this composite compared to the pure ZNWs (1.41 s−1 and 47400 Ω). In2O3 had a bandgap of 2.8 eV, with both its VB and CB situated slightly above the corresponding energy levels of ZnO.252 Under visible light, electrons are excited from VB to CB of In2O3, and a transient photocurrent is generated, leaving the holes to participate in glucose oxidation. Due to the high electron mobility and geometry of vertically aligned ZNWs, the injected electrons are effectively collected as a result of the reduction in the electron transport resistance distance in the photoanode. However, a high loading of In2O3 increases the shell thickness on ZNWs and creates a large number of grain boundaries. Thus, electrons in In2O3 have to travel a longer distance to reach the ZNWs, resulting in an increased charge transfer resistance and lower separation efficiency.251
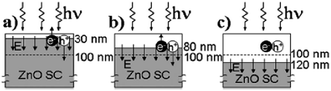 |
| | Fig. 16 The relationship between IEF and the effective UV-light absorption layer (about 100 nm, from the surface to the dotted line), in which the thickness of AZO TF is about (a) 30 nm, (b) 80 nm and (c) 100 nm (reprinted with permission from ref. 249; Copyright 2011 @ Royal Society of Chemistry). | |
The small size and high surface-to-volume ratio of the mesoporous spindle-like Fe2O3–ZnO core–shell contributed to a faster RhB degradation under UV/visible light.253 The electrons in the CB of the α-Fe2O3 core transfer to the ZnO CB shell, driven by the decrease in the potential energy. Thus, the confined potential of the excited electron–hole pair reduces their interaction with traps on the surface and in the environment. This heterostructure was obtained by the seed mediated growth route and the ZnO shell formation involved two steps: adsorption on the surface of the monodispersed seeds and growth under heat treatment (Fig. 17). The presence of spindle-like hematite seeds favours the growth of ZnO via heterogeneous nucleation, rather than forming separate nuclei by homogeneous nucleation. The gaps in the single α-Fe2O3 seed are distributed between both the grain boundaries, and the spindle-like particle is composed of small nanocrystals, while the overall particle surface is quite rough. This feature endows α-Fe2O3 seeds with a high adsorption capacity for zinc ions. The adsorbed zinc precursor reacts with NH3 to form Zn(OH)2, which then transforms to a layer ZnO shell during thermal treatment (550 °C, 2 h).253 The ZnO–NiO (cubic) NFs exhibits a high activity for RhB degradation compared to the individual components.254 Under UV light, an electron transfers from the CB of NiO to the ZnO CB, while a hole migrates in the opposite direction. The p-NiO (Eg = 3.5 eV) has a high hole mobility and low lattice mismatch with ZnO, which benefits the formation of a stable p–n heterojunction.255 In addition, PL studies indicate a high concentration of oxygen vacancy defects for the composite, which improves the charge carrier separation.254
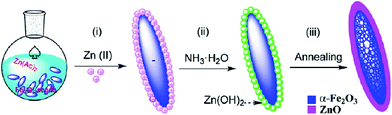 |
| | Fig. 17 Stepwise formation of a Fe2O3@ZnO heterostructure; (i) adsorption of zinc species on hematite seeds; (ii) addition of ammonia to form Zn(OH)2; (iii) decomposition of the Zn(OH)2 to form a ZnO layer (reprinted with permission from ref. 253; Copyright 2012 @ American Chemical Society). | |
7.2 Photocatalytic activity of ZnO–Metal sulphide
The sensitization of ZnO via electronic coupling with a narrow bandgap photosensitizer is a viable approach to exploit the utilization of the solar spectrum. The conventional charge transfer between narrow and wide bandgap semiconductors is beneficial for isolating the oxidation and reduction reaction, as the electrons and holes dwell on different semiconductor surfaces. For an efficient transfer of electrons between the sensitizer and ZnO, the CB energy level of ZnO must be lower than the excited state or the CB edge of the sensitizer. In this perspective, CdS with wurtzite polymorph is more desirable because of its similar lattice structure with ZnO, thus establishing a close interaction between these two semiconductors.
ZnO–CdS displayed an appreciable activity for the degradation of RhB under the simulated sunlight, attributed to its extended photoresponse and increased charge carrier separation.256 The nanostructures were composed of 3D flower-like ZnO microstructures coated with cubic CdS NPs. The composite preparation involved two stages: first, flower-like ZnO was dispersed in an aqueous solution of CdCl2 by ultrasonic agitation to facilitate the adsorption of Cd2+ ions on the ZnO surface, followed by the addition of sodium sulphide, resulting in the uniform coating of CdS. Due to the rapid precipitation and low reaction temperature, small size CdS NPs were formed. The coarse–surface structure enhanced the dye adsorption and reduced the reflection of incident light, thus benefiting the photocatalytic reactions.257 The ZnO–CdS system has a “staggered type” type-II band alignment, which means that ZnO CB is located between the CB and VB of CdS.258 Under visible light, an electron migrates from the CB of CdS to the ZnO CB, while holes do not transfer in the opposite direction, as the CdS VB is more cathodic than ZnO. The electrons accumulated on ZnO initiate dioxygen reduction, whereas the holes in CdS react with water adhering to its surface to generate hydroxyl radicals or they can also directly oxidize organic molecules. The performance was found to depend on the CdS loading; with CdS prepared by 2.5 g L−1 CdCl2 and 1.25 g L−1 of thiourea showing a high activity compared to other conditions.259 The reaction rate was drastically suppressed in the presence EDTA and slightly with dimethyl sulfoxide (electron quencher), indicating that the holes are the main active species in the degradation mechanism. For a higher CdS loading (5.0 g L−1 CdCl2 and 2.5 g L−1 of thiourea), the activity was reduced due to the formation of Zn1−xCdxS ternary compound, which hinders the sensitization process by transporting the electrons from CdS to this transition layer without being transported to ZnO due to the lower position of Zn1−xCdxS CB.260 In this study, the composite was prepared with the aid of ultrasound irradiation. When a ZnO microsphere is added to the reaction system, the hydrogen and hydroxyl radicals are formed from the ultrasound induced dissociation of water molecules. The hydrogen radicals act as a reducing species and trigger the decomposition of thiourea to form S2−, which reacts with Cd2+ to form the CdS NPs. The ultrasound induced cavitations provide a clean ZnO microsphere surface for the adhesion of S2− (or CdS), thereby facilitating an intimate contact between the two semiconductors.
| | |
2H˙ + RS → H2S + R˙ (RS = H2NCSNH2)
| (59) |
| | |
n(CdS) + ZnO → ZnO–CdS
| (61) |
Kundu et al.260 also reported that a minimum CdS loading (4.1 wt%) resulted in faster degradation of MB, whereas a large bandgap of CdS and a decrease in the surface hydroxyl groups at the ZnO surface at high loading (5.4–11.7 wt%) was detrimental. In the synthetic route, cadmium nitrate was rapidly evaporated on ZnO NRs, followed by the addition of Na2S with varied concentrations. The formation of the composite was controlled by two competing processes: the dissolution of the precursor phase in aqueous solution and their conversion to sulfide. At a low concentration of Na2S, the rate of sulfiding is considerably slower compared to the rate of dissolution, and therefore most of the precursor film dissolves before being converted to the sulfide phase. Conversely, at high concentrations, most of the film is converted to the sulfide phase as the conversion rate is considerably faster compared to the dissolution rate, whereas fine CdS NPs are formed on the substrate at an optimum concentration of Na2S.260 The controlled heterogeneous nucleation of the CdS NPs on the ZnO TPs resulted in a stable heterojunction that was active for the degradation of MB under visible light.261 CdS formation on ZnO–TPs proceeded via an ion-by-ion mechanism when DMF was used as the solvent for the growth of CdS in the neutral environment. It is likely that the Cd2+ ions adsorbed on the ZnO surface act as “nucleation seeds” for CdS formation. The slow dissociation of thiourea with DMF enables the control of the S2− concentration through temperature setting and also avoids the bulk precipitation of CdS.262 This synthetic route is an alternative method to preserve the ZnO nanostructured morphology from the damages induced by the standard alkaline chemical bath deposition process and does not require the addition of any surfactants or passivating agents. Thus, superior activity originates from a well dispersed and relatively suitable coverage of CdS NPs on the ZnO surface.
The core–shell NRA of ZnO–CdS, with a shell thickness of 30 nm, showed a high activity under simulated solar light for the degradation of RhB.263 Study of the decay kinetics indicated that the average carrier lifetime of the core–shell NRA is longer than that of uncoated ZnO NRs, due to the charge separation, as the electrons resides in the core and the holes accumulates in the shell. The time required for an electron transfer is about 18 ps, which is shorter than the electron lifetime in CdS.264 Once the electron diffuse into the CB of ZnO, the probability of its decay is small because there are no free holes in the VB of ZnO under visible light. This heterostructure was designed by the surface functionalization of ZnO by citric acid, followed by the slow addition of Cd2+ and S2−, because the normal CdS exchange process is so fast that the ZnO core can be destroyed during the deposition process. During the synthesis, citric acid adsorbs on the surface of ZnO NRs via a carboxyl group with an outward orientation of –OH functional groups. The negative charges on the functionalized surfaces of ZnO attract Cd2+, and thin Cd(OH)2 layers are formed. After the addition of sodium sulphide, a CdS shell layer is formed on the surface of ZnO NRs via the substitution of OH− by S2− ions. With an increase in the amount of citric acid, CdS shell layers gradually become uniform, along with an increase in the shell thickness (Fig. 18). The well-defined ZnO–CdS grown on the indium tin oxide substrate was active for 3,4-dihydroxy benzoic acid degradation under visible light.265 In this study, the CdS layer was thinner than the room temperature diffusion length of electrons, which facilitated the ballistic process of the charge transfer pathways. Also, the CdS layer was discontinuous and too thin to support the interfacial depletion layer. Hence, a macroscopic potential barrier does not exist at the ZnO–CdS interface. The ZnO–CdS was active for the degradation of Eriochrome Black T under visible light, wherein ZnO NRAs were obtained by electrochemical deposition on the FTO substrate and CdS was then coated by the chemical bath deposition.266 The optimal loading of CdS corresponds to a deposition time of 40 min, at which time ZnO–CdS showed a significant visible light response compared to at a low deposition time (3 min) where the visible light response was not observed. Interestingly, a blue shift in CdS absorption was observed for a deposition time of 10–20 min, indicating that the NPs were subjected to strain during the growth on ZnO. It was proposed that an initial voltage pulse and the time are very important for obtaining a homogeneously distributed seed layer of ZnO for the subsequent growth of ZnO NRs on the FTO substrate. In other words, ZnO NRs were grown in short clusters and were deposited on primary ZnO crystals in the absence of a voltage pulse. A shorter time (0.5 h) leads to a low seed density, while a longer time (3 h) results in the formation of a continuous layer of ZnO on FTO with an initial voltage pulse. Contrarily, ZnO NRs deposited for 1.5 h had a high density, long length, small diameter with narrow size distribution and a smooth surface. Unfortunately, this composite was susceptible to photocorrosion after 2 h of degradation reactions.
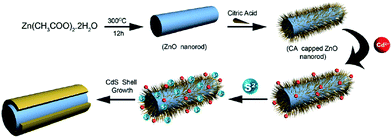 |
| | Fig. 18 Mechanism of formation of ZnO–CdS core–shell nanorod arrays by the surface-functionalization method (reprinted with permission from ref. 263; Copyright 2012 @ American Chemical Society). | |
The ZnO–ZnS core–shell NRs (with a molar ratio of ZnS/ZnO of 0.6) synthesized by the water bath route (ZnO conversion to ZnO–ZnS via surface sulfidation) showed a maximum hydrogen production under UV and solar light with quantum efficiencies of 22% and 13%, respectively, from a water–glycerol mixture.267 The CB of ZnS is more negative than ZnO, while the VB of ZnO is more positive than ZnS, thereby facilitating electron and hole transfer in the opposite direction. The hole can be trapped by surface hydrdoxyl groups, which later oxidizes glycerol to CO2, and the electron on the ZnO surface reduces protons to hydrogen. The small energy gap difference between the ZnS VB and O2/H2O energy levels, together with the high oxidative power of the surface chemisorbed hydroxyl groups on ZnS, contributes to the overall efficiency. In addition, this composite was found be thermally active (40–80 °C) under UV light.267 The ZnO–CuS obtained by decorating CuO on the ZnO NTs, followed by the addition of thioacetamide (80 °C, 1 h) was active for decomposing MB at neutral pH conditions under visible light.268 The decline in degradation with silver nitrate and the increase in the reaction with ammonium oxalate (i.e. a hole scavenger) suggests that the electron-driven hydroxyl radicals were found to be the major pathway in the photocatalysis. The type-II band alignment between p-CuS and n-ZnO leads to the transfer of electrons from the CB of CuS to the ZnO CB, while holes in the VB of CuS oxidize water to generate hydroxyl radicals.
The heterostructuring of ZnO with lattice matched or dissimilar materials passivate the surface recombination centers and help it resist photocorrosion. The versatility of ZnO to be decorated by a variety of semiconductors with distinctive morphologies under different experimental conditions highlights its stable and suitable surface properties. In addition, the formation of impurity phases, interdiffusion among the coupled components resulting in an alteration of the electronic energy levels, and the change in its interfacial defect structure remain great challenges to overcome to fabricate impurity free heterojunctions. The organization pattern, crystallographic structures, electron affinity/work function, charge carrier mobility, mechanical adhesion, band bending, the nature of the surface/interface/bulk defects, and the shape of each component in an integrated heterostructure determine their physical–chemical–electronic properties and performance.216c The formation of heterostructure is limited by the band edge positions and a poor interfacial structure of the composite. While the former can be overcome by a suitable choice of material or by tuning the bandgap of the sensitizer, the latter still remains a great challenge from the point of view of creating a favourable interface structure. Because much literature discusses the growth of a secondary phase semiconductor on prenucleated ones, the one-pot synthesis involving the simultaneous nucleation of both the semiconductors with a modified surface/interface needs to be further investigated for developing cost-effective approaches.
8. Modification with carbon structures
8.1 Doping with carbon (C–ZnO)
Carbon doping is reported to be very active for visible light induced photocatalysis and promotes charge carrier separation by channelling the photoexcited electrons to nanosized carbons on the surface of catalyst.269 The heating of zinc chloride and EG (160 °C), followed by calcination (600 °C, 2 h) results in C–ZnO, which can mineralize HCHO under the irradiation of an indoor fluorescent lamp.270 After 60 min of irradiation, the CO2 concentration increased to 780 ppm for C–ZnO, whereas only 595 ppm was achieved for N–TiO2. This difference was attributed to the extended visible absorption edge (400–700 nm) for C–ZnO, while N–TiO2 showed weak absorption at ∼500 nm. In addition, PL studies revealed a lower intensity signal for C–ZnO compared to N–TiO2, suggesting effective charge carrier separation in the former case. The flower-like C–ZnO obtained from Zn5(CO3)2(OH)6 was active towards the degradation of RhB under visible light. This was attributed to the high oxidative power of C–ZnO as a result of the downward shift in the energy band structure and to its organized morphological features, which enhance the light absorption due to multiple reflection of the trapped incident light. The decline in the activity at higher thermal treatments (>500 °C) was due to the oxidation of the lattice carbon and the collapse of the mesoporous structure.271 The C–ZnO obtained from a solution of zinc nitrate, HMT and Vitamin-C through heating (95 °C, 1 h) and calcination (500 °C, 2 h) exhibited visible light activity for the degradation of Orange II.272 A rod-like structure for ZnO was observed with HMT, while the aspect ratio of the rods decreased with the addition of Vitamin-C, indicating a suppressed growth along [001] direction.273 The –COO− and –OH groups of Vitamin-C adsorb onto the positively charged Zn2+ (0001) surface, thus blocking contact between the growth units and the (0001) crystal surface. Under these conditions, growth along the six symmetric directions (non-polar plane) was enhanced. During calcination, ZnO crystallizes and the combustion of carbon takes place, followed by the rearrangement of the host–guest atoms to render the substitution of carbide ions at lattice oxygen sites.272 The visible light performance of carbon doped strings of ZnO was demonstrated in the degradation of 4-NP and in photoelectrochemical water splitting.274 The efficiency of bare ZnO (0.016%) was lower than for C–ZnO (0.18%). The efficiency of water splitting was calculated using the formula;| |
 | (62) |
where Vapp is the applied voltage, I is the measured current density, and Plight is the power density of illumination. In this study, the surface reconstructed monolayers of poly(styrene-block-2-vinylpyridine) diblock copolymer (PS-b-P2VP) inverse micelles were used as a template to obtain the ZnO NPs under oxygen plasma. Then, the partially removed polymer was cross-linked by UV light under vacuum, followed by carbonization (600 °C, 1 h) in an inert atmosphere, leading to carbon doping as a carbonate species in the ZnO lattice. This creates a new energy level above the VB via hybridization of the O 2p and C 2p orbitals to give a visible light response, and furthermore the carbon network at the surface may act as a channel to divert excitons, consequently supressing the recombination pathways (Fig. 19). The 3D mesoporous C–ZnO obtained by the solvothermal route (160 °C, 24 h) in EG using glucose as a carbon source showed a maximum adsorption capacity of CR (162 mg g−1) and a high visible light activity for the degradation of CR and RhB.275 Based on experimental studies, it was concluded that both glucose and EG played an important role in the formation of a hamburger-like nanoarchitecture for C–ZnO. The anionic functional group of the EG and glucose interact with the (0001) plane of the ZnO to lower its surface free energy, resulting in an equiaxed or spherical morphology. The complex mixture of the aromatic compounds and the oligosaccharides formed as a result of the chemical reactions of glucose under the solvothermal condition increase the viscosity of the resulting solution (i.e. a polymerization step). The growth of a 3D “twinning superstructure” occurs at this juncture, which takes place by the continuous adjoining of additional oligosaccharides at the surface of the ZnO subunits. This type of twinning is allowed due to the adsorption of polymers, which counterbalance and perhaps overcompensate the like charges on these faces, and then the self-assembly of these subunits may continue via stacking of their polar faces of the opposite charges facing each other.276 As the heating time is extended, the carbonaceous polysaccharides adhered on the Zn2+ transform to C–ZnO with a carbon atom occupying the interstitial sites in the ZnO. Thus, the glucose confines the growth of ZnO and also serves as an assembly agent to construct NPs into a 3D hamburger-like mesoporous superstructure.
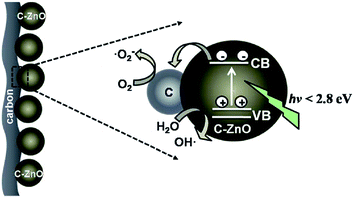 |
| | Fig. 19 Schematic presentation of doped and surface carbon in C–ZnO. The grey colour part indicates that the polymer nanowires are turned into carbon and olive colour spheres, representing C–ZnO (reprinted with permission from ref. 274; Copyright 2013 @ Royal Society of Chemistry). | |
The doping of carbon at the substitutional/interstitial site or as a carbonate ion results in only visible light absorption, and its role as a charge carrier trap is still far from over as a critical discussion. Similar to nitrogen dopants, the expulsion of doped carbon from the ZnO lattice in high temperature treatment is the major drawback for this approach. Therefore, extra caution is required to balance between carbon doping and crystallization during the preparative step.
8.2 Modification with other carbon forms
Highly crystalline 1D ZnO grown on CNFs substrate without any aggregation have shown better performance for the degradation of RhB.277 Here, the photogenerated electron may move freely towards the surface of the CNFs, and then the VB holes migrate to the surface and react with the surface adsorbed water or hydroxyl groups to produce the hydroxyl radicals. PL studies also revealed the efficient charge carrier separation in this heterostructure, as conductive 1D CNFs serve as ideal electron pathways.278 The coverage density of the ZnO NPs coating on the CNFs surface was tuned by adjusting the molar ratio of zinc precursor to CNFs during the hydrothermal synthesis (180 °C, 12 h). The solar light activity of ZnO-activated carbon increased with the increase in pH up to 9, and remained constant, even though the adsorption of Direct Blue 53 molecules is negligible in an alkaline medium.279 It was concluded that the degradation not only takes place on the catalyst surface but also near to its vicinity. This synergistic effect was attributed to the adsorption of dye on the activated carbon, which later diffuses to ZnO located at the pores, followed by a subsequent degradation (Fig. 20). The carbon-coated ZnO NRs obtained by the microwave-assisted method resulted in a faster decomposition of MB under visible light.280 This was achieved by modifying the surface of ZnO NRs by amino groups through the interaction with (3-aminopropyl)triethoxysilane, followed by grafting by glucose, and then microwave irradiation to stimulate the transformation of glucose into carbon (Fig. 21). The high activity was attributed to the following reasons: (i) amorphous carbon has a good adsorptive capacity, and hence the concentration of MB can be enriched on the ZnO surface for photocatalytic reactions. The driving force for dye adsorption is mainly through the interaction between graphitic carbon and the aromatic rings of the dye molecules.281 (ii) Coating of the low work function amorphous carbon (3.5–4.0 eV) improved the light absorption properties of ZnO. The CB electrons of ZnO could move freely to the surface of the carbon particles, thus directing the charge carriers to flow in the opposite direction. (iii) The uniform and well-dispersed carbon coating increased the corrosion resistance for ZnO, thus giving a better stability.280 The bare ZnO lost its photocatalytic activity after calcining at ≥700 °C, whereas ZnO–C retained its activity even at 1000 °C, indicating that surface carbon improved the thermal stability of ZnO.282 The ZnO–C prepared by a simple solvothermal treatment (160 °C, 24 h) of zinc acetate and ethanol with iodine was active for the degradation of RhB under visible light.283 Iodine promoted the oxidation of ethanol and resulted in the formation of carbon, which self-assembled with ZnO to form composite spheres. The extent of carbonization, and hence the photocatalytic activity was influenced by the amount of iodine used, and the optimum was determined to be 0.2 g (carbon fraction of 30%). During the process, iodine catalyzed both the alcoholysis of zinc ions and the carbonization of ethanol. A controlled experiment with only iodine and ethanol did not initiate any carbon formation, signifying that the carbonization of ethanol occurred due to the combining effect between the iodine and zinc ions. The surface hybridization of ZnO with graphite-like carbon layers suppressed grain coalescence and the crystal growth, giving exceptional photostability and thermal stability.284 The degree of graphitization was dependent both on the graphitization temperature and on the mass ratio of carbon in the synthetic procedure, and the best ratio of 16.9 wt% was reported for the degradation of MB even under extreme pH conditions. At lower carbon content, coalescence and crystal growth were not efficiently suppressed, whereas higher carbon content shielded the light reaching the catalyst surface, with both effects hampering the photoactivity. The degradation mechanism was mediated by the hydroxyl radicals on the ZnO surface, while it was mediated by direct reaction with the holes on the surface and in the solution bulk, which were attributed to an enhanced adsorption of MB on the surface of ZnO–C.
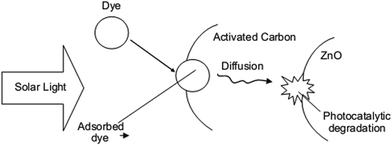 |
| | Fig. 20 Adsorption and diffusion of dye molecules towards ZnO and their degradation (reprinted with permission from ref. 279; Copyright 2007 @ Elsevier). | |
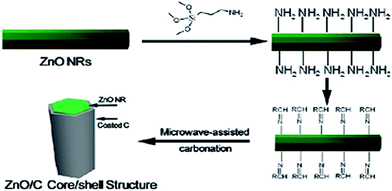 |
| | Fig. 21 Mechanism of formation of a carbon-coated ZnO core–shell structure (reprinted with permission from ref. 280; Copyright 2009 @ American Chemical Society). | |
The coating of carbon enhances the adsorption of pollutants on the catalyst surface, simultaneously facilitating both adsorption and photocatalysis. It is interesting to note that a carbon coating on the ZnO surface can be achieved by cheaply available precursors such as citric acid, alcohols and polysaccharides. The change in surface structure of ZnO after carbon coating and their influence in degradation reactions require a more in-depth study.
8.3 Modification with graphene and g-C3N4
Materials with delocalized conjugate π-structures have been extensively investigated in electron transfer processes, as they reduce recombination via capturing the photogenerated electrons from the CB of a semiconductor and transport them to adsorbed oxygen for the production of reactive oxygen species. A delocalized π-structure material coupled with a semiconductor serves as a unique “dyade” type of structure, forming a common conjugated system, which improves the photocatalytic performance in the entire spectral range.285 As a rising star of the carbon family, graphene composed of single, bi and a few layers of carbon atoms forming six membered rings has gained significant attraction because of its multifaceted physico-chemical and optoelectronic properties. In particular, its unique 2D structure and plenty of π-electrons provide a suitable platform for the growth of stable inorganic semiconductors.286 It is reported that the confinement effect of the graphene plane would favour the dispersion and stability of ZnO, whereas ZnO particles on a graphene sheet will act as a type of chemical obstacle to prevent the graphene from agglomeration, and thus help to retain its high specific surface area.287
ZnO–FGS in which ZnO NPs were homogeneously dispersed on FGS showed a high activity for the decomposition of R6G.288 This composite was prepared by a thermal treatment method, wherein the addition of PVP stabilized the GO sheets and promoted the nucleation of ZnO, followed by their deposition on the carbon sheets. An increase in the photon absorbance caused by FGS and the electron transfer pathways between the highly crystalline ZnO and FGS contribute to its overall efficiency. The degradation of MB and MO with ZnO–GO under visible light was further improved by annealing under N2 atmosphere (400 °C, 2 h).289 The composite preparation involved two distinctive stages: (i) the dissolution of zinc chloride in GO suspension, which renders Zn2+ ions available to get adsorbed at the surface of GO sheets due to their bonding with negatively charged oxygen containing functional groups via electrostatic forces; (ii) the combination of the crystal growth units of Zn(OH)42− and ZnO2− formed under alkaline medium with the functional groups of GO sheets via intermolecular hydrogen bonding or coordinate bonds that act as anchor sites for ZnO NPs. With subsequent heating at 90 °C (6 h), the ZnO nuclei are formed. The degradation mechanism initially involves the excitation of MB, followed by an electron transfer to ZnO CB via the GO sheets, which are later captured by adsorbed oxygen to produce reactive oxygen radicals. Finally, MB*+ is degraded by itself or by the attack of reactive oxygen species (Fig. 22). Furthermore, the large surface area and hierarchical porosity of flower-like ZnO anchored onto GO enhance the adsorption and mass transfer of dyes and oxygen species.289 The fabrication of ZnO–graphene via the solvothermal reaction (150 °C, 8 h) of Zn–EG–Ac complex and GO in ethanol solvent is beneficial for the decomposition of RhB.287 The reaction system possessed the following characteristics: (i) the abundant functional groups in the Zn–EG–Ac complex interact with the functional groups (–COOH, –OH) of GO and hydrolyse in situ on GO to form well-dispersed ZnO NPs without the need for using an external alkali; (ii) Zn–EG–Ac has a relatively high stability in ethanol, which promotes the growth of ZnO on graphene rather than homogeneous nucleation in the reaction medium; (iii) ethanol facilitates GO reduction and also slows down the liberation of Zn2+ to produce fine ZnO NPs tightly decorated on graphene. In another study, ZnO–graphene composite prepared by the chemical deposition route was also active for the degradation of RhB under UV/visible light.290 The redox potentials of RhB and RhB* are 0.95 V and −1.42 V (vs. NHE), while the work functions are −5.45 eV and −3.08 eV (vs. vacuum), respectively. Comparing the potential of the CB (−4.05 eV) and VB (−7.25 eV) of ZnO with the work function of graphene (−4.42 eV),291 direct electron transfer from RhB* to graphene seems to be thermodynamically more favorable (Fig. 23).292 In addition, the composite also served as a filtration membrane to remove the dye molecules from water. Graphene sheets densely decorated by the ZnO NSs resulted in a good combination for the reduction of Cr(VI),293 in which the photogenerated electron reduces Cr(VI), while the hole oxidizes water molecules.
| | |
Cr2O72− + 14H+ + 6e− → 2Cr3+ + 7H2O
| (63) |
| | |
2H2O + 4h+ → O2 + 4H+
| (64) |
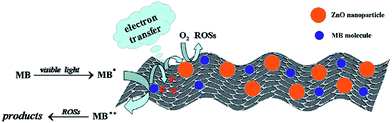 |
| | Fig. 22 Photosensitized degradation of MB over ZnO–GO under visible light (reprinted with permission from ref. 289; Copyright 2012 @ Elsevier). | |
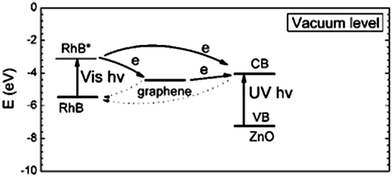 |
| | Fig. 23 Charge transfer mechanism for ZnO–graphene sensitized by a dye molecule (reprinted with permission from ref. 290; Copyright 2011 @ Royal Society of Chemistry). | |
The presence of graphene increases the light absorption intensity, as a result of the increase in the surface electric charge of the oxides in the composite, and modifies the charge carrier dynamics during irradiation, thus promoting the activity. The activity decreases at a higher content of graphene (>1.0 wt%) due to following reasons: (i) graphene absorbs the UV light, and thus there exists a light harvesting competition between ZnO and graphene; (ii) excess graphene acts as recombination centres.294 The ZnO–graphene obtained via the heat treatment of the ZnO–GO (200 °C, 2 h) containing 2.5% graphene exhibited higher visible light activity for the degradation of MB compared to the ZnO–MWCNTs and retains 80% of the initial activity, even after recycling five times.295 TEM images revealed that both sides of the GO sheets are closely coated with ZnO particles (average size of 5 nm) in relatively high density to form a sandwich-structure (ZnO/GO/ZnO), which favoured strong electronic interactions. The PL studies revealed that the presence of graphene eliminates the surface defects of ZnO particles. However, the ZnO particles will peel off the graphene sheets and the graphene structure will be slowly destroyed during the durability test. In addition, the impurity or inorganic anions occupy the active centers and hampers the photocatalyst performance.295
The ZnO–graphene hybrid containing 2 wt% of graphene obtained via the reduction of GO with hydrazine was visible light active for the degradation of MB, accompanied by the participation of both the superoxide and hydroxyl radicals in the solution, as well as in direct hole oxidation.296 By this in situ reduction method, ZnO NPs could efficiently separate the graphene sheets and retard the restacking of graphene sheets. The defect sites on GO served as nucleation sites for ZnO, resulting in less agglomerated particles. For this method, Gayathri et al.297 reported that the optimum amount of ZnO (600 mg of Zn2+ ion precursor in the synthesis step) showed a higher activity compared to a low (300 mg) or high (900 mg) amount, despite the other catalysts showing very low PL intensity. The increase in Zn2+ concentration makes the ZnO NPs stick to the surface of the graphene sheets and prevent the graphene sheets from combining as a result of weak van der Waals forces between the graphene layers. However, graphene sheets stack together at a low concentration of Zn2+ and appear like ZnO on multilayered graphene, due to the loss of the 2D planar structure.297 The microwave assisted irradiation method to synthesize ZnO–RGO in diethylene glycol medium was powerful for the decolorization of self-photosensitized dyes (RhB and MB) under visible light, but was ineffective for the degradation of benzoic acid.298 This result indicated that the decolorization of dyes is mainly due to the photosensitization process, rather than excitation of the ZnO–RGO nanohybrids. In this case, electron transfer takes place from the dye* to ZnO or through RGO, which contributes to an effective charge carrier separation. A high loading of ZnO was detrimental (0.0092–0.0115 M zinc acetate in the preparation step), as it may reduce the quantity of photogenerated charges, due to an unfavourable morphology and poor charge carrier transfer through the ZnO nanocrystals. The ZnO–RGO synthesized by the UV-assisted photocatalytic reduction of graphite oxide by ZnO in ethanol was efficient for the removal of Cr(VI) ion, due to the increased light absorption intensity and range, with reduced carrier recombination.299 In addition, this composite also served as a good adsorbent for the removal of colour removal under dark conditions.300
The soft material g-C3N4 (Eg 2.7 eV) as an analogue of graphite possesses a stacked 2D structure, with tri-s-triazine building units connected with planar amino groups in each layer and weak van der Waals forces between the layers. It is exceptionally stable under light illumination in a wide pH range (0–14), thus making it a potential candidate for many green energy applications.301,302
The ZnO hybridized with g-C3N4 (ZnO:g-C3N4) via the monolayer dispersion method showed a high activity for the degradation of MB under UV/visible light at a 3% loading of g-C3N4.303 This enhancement was mainly due to the π-stacking type adsorption between MB and g-C3N4, which was similar to the conjugation between graphene and aromatic molecule. The MB can be adsorbed on the g-C3N4 surface with an offset face to face orientation via π–π conjugation until an adsorption–desorption equilibrium is reached. The superior adsorption capacity was a worthy complement for the high activity of hybridized g-C3N4:ZnO. The suppression of the degradation of dye with EDTA and TBA confirms the dominant involvement of holes and hydroxyl radicals under UV and visible light photocatalysis, respectively. Under UV light, the hole transfers from ZnO to the HOMO of g-C3N4, resulting in an efficient charge separation and it also suppresses the photocorrosion of ZnO. The g-C3N4 absorbs visible light to induce π–π* transition from the HOMO to LUMO. The LUMO potential of C3N4 is −1.12 eV,304 which is more negative than the CB of ZnO (−0.5 eV), and the excited electrons are directly injected to the CB of ZnO. The g-C3N4+ radical can accept electrons from the fragments of the degradation of MB and return to the ground state.303 The formation of a chemically bonded interface between g-C3N4 and ZnO originates from the condensation reaction between the triazine amino group and the surface hydroxyl groups of ZnO. The concentration of the Zn2+ ions after the degradation of MO and 4-NP under visible light using this composite was found to be considerably lower than for pure ZnO, attesting to its long-term stability in aqueous solution.305 The activity increased with an enhancement in the ZnO content upto 15.6 wt% and reduced at higher values (58.1 wt%), which was attributed to the unsuitable ratio between the coupled semiconductors and to the poor visible light response of the composite. In another study, Shifu and co-workers reported that the optimum amounts of ZnO and g-C3N4 were 15 and 5 wt% for ZnO:g-C3N4 prepared from ball-milling and from the deposition–precipitation methods, respectively.306,307 This was attributed to the balanced introduction of the trap sites for the charge carriers, which prolongs the carrier lifetime. The reaction kinetics with different radical scavengers declined in the order: benzoquinone > isopropyl alcohol > ammonium oxalate, confirming the pivotal role of the superoxide radicals in the decomposition mechanism of RhB. However, the hydroxyl radical generation, as evidenced by PL spectroscopy, indicates that the superoxide radical, thus formed transforms to the hydroxyl radicals by a series of reactions, and contributes to complete mineralization.306,307 The holes generated in g-C3N4 cannot oxidize OH− or H2O to hydroxyl radicals, because the position of the VB of g-C3N4 (1.57 eV vs. NHE) is more negative compared to the standard redox potential of E (OH˙/OH−) = 1.99 eV vs. NHE.
The ZnO–mesoporous g-C3N4 core–shell structure was also active for the degradation of MB under UV/visible light.308 The activity was closely related to the thickness of the shell, heterojunction interfaces and the core–shell structure induced by the lattice match between the two components. The processes of formation for the core–shell structure takes place through two steps: mesoporous g-C3N4 is exfoliated into sheet-like nanostructures via ultrasonication in methanol solvent, and later these sheets are uniformly decorated on the ZnO surface to minimize the surface energy. The photoactivity increased with the loading of g-C3N4 up to 4 wt% under UV light, whereas the activity increased up to 20 wt% under visible light. The increase in shell thickness shields the UV light photons from reaching the ZnO surface, while it improves the visible light absorption of the composite, benefiting charge transfer at the heterojunction interfaces. The mesoporous structure easily permits the UV light to reach ZnO, compared to bulk g-C3N4. The optimal amount of ZnO in ZnO: mesoporous g-C3N4 was found to be 24.9 wt% under visible and solar light for the degradation of MB.309 In this case, ZnO was allowed to nucleate and uniformly disperse on the mesoporous g-C3N4 sheets under solvothermal conditions (160 °C, 24 h). The holes were found to be the major oxidant responsible for the degradation of pollutant compared to hydroxyl and superoxide radicals. Instead of bulk g-C3N4 with a big sheet of a lamellar structure, mesoporous g-C3N4 with an open crystalline pore wall and large surface area will be more beneficial in photocatalysis.308,309 The C–ZnO:g-C3N4 showed high activity for the degradation of MB under visible light, at 50.7 wt% g-C3N4, corresponding to the homogeneous hybridization between ZnO and g-C3N4 (Fig. 24).310 This is the first report on simultaneous carbon doping and heterojunction formation of ZnO with g-C3N4. In this work, high temperature treatment of zinc nitrate and dicyandiamide initiated the formation of g-C3N4 and ZnO, respectively, through the thermal decomposition. Moreover, dicyandiamide adhering to the surface of ZnO NPs undergoes combustion and subsequent atomic rearrangements (Zn, C and O), enabling the substitution of carbon for oxygen in the bulk of the lattice.
 |
| | Fig. 24 Illustration of the electron–hole separation, transport, and photocatalytic progress of C–ZnO:g-C3N4 under visible light (reprinted with permission from ref. 310; Copyright 2014 @ American Chemical Society). | |
The electronic coupling of ZnO with g-C3N4 may be an ideal system to promote the charge carrier separation, due to their well-matched overlapping band structure. The activity seems to be very sensitive to the content of both ZnO and g-C3N4 in the composite, and the preparative method used seems to be a decisive factor in governing the photocatalytic pathways. The relative tuning of the individual components and the decoration of g-C3N4 on various morphologies of ZnO still requires further research.
8.4 Modification with CNTs, fullerenes and CQDs
The favourable bandgap energy level alignment in ZnO–MWCNTs–CdS resulted in its high photocurrent density (0.25 mA cm−2) compared to ZNWs–MWCNTs (0.10 mA cm−2) and CdS–ZNWs (0.20 mA cm−2) under solar light.311 In this bandgap configuration, unidirectional electron transfer takes place from CdS to ZNWs to MWCNTs to favour the separation and the photocurrent collection. The CdS serves as an electron sensitizer or relay, while ZNWs and MWCNTs act as an electron sink (Fig. 25). The electrons from the ZnO CB are trapped by MWCNTs via an inductive effect that promotes the interfacial electron transfer process. Such as the growth of ZNWs on MWCNTs improves the active surface area, thus enhancing the photon absorption, while the NW configuration affords conductive pathways to the charge carrier transfer process. The MWCNTs possess more landing sites for the adhesive growth of NWs and the space between each NT would be beneficial for its dispersion. The photocurrent of the ZNWs–MWCNTs was higher under UV light compared to solar light, as the extra excitons photogenerated at the nanocomposite–electrolyte interface intensifies the electric field produced in the depletion layer, which further assists the electron transport processes.312 The ZnO (34%)–MWCNTs were effective in the removal of cyanide under a 355 nm UV light generated by the third harmonic of Nd:YAG laser.313 The structural model proposed indicates that the zinc atom is aligned closer to the oxygen atom of the carbonyl group, whereas an oxygen atom of ZnO remains close to the hydrogen atom of the hydroxyl group on the functionalized MWCNTs (Fig. 26). The former is possible as the electron density on oxygen atoms is shared with zinc, which has an electron affinity, while the latter is favoured because of the hydrogen bond formation. In such types of hetero-interphase interactions, attractive van der Waals forces play a significant role. The rate of removal of cyanide was enhanced with the laser energy (60 mJ to 200 mJ), but declined with an increase in the pH values (4–12). When the laser energy is increased, the incident photon flux increases in the solution and facilitates the generation of more charge carriers. The PZC of functionalized MWCNTs was 4.9, and hence an efficient adsorption-degradation was observed at acidic pH. With an increase in pH, both MWCNTs and ZnO become negatively charged and the cyanide experiences repulsion from the catalyst surface.313
 |
| | Fig. 25 Charge carrier separation and transportation in a CdS–ZNWs–MWCNTs heterojunction (reprinted with permission from ref. 311; Copyright 2011 @ Elsevier). | |
 |
| | Fig. 26 Ball and stick representative diagram of MWCNT–ZnO composite (reprinted with permission from ref. 313; Copyright 2010 @ IOP Science). | |
The increased light absorption and high surface energy of the ZnO–TiO2–CNTs facilitated both the adsorption and reduction of the Cr(VI) ion.314 The CNTs served as wires connecting different ZnO NSs and the TiO2 NPs, and also promotes the electrons to freely flow throughout the structure without any scattering from atoms or defects (ballistic transport).315 The IEF at the surface of the composite and the constructed stepwise structure of the band edge levels leads to electron transfer from ZnO to TiO2 to the CNTs, whereas the hole traverses from TiO2 to ZnO. The increase in the loading of CNTs (4 wt%) competes with photon absorption with TiO2 or ZnO and hampers the dispersion of the metal oxides on the CNTs surface, in addition to promoting the recombination process.316 The fabrication of MWCNTs-doped ZnO (Zn–O–C bond) by a electrospinning method was positive for the degradation of MB under UV/visible light. Under UV light, electrons from the ZnO CB move freely to the MWCNTs, while MWCNTs serve as a sensitizer by absorbing the visible light and transferring the electrons to the ZnO CB.317 The positively charged MWCNTs remove an electron from the VB of ZnO leaving a hole. The oxidation of MWCNTs with concentrated acid and the presence of surface alcohol, ketone and the acid groups enhance the adsorption of dye on the composite surface, thus favouring an efficient photocatalysis. The ZnO:N–CNTs exhibit better activity compared to ZnO–CNTs for the degradation of benzoic acid and RhB under visible light.318 The composite activity, with respect to the crystallite size of ZnO, had the following sequence: 9.5 > 7.3 > 5.4 > 4.2 nm. The relative position in the CB of the N–CNTs allowed the transfer of electrons to ZnO, and shows that defects introduced by nitrogen doping might have separated the charge carriers. Pd–ZnO:MWCNTs at an optimum ratio of 0.08:1.0:0.04 were successful in reducing Hg(II), with excellent recyclability under visible light.319 The introduction of CNT and Pd prevented the ZnO NPs from agglomeration, showing that ZnO was strongly attached to the surface of the CNTs network. The Pd doping and CNT incorporation induced a visible light response, while the large surface area of the composite provided more active sites for the reduction of Hg(II).
The hybridization of C60 at a loading of 1.5% on the ZnO surface enhanced the activity for the degradation of MB and the photocurrent generated remained constant even after prolonged UV illumination (20–35 h).320 The CB energy level of ZnO (−0.5 V vs. NHE at pH 7) thermodynamically favours the electron transfer to the C60 molecules (E C60/C60− is −0.2 V), and the delocalized conjugated π-system of C60 promotes the facile migration of electrons because of the minimal changes in the structure and in the solvation associated with the process.321,322 The crystal structure of ZnO was changed to Zn5(OH)6(CO3)2 after prolonged illumination, whereas the pristine phase was retained for the composite, suggesting that C60 inhibited the photocorrosion of ZnO via deactivating the surface oxygen atoms. ZnO–CQDs were beneficial for the degradation of toxic gases (i.e. benzene and methanol) under visible light.323 The CQDs loaded on the ZnO surface form a “dyade” structure, providing access to the photoinduced charge transfer pathways. At the dyade structure, the photoinduced electrons are transferred to joint charge transfer states located on the CQDs, while holes accumulate electrically and structurally near ZnO. The electrons can be freely shuttled along the conducting network of CQDs and combine with adsorbed oxygen to produce superoxide and hydroxyl radicals in the solution. Moreover, the π-interaction between the conjugated structure of the CQDs and the aromatic pollutant is beneficial to enrich its concentration on the composite surface.324 In addition, CQDs with upconversion PL properties convert the longer wavelength light to a shorter wavelength, resulting in a bandgap excitation of ZnO, and consequently facilitating the generation of more charge carriers (Fig. 27).
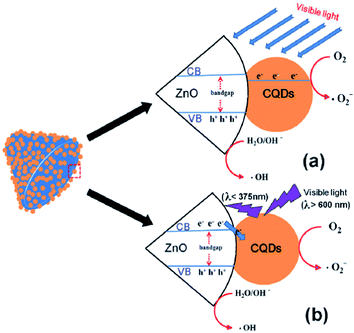 |
| | Fig. 27 Photocatalytic process of ZnO–CQDs under visible light (reprinted with permission from ref. 323; Copyright 2012 @ Royal Society of Chemistry). | |
The modification of ZnO with various carbon nanostructures, such as activated carbon, amorphous carbon, graphitic carbon, CNFs, graphene, RGO, g-C3N4, fullerenes, CNTs and CQDs, improves both the photoactivity and the stability of ZnO during photocatalytic reactions. In a few cases, these carbon nanostructures serve as a support for the growth of ZnO nanocrystallites or ZnO can act as substrate for the growth of these carbon forms. The excellent capacity of carbon nanostructures to enrich pollutant concentration on the catalyst surface and their robust capacity as electron conducting channels allows more carriers to participate in degradation reactions. These semiconductor–carbon composite nanostructures are expected to dominate in waste water purification in the near future.
9. Reactivity of exposed polar and non-polar facets
It is widely accepted that the photocatalytic reaction occurs at the catalyst surface–pollutant interface. Thus, the morphology of the catalyst affects the surface atomic rearrangements and coordination, which thus modulates the crystal facets in different orientations and plays an important role in the photocatalysis.325 The difference in the surface atomic configuration results in a distinct capacity to adsorb oxygenated species and the target molecules. Such speculation has given the impetus to explore the relationship between the surface orientation of ZnO and their photocatalytic efficiency.
Compared with non-faceted ones, ZnO with a hexagonal faceted prism (3–4 nm) showed a high performance for the degradation of MB, which was attributed to the large surface area and the oxygen vacancy defects that promoted an efficient charge transfer process.326 ZnO with a high degree of exposed {0001} facets embedded in a flower-like matrix was active for the degradation of RhB compared to ZnO NRs (with fewer exposed {0001} facets) and platelets (with an equal number of Zn-{0001} and O-{000} facets) under visible light.327 The origin of the visible light activity was attributed to the polar surface induced oxygen defects such as oxygen vacancies or interstitial oxygen.31 Furthermore, the flower-like matrix possess a high degree of dispersion and prevents the aggregation of ZnO NPs, thereby improving the stability. The flower-like ZnO microarchitecture built by the self-assembly of dense NSs with an ultrathin thickness of 5 nm and a large population of polar (0001) planes exhibit a high performance for the degradation of MO compared to other morphologies such as NPs, NRs and NSs.328 These NSs were obtained by a citrate-mediated hydrothermal treatment in the presence of zinc nitrate and HMT. The addition of citric acid changes the orientation of ZnO from (0001) to (100), followed by a morphological transformation from NRs to NSs and a change in the main exposed surface from non-polar to polar facets, attributed to its complexation on the (0001) surface. These size-quantized NSs promote dye adsorption, enhance the light harvesting capacity and efficiently channelize the carriers from the bulk to the surface.329 The grid-like or net-like arrangement of NSs retain their structural integrity even after vigorous ultrasonication (50 min).
The influence of the ZnO morphology for the degradation of RhB followed the following order: cones > bullets > plates > rods, whereas the activity of the exposed crystal facet showed the following trend: {101} ≫ {0001}, {100}.330 The morphologies and crystal facets were controlled by varying the reaction temperature and the benzylamine/Zn2+ ratio, with benzylamine serving as a reactant and capping agent to stabilize the (101) planes. On the basis of a model, it was proposed that the {101} facets possess a ridge-and-valley topography with oxygen termination, whereas the surface oxygen atoms alternate between two-coordinated and three-coordinated in nature. Because low coordinated oxygen atoms are generally saturated by hydrogen atoms via O⋯H–O–H hydrogen bond formation, they generate dense hydroxyl radicals in the solution. The ZnO nanoplatform with an exposed {0001} facet showed a higher photocatalytic activity compared to a hamburger-like morphology with a {100} facet.331 The former was obtained by adding an aqueous solution of NaOH to zinc acetate, whereas adding sodium citrate resulted in the latter morphology under hydrothermal conditions (160 °C, 5 h). The concentration of growth units involved in the nucleation and growth was reduced by the complexation of citrate ions with Zn2+ ions.332 In addition, the longer organic molecular chain of citrate anions allows the ZnO growth units loosely spread in the reaction solution, leading to the formation of a bigger and looser hamburger-like morphology. ZnO nanoplates with a higher proportion of polar to non-polar surfaces induce a rapid generation of H2O2 under UV irradiation, lasting for 730 min, compared to NRs, microrods and dumb-bell shaped, which is consistent with their respective area of polar faces.107 Recently, Bopella et al.333 also reported that the performance of ZnO for the degradation of MB increased with the increase in the proportion of polar to non-polar facets. The ZnO NRs with a cone and small aspect ratio showed a superior performance for the degradation of MO compared to the short-and-fat microrods and NRs with a cone and large aspect ratio.334 Control over the shape was achieved by varying the reaction temperature and time in the hydrothermal reaction of the zinc foil–zinc acetate–H2O system. The formation of NRs with a cone originates from the change of V(0001) and V(010) during the growth. The crystalline nuclei grow into a rod-like structure (120 °C, 2 h), as V(0001) is larger than V(010). The diffusion rate of zinc–hydroxyl complexes and zinc acetate in the solution will be slow and results in a concentration gradient between the Zn-(0001) and O-(000) planes. As a consequence, the concentration of zinc–hydroxyl complexes and zinc acetate will be maximum around the top of a NR and lower around the root. Thus, V(010) at the top of a NR is lower than that of its root, resulting in a cone-like structure. With an increase in temperature (185 °C, 2 h), the diffusion rate between the reactants increases and the concentration gradient decreases, resulting in a NR with a cone and large aspect ratio. The formation of a short-and-fat microrod (120 °C, 6 h) originates from the small difference between V{0001} and V(010).334
The prismatic aggregates of ZnO calcined at 800 °C (4 h) with a {100} facet demonstrated the highest activity for the decomposition of CH3CHO.335 The SEM results revealed that the surface exposed crystal faces or the ratios among the exposed crystal faces were evidently different following self-assembly of the particles towards prismatic aggregates with an increase in the calcination temperature. The driving force for the self-assembly originated from the solid state diffusion and migration of the grain boundaries in the ZnO. In addition, the ZnO single crystal with non-polar {100} or {11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0} facets having O–Zn alternating atomic outermost layers performed better compared with a polar {000} or {0001} single crystal with an O–O or Zn–Zn monotonous atomic outermost layer.335,336 This prismatic morphology transformed to a sheet-shape after coupling with VOx, which facilitated more exposure of {100} and consequently enhanced the activity. However, rod-like ZnO and no change in morphology was observed after coupling with CeOx and WOx, respectively, which were not beneficial to photoactivity.337
0} facets having O–Zn alternating atomic outermost layers performed better compared with a polar {000} or {0001} single crystal with an O–O or Zn–Zn monotonous atomic outermost layer.335,336 This prismatic morphology transformed to a sheet-shape after coupling with VOx, which facilitated more exposure of {100} and consequently enhanced the activity. However, rod-like ZnO and no change in morphology was observed after coupling with CeOx and WOx, respectively, which were not beneficial to photoactivity.337
The conical-like ZnO showed better photocatalytic activity compared to hourglass-like ZnO, although these morphologies exhibited similar crystal facets. This was attributed to the following: (i) the conical-like ZnO exhibits large areas of (0001) planes because the two halves of the hourglass-shaped crystals are attracted through the (0001) planes; (ii) the interior cavities in the conical-like ZnO allows multiple reflections of UV light, which facilitates an efficient use of the incident light. The ZnO with {101} and {100} facets showed the very least activity.128 The activity of ZnO with a hexagonal plate-like particle size was >5 fold for the degradation of MB compared to the rod-shaped particles. The surface of c-axis aligned rods mostly consists of non-polar facets, whereas the dominant surfaces exposed on a hexagonal plate of the same diameter are the polar facets.127
The ZnO superstructures for the degradation of Orange-II showed the following order: structure-I > structure-IV > structure-III > structure-II (Fig. 28).338 The structure-II and structure-III had six symmetric side walls enclosed by {100} surfaces, whereas structure-IV was composed of cone-like building blocks, of which the exposed crystal faces were {101}, {0001} and {100}. Because of the reduced areal proportion of low active {100} facets, structure-IV exhibited a better performance compared to structure II and III. Interestingly, size quantized structure-I had a main exposed plane {210}, which has higher in surface energy than the {100} surface and close to the polar (0001) surface.339 These 3D superstructures were tailored by varying the concentration of zinc acetate and Na2O2, along with the reaction temperature without an organic additive. Under the reaction conditions (0.03 M zinc acetate and 0.1 M Na2O2 at pH 12.9), the adsorption of acetate anion and Zn(OH)3− in the solution compete with Zn(OH)42− for the occupation of the (0001) plane. Thus, the surplus non-growth unit anions stabilized the surface charge and the structure of the (0001) surfaces to some extent, allowing a fast growth along 〈100〉. Hence, the ZnO crystals grew along 〈0001〉 and 〈100〉 directions (both are fast growth directions), resulting in ZnO NSs with an exposed {210} surface.
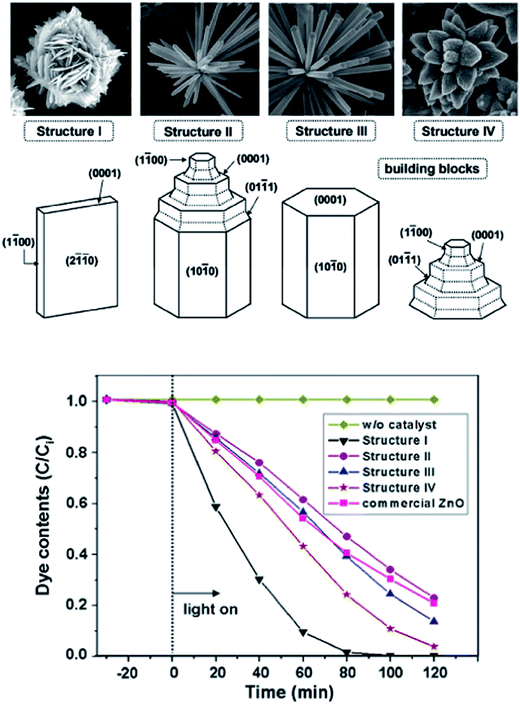 |
| | Fig. 28 The 3D ZnO superstructures with their building blocks and photocatalytic activity (reprinted with permission from ref. 338; Copyright 2010 @ American Chemical Society). | |
The ZnO nanodisks with a {0001} facet showed a high activity for the degradation of dye compared to the ZNWs with {010} planes.340,341 Although NWs had a higher activity over nanodisks in the initial 4 h of illumination, the final degradation after 5 h was slower than for nanodisks, which may be related to the formation of some long lived by-products covered at the surface of NWs, which are resistant to react with hydroxyl radicals.340 The heterostructure of the ZnO-disks with CdS improved the hydrogen evolution rate under visible light by a factor of 2.8 times higher than the non-polar ZnO–CdS.342 Thus, it was concluded that the metallic features of polar surfaces promote easy charge transfer at the interface between ZnO and CdS, whereas non-polar (010) surfaces remain as semiconducting in nature. In addition, the XPS VB spectra revealed that the VBM of the disk is lower compared to the rods, which reflects the strong oxidative power of photogenerated holes on the disks. Surprisingly, there was only a marginal difference in the hydrogen evolution rates between ZnO rods and the disk samples. This result was against the hypothesis that the polar surfaces significantly promote the photodecomposition of organic pollutants.127,340 This contradiction was due to the following reasons: (i) the polar function of the disk being restrained as they adsorb ions in the solution (Na+, S2− or SO32); (ii) ZnO itself not being a good candidate for hydrogen evolution, as the bottom of its CB is little higher than the H+/H2 potential.343 The Z-scheme transfer of photogenerated charge carriers between ZnO and CdS mediated by Au NPs at the interface resulted in significant hydrogen evolution (Fig. 29).344 Both Au and CdS were selectively deposited at the oxygen-terminated {000} surface, based on the rationale that the Au3+- and Cd2+-based complexes are electrostatically adsorbed on negatively charged surfaces. Thus, the CdS serves as a reductive site for hydrogen evolution, whereas ZnO serves as an oxidative site. In the Z-scheme, the recombination of photoexcited electrons from the ZnO CB and holes from the CdS VB occur at the interface, and thus photoexcited electrons and holes with high CB and VB potential, respectively, can be retained for redox reactions. Huang et al.345 suggested that Bi2WO6 crystal seeds can be used to tune the polar facets of ZnO, and hence the photocatalytic activity. The ZnO morphology gradually changed from NRs to NSs after the addition of Bi2WO6, due to the electrostatic interaction between these two semiconductors, consequently changing the ratio of the polar facets. Compared with previous reports that extensively used organic additives to tune the polar facets, this method has outlined the heterogeneous nucleation of ZnO on Bi2WO6 with specific crystal planes with a minimal lattice mismatch (2.5%). The (100) planes of ZnO stack parallel to the (200) planes of Bi2WO6, forming the heterojunction, indicating that ZnO NSs grow epitaxially along the normal direction of the (200) plane. In addition, the polar surfaces of ZnO were also found to be catalytically active for the N-formylation of aniline and for the thermal decomposition of ammonium perchlorate.346,347
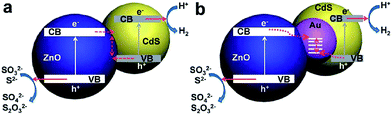 |
| | Fig. 29 Schematic presentation of: (a) the direct Z-scheme charge-carrier transfer process in CdS–ZnO and (b) the vectorial Z-scheme charge-carrier transfer process in CdS–Au–ZnO (reprinted with permission from ref. 344; Copyright 2013 @ Royal Society of Chemistry). | |
The internal electric field between the polar faces of Zn-{0001} and O-{000} arising from the spontaneous polarization drives the electrons and holes to migrate towards the positive and negative polar planes, facilitating the reduction and oxidation to operate at different sites, respectively.348 Theoretically, the strong preference of zinc ions for a tetrahedral coordination geometry and the coordinatively unsaturated environment of zinc ions on the (0001) plane facilitates more generation of H2O2, and adsorbs atmospheric oxygen through chemical or physical adsorption and hydroxyl anions (i.e. via electrostatic attraction) to a larger extent on its surface. The Zn-{0001} facet holds the highest chemisorption ability and surface energy, which enrich the dye adsorption at the surface and makes it inherently more reactive than the O-{000} or non-polar {100} plane.107,331,340,349
There is an urgent need to optimize the design and a synthetic route to obtain the ZnO nanocrystallite with well-defined facets. If the particle size with specific exposed crystal facets is tailored, the carrier separation efficiency could be maximized by considering the interdependence between the surface structure and the diffusion length of the charge carriers. The major drawback of particle aggregation during the photocatalytic reactions leads to a low dispersion and masks the exposed active facets.
10. Conclusion and future prospects
A significant challenge in the research of solar energy conversion and photocatalysis is the rational design of materials that can efficiently trap solar energy and facilitate the active involvement of photogenerated charge carriers in redox reactions. Intense research efforts have led to significant progress and as a result ZnO has emerged as a robust nanomaterial for the complete mineralization of organic pollutants under light illumination. However, the fast recombination of photogenerated excitons, zero resistance to photoinduced corrosion and dissolution at extreme pH values, aggregation of particles because of structural instability, and a large bandgap that remains inert in the visible light response remain as barriers to its extended applicability.
In this context, this review aims to recognize the impressive strategies and modifications developed to date to overcome the aforementioned drawbacks (Fig. 30), besides discussing the mechanism of charge transfer, preparative methods, growth and their relative effects on the degradation reactions. Irrespective of the modifications, the efficient interaction between the charge carriers and free radicals with the pollutants is what ultimately determines photocatalytic efficiency. It is clearly demonstrated that the photocatalytic activity is a complex function of several interdependent parameters and that each modification has its own merits and demerits. Although, the modified ZnO performed well against commercial ZnO or Degussa P25, the existing gap among the issues such as multifunctional properties, photocatalytic performance, problems associated with their durability and the reaction selectivity for ZnO systems still needs to be bridged. The photoactivity of ZnO with a biphasic crystal structure is largely unexplored in photocatalysis to date. Further studies are still needed to resolve the aforementioned drawbacks in a single approach; achieving this could boost the application of ZnO-based nanomaterials in many areas.
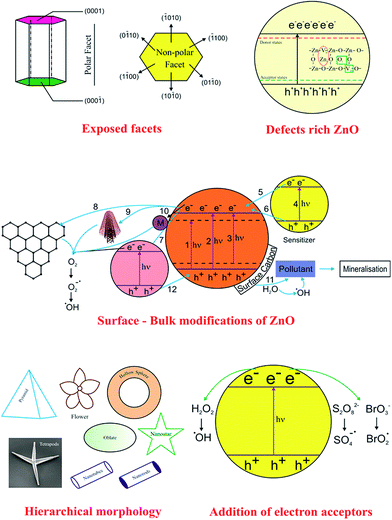 |
| | Fig. 30 Overall view of ZnO-based photocatalysis. The scheme at the four corners shows the reactive facets (top left), intrinsic defects (top right), nanostructured morhopology (bottom left) and the generation of free radicals with electron acceptors (bottom right). The centre image represents the charge transfer pathways in the surface-bulk modification of zinc oxide that are reported to improve the photoactivity. Path 1-excitation from VB to the dopant level (metal ions); Path 2-bandgap excitation of ZnO; Path 3: excitation from the impurity level (nitrogen or carbon doping) to CB; Path 4: excitation of the sensitizer (for instance, CdS, CuS, g-C3N4, inorganic complexes); Path 5: transfer of the excited electrons to the ZnO CB; Path 6: direct excitation of the electron from the sensitizer to the CB of ZnO (as observed in the surface complex formation between 4-CP and ZnO); Path 7 and 12: transfer of the electrons and holes in a bicomponent system (for instance ZnO–SnO2); Path 8–10: electron trapping from graphene, MWCNTs and the metal islands on ZnO surface, respectively; Path 11: surface carbon enriches pollutant adsorption on the ZnO surface, thus facilitating the direct attack of the hole or the hole oxidizes water molecules to generate hydroxyl radicals. | |
Acronyms
| Aluminium-doped ZnO |
AZO |
Nanoparticle(s) |
NP(s) |
| Carbon nanofibers |
CNFs |
Nanorod(s) |
NR(s) |
| Carbon nanotubes |
CNTs |
Nanosheet(s) |
NS(s) |
| Carbon quantum dot(s) |
CQD(s) |
Nanotube(s) |
NT(s) |
| 4-Chlorophenol |
4-CP |
Nanowire(s) |
NW(s) |
| Conduction band |
CB |
4-Nitrophenol |
4-NP |
| Conduction band offset |
ΔEc |
Orange G |
OG |
| Critical micelle concentration |
CMC |
Photoluminescence |
PL |
| Crystal violet |
CV |
Polyaniline |
PANI |
| 2,4-Dichlorophenol |
DCP |
Polyvinylpyrrolidone |
PVP |
| N,N-Dimethylformamide |
DMF |
Pre-micelle concentration |
PMC |
| Electrochemical impedance spectroscopy |
EIS |
Reduced graphene oxide |
RGO |
| Energy dispersive X-ray spectroscopy |
EDS |
Rhodamine B |
RhB |
| Ethylenediaminetetraacetic acid |
EDTA |
Rhodamine 6G |
R6G |
| Ethylene glycol |
EG |
Salicylic acid |
SA |
| Ethyl violet |
EV |
Scanning electron microscopy |
SEM |
| Fluorine doped tin oxide |
FTO |
Sulfur mustard |
HD |
| Fluorescence spectroscopy |
FS |
Tetrapod(s) |
TP(s) |
| Functionalized graphene sheet(s) |
FGS(s) |
t-Butyl alcohol |
TBA |
| Graphene oxide |
GO |
t-Butylphosphonic acid |
TBPA |
| Hexadecylamine |
HCA |
2,4,6-Trichlorophenol |
TCP |
| Hexamethylenetetramine |
HMT |
Triethanol amine |
TEA |
| Highest occupied molecular orbital |
HOMO |
Tetraethylammonium hydroxide |
TEAOH |
| Internal electric field |
IEF |
Tetrahydrofuran |
THF |
| Lowest unoccupied molecular orbital |
LUMO |
Transmission electron microscopy |
TEM |
| Malachite green |
MG |
Tetramethylammonium hydroxide |
TMAOH |
| Mars–van Krevelen |
MVK |
Valence band |
VB |
| Methylene blue |
MB |
Valence band offset |
ΔEv |
| Methyl orange |
MO |
Water contact angle |
WCA |
| Methyl red |
MR |
X-ray photoelectron spectroscopy |
XPS |
| Monocrotophos |
MCP |
Zinc–ethylene glycol–acetate |
Zn–EG–AC |
| Multiwalled carbon nanotube(s) |
MWCNT(s) |
Zinc oxide nanowire(s) |
ZNW(s) |
| Nanorod array(s) |
NRA(s) |
Zero point charge |
ZPC |
| Nanofiber(s) |
NF(s) |
|
|
Acknowledgements
Dr S. Girish Kumar acknowledges financial support from Dr Daulat Singh Kothari Post Doctoral Fellowship (University Grants Commission, New Delhi, INDIA) and the Department of Physics, IISc, Bangalore for providing the research facilities.
References
-
(a) L. G. Devi, S. G. Kumar, K. M. Reddy and C. Munikrishnappa, J. Hazard. Mater., 2009, 164, 459–467 CrossRef PubMed;
(b) L. G. Devi, K. E. Rajashekhar, K. S. A. Raju and S. G. Kumar, J. Mol. Catal. A: Chem., 2009, 314, 88–94 CrossRef CAS PubMed;
(c) L. G. Devi, S. G. Kumar and K. M. Reddy, Cent. Eur. J. Chem., 2009, 7, 468–477 CrossRef CAS PubMed;
(d) A. S. Giri and A. K. Golder, RSC Adv., 2014, 4, 6738–6745 RSC.
-
(a) P. V. Nidheesh, R. Gandhimathi, S. Velmathi and N. S. Sanjini, RSC Adv., 2014, 4, 5698–5708 RSC;
(b) T. Han, L. Qu, Z. Luo, X. Wu and D. Zhang, New J. Chem., 2014, 38, 942–948 RSC;
(c) L. G. Devi, K. S. A. Raju and S. G. Kumar, J. Environ. Monit., 2009, 11, 1397–1404 RSC;
(d) I. Muthuvel and M. Swaminathan, Catal. Commun., 2007, 8, 981–986 CrossRef CAS PubMed.
-
(a) L. G. Devi, S. G. Kumar, K. S. A. Raju and K. E. Rajashekhar, Chem. Pap., 2010, 64, 378–385 CrossRef CAS PubMed;
(b) K. R. Naqvi, J. Marsh and V. Chechik, Dalton Trans., 2014, 4745–4751 RSC;
(c) L. Zhou, H. Zhang, L. Ji, Y. Shao and Y. Li, RSC Adv., 2014, 4, 24900–24908 RSC;
(d) A. C. Pradhan, B. Nanda, K. M. Parida and M. Das, Dalton Trans., 2013, 558–566 RSC.
-
(a) Z. Huang, P. Wu, H. Li, W. Li, Y. Zhu and N. Zhu, RSC Adv., 2014, 4, 6500–6507 RSC;
(b) J. M. Sanchez, L. H. Reyes, J. L. G. Mar, J. M. P. Hernandez and A. H. Ramirez, Photochem. Photobiol. Sci., 2011, 10, 332–337 RSC;
(c) P. V. Nidheesh and R. Gandhimathi, Desalination, 2012, 299, 1–15 CrossRef CAS PubMed;
(d) A. M. de Freitas, C. Sirtori, C. A. Lenz and P. G. P. Zamora, Photochem. Photobiol. Sci., 2013, 12, 696–702 RSC.
- R. Vinu and G. Madras, J. Indian Inst. Sci., 2010, 90, 189–230 CAS.
-
(a) M. Wang, J. Ioccozia, L. Sun, C. Lin and Z. Lin, Energy Environ. Sci., 2014, 7, 2182–2202 RSC;
(b) G. Zhang, G. Kim and W. Choi, Energy Environ. Sci., 2014, 7, 954–966 RSC;
(c) K. Huo, B. Gao, J. Fu, L. Zhao and P. K. Chu, RSC Adv., 2014, 4, 17300–17324 RSC;
(d) J. B. Joo, M. Dahl, N. Li, F. Zaera and Y. Yin, Energy Environ. Sci., 2013, 6, 2082–2092 RSC.
-
(a) S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS PubMed;
(b) L. G. Devi and S. G. Kumar, Cent. Eur. J. Chem., 2011, 9, 959–961 CrossRef PubMed;
(c) K. Bourikas, C. Kordulis and A. Lycourghiotis, Chem. Rev., 2014, 114, 9754–9823 CrossRef CAS PubMed;
(d) L. Sang, Y. Zhao and C. Burda, Chem. Rev., 2014, 114, 9283–9318 CrossRef CAS PubMed.
-
(a) K. Lv, B. Cheng, J. Yu and G. Liu, Phys. Chem. Chem. Phys., 2012, 14, 5349–5362 RSC;
(b) C. Aprile, A. Corma and H. Garcia, Phys. Chem. Chem. Phys., 2008, 10, 769–783 RSC;
(c) G. Liu, L. Wang, H. G. Yang, H. M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
-
(a) H. Zheng, J. Z. Ou, M. S. Strano, R. B. Kaner, A. Mitchell and K. K. Zadeh, Adv. Funct. Mater., 2011, 21, 2175–2196 CrossRef CAS;
(b) C. Janaky, K. Rajeshwar, N. R. de Tacconi, W. Chanmanee and M. N. Huda, Catal. Today, 2013, 199, 53–64 CrossRef CAS PubMed.
-
(a) L. Zhu, B. Wei, L. Xu, Z. Lu, H. Zhang, H. Gao and J. Che, CrystEngComm, 2012, 14, 5705–5709 RSC;
(b) D. P. Dutta, M. Roy and A. K. Tyagi, Dalton Trans., 2012, 10238–10248 RSC;
(c) S. Balachandran and M. Swaminathan, J. Phys. Chem. C, 2012, 116, 26306–26312 CrossRef CAS.
-
(a) T. K. Townsend, E. M. Sabio, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2011, 4, 4270–4275 RSC;
(b) D. K. Bora, A. Braun and E. C. Constable, Energy Environ Sci., 2013, 6, 407–425 RSC;
(c) J. Sun, K. L. Wu, X. Z. Li, C. Dong, X. W. Wei, X. W. Wang, B. Zhang, Z. X. Zhang and J. R. Huang, CrystEngComm, 2014, 16, 6873–6881 RSC;
(d) K. Sivula, F. L. Formal and M. Gratzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
-
(a) K. Pirkanniemi and M. Sillanpaa, Chemosphere, 2002, 48, 1047–1060 CrossRef CAS;
(b) H. Zhang, G. Chen and D. W. Bahnemann, J. Mater. Chem., 2009, 19, 5089–5121 RSC.
-
(a) L. Ye, Y. Su, X. Jin, H. Xie and C. Zhang, Environ. Sci.:Nano, 2014, 1, 90–112 RSC;
(b) J. Li, Y. Yu and L. Zhang, Nanoscale, 2014, 6, 8473–8488 RSC;
(c) H. Cheng, B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC.
-
(a) F. Dong, Q. Li, Y. Zhou, Y. Sun, H. Zhang and Z. Wu, Dalton Trans., 2014, 9468–9480 RSC;
(b) F. Dong, Y. Sun, W. K. Ho and Z. Wu, Dalton Trans., 2012, 8270–8284 RSC;
(c) F. Dong, A. Zheng, Y. Sun, M. Fu, B. Jiang, W. K. Ho, S. C. Lee and Z. Wu, CrystEngComm, 2012, 14, 3534–3544 RSC.
-
(a) D. Zhang and F. Zeng, Res. Chem. Intermed., 2010, 36, 1055–1063 CrossRef CAS PubMed;
(b) D. Zhang, Transition Met. Chem., 2010, 35, 689–694 CrossRef CAS;
(c) D. Zhang and F. Zeng, J. Mater. Sci., 2012, 47, 2155–2161 CrossRef CAS;
(d) D. Zhang, Russ. J. Phys. Chem. A, 2013, 87, 137–144 CrossRef CAS.
- P. V. Kamat, R. Huehn and R. Nicolaescu, J. Phys. Chem. B, 2002, 106, 788–794 CrossRef CAS.
-
(a) D. C. Look, D. C. Reynolds, J. R. Sizelove, R. L. Jones, C. W. Litton, G. Cantwell and W. C. Harsch, Solid State Commun., 1998, 105, 399–401 CrossRef CAS;
(b) A. K. Chandiran, M. A. Jalebi, M. K. Nazeeruddin and M. Gratzel, ACS Nano, 2014, 8, 2261–2268 CrossRef CAS PubMed.
-
(a) M. Quintana, T. Edvinsson, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2007, 111, 1035–1041 CrossRef CAS;
(b) J. A. Anta, E. Guillen and R. T. Zaera, J. Phys. Chem. C, 2012, 116, 11413–11425 CrossRef CAS;
(c) S. Cho, J. W. Jang, S. H. Lim, H. J. Kang, S. W. Rhee, J. S. Lee and K. H. Lee, J. Mater. Chem., 2011, 21, 17816–17822 RSC.
-
(a) G. Marci, V. Augugliaro, M. J. L. Munoz, C. Martin, L. Palmisano, V. Rives, M. Schiavello, R. J. D. Tilley and A. M. Venezia, J. Phys. Chem. B, 2001, 105, 1033–1040 CrossRef CAS;
(b) P. V. Kamat, I. Bedja and S. Hotchandani, J. Phys. Chem., 1994, 98, 9137–9142 CrossRef CAS.
- N. Daneshvar, D. Salari and A. R. Khataee, J. Photochem. Photobiol., A, 2004, 162, 317–322 CrossRef CAS.
-
(a) A. A. Khodja, T. Sehili, J. F. Pilichowski and P. Boule, J. Photochem. Photobiol., A, 2001, 141, 231–239 CrossRef CAS;
(b) C. Tian, Q. Zhang, A. Wu, M. Jiang, Z. Liang, B. Jiang and H. Fu, Chem. Commun., 2012, 2858–2860 RSC.
-
(a) J. P. Percherancier, R. Chapelon and B. Pouyet, J. Photochem. Photobiol., A, 1995, 87, 261–266 CrossRef CAS;
(b) C. Hariharan, Appl. Catal., A, 2006, 304, 55–61 CrossRef CAS PubMed.
-
(a) I. Poulios, M. Kositzi and A. Kouras, J. Photochem. Photobiol., A, 1998, 115, 175–183 CrossRef CAS;
(b) A. Akyol, H. C. Yatmaz and M. Bayramoglu, Appl. Catal., B, 2004, 54, 19–24 CrossRef CAS PubMed.
-
(a) P. R. Shukla, S. Wang, H. M. Ang and M. O. Tade, Sep. Purif. Technol., 2010, 70, 338–344 CrossRef CAS PubMed;
(b) G. Colon, M. C. Hidalgo, J. A. Navio, E. P. Melian, O. G. Diaz and J. M. D. Rodriguez, Appl. Catal., B, 2008, 83, 30–38 CrossRef CAS PubMed.
-
(a) C. Lizama, J. Freer, J. Baeza and H. D. Mansilla, Catal. Today, 2002, 76, 235–246 CrossRef CAS;
(b) J. Fenoll, P. Sabater, G. Navarro, G. P. Lucas and S. Navarro, J. Hazard. Mater., 2013, 244–245, 370–379 CrossRef CAS PubMed.
-
(a) V. Kandavelu, H. Kastien and K. R. Thampi, Appl. Catal., B, 2004, 48, 101–111 CrossRef CAS PubMed;
(b) Y. Liao, C. Xie, Y. Liu and Q. Huang, J. Alloys Compd., 2013, 550, 190–197 CrossRef CAS PubMed.
- J. Liqiang, W. Dejun, W. Baiqi, L. Shudan, X. Baifu, F. Honggang and S. Jiazhong, J. Mol. Catal. A: Chem., 2006, 244, 193–200 CrossRef PubMed.
-
(a) S. S. Warule, N. S. Chaudhari, B. B. Kale and M. A. More, CrystEngComm, 2009, 11, 2776–2783 RSC;
(b) E. Evgenidou, K. Fytianos and I. Poulios, Appl. Catal., B, 2005, 59, 81–89 CrossRef CAS PubMed.
-
(a) S. Baruah, S. S. Sinha, B. Ghosh, S. K. Pal, A. K. Raychaudhuri and J. Dutta, J. Appl. Phys., 2009, 105, 074308 CrossRef PubMed;
(b) Y. Lv, C. Pan, X. Ma, R. Zong, X. Bai and Y. Zhu, Appl. Catal., B, 2013, 138–139, 26–32 CrossRef CAS PubMed.
-
(a) D. Chen, Z. Wang, T. Ren, H. Ding, W. Yao, R. Zong and Y. Zhu, J. Phys. Chem. C, 2014, 118, 15300–15307 CrossRef CAS;
(b) Z. Pei, L. Ding, J. Hu, S. Weng, Z. Zheng, M. Huang and P. Liu, Appl. Catal., B, 2013, 142–143, 736–743 CrossRef CAS PubMed.
-
(a) F. Kayaci, S. Vempati, I. Donmez, N. Biyikli and T. Uyar, Nanoscale, 2014, 6, 10224–10234 RSC;
(b) Y. Lv, W. Yao, X. Ma, C. Pan, R. Zong and Y. Zhu, Catal. Sci. Technol., 2013, 3, 3136–3146 RSC;
(c) Y. Zheng, C. Chen, Y. Zhan, X. Lin, Q. Zheng, K. Wei, J. Zhu and Y. Zhu, Inorg. Chem., 2007, 46, 6675–6682 CrossRef CAS PubMed.
-
(a) H. W. Jeong, S. Y. Choi, S. H. Hong, S. K. Lim, D. S. Han, A. A. Wahab and H. Park, J. Phys. Chem. C, 2014, 118, 21331–21338 CrossRef CAS;
(b) X. Xu, X. Duan, Z. Yi, Z. Zhou, X. Fan and Y. Wang, Catal. Commun., 2010, 12, 169–172 CrossRef CAS PubMed;
(c) M. Y. Guo, A. M. C. Ng, F. Liu, A. B. Djurisic and W. K. Chan, Appl. Catal., B, 2011, 107, 150–157 CrossRef CAS PubMed.
-
(a) J. Han, Y. Liu, N. Singhal, L. Wang and W. Gao, Chem. Eng. J., 2012, 213, 150–162 CrossRef CAS PubMed;
(b) Y. Peng, Y. Wang, Q. G. Chen, Q. Zhu and A. W. Xu, CrystEngComm, 2014, 16, 7906–7913 RSC.
-
(a) S. C. Pillai, J. M. Kelly, R. Ramesh and D. E. McCormack, J. Mater. Chem. C, 2013, 1, 3268–3281 RSC;
(b) J. Li, Q. Sun, C. Jin and J. Li, Ceram. Int., 2015, 41, 921–929 CrossRef CAS PubMed.
- A. B. Djurisic, A. M. C. Ng and X. Y. Chen, Prog. Quantum Electron., 2010, 34, 191–259 CrossRef CAS PubMed.
- Y. Tu, L. Zhou, Y. Z. Jin, C. Gao, Z. Z. Ye, Y. F. Yang and Q. L. Wang, J. Mater. Chem., 2010, 20, 1594–1599 RSC.
-
(a) Z. L. Wang and J. Song, Science, 2006, 312, 242–246 CrossRef CAS PubMed;
(b) L. S. Mende and J. L. M. Driscoll, Mater. Today, 2007, 10(5), 40–48 CrossRef.
-
(a) A. E. Lavat, C. C. Wagner and J. E. Tasca, Ceram. Int., 2008, 34, 2147–2153 CrossRef CAS PubMed;
(b) K. Ghule, A. V. Ghule, B. J. Chen and Y. C. Ling, Green Chem., 2006, 8, 1034–1041 RSC.
-
(a) G. Heiland and H. Ibach, Solid State Commun., 1966, 4, 353–356 CrossRef CAS;
(b) H. Liu, F. Zeng, S. Gao, G. Wang, C. Song and F. Pan, Phys. Chem. Chem. Phys., 2013, 15, 13153–13161 RSC.
-
(a) P. Spathis and I. Poulios, Corros. Sci., 1995, 37, 673–680 CrossRef CAS;
(b) J. Han, W. Qiu and W. Gao, J. Hazard. Mater., 2010, 178, 115–122 CrossRef CAS PubMed.
- A. L. Rudd and C. B. Breslin, Electrochim. Acta, 2000, 45, 1571–1579 CrossRef CAS.
-
(a) A. Moezzi, A. M. McDonagh and M. B. Cortie, Chem. Eng. J., 2012, 185–186, 1–22 CrossRef CAS PubMed;
(b) J. C. Fan, K. M. Sreekanth, Z. Xie, S. L. Chang and K. V. Rao, Prog. Mater. Sci., 2013, 58, 874–985 CrossRef CAS PubMed;
(c) J. L. Gomez and O. Tigli, J. Mater. Sci., 2013, 48, 612–624 CrossRef CAS.
-
(a) A. Janotti and C. G. V. de Walle, Rep. Prog. Phys., 2009, 72, 126501 CrossRef;
(b) Y. Kozuka, A. Tsukazaki and M. Kawasaki, Appl. Phys. Rev., 2014, 1, 011303 CrossRef PubMed;
(c) J. J. Chen, X. R. Deng and H. Deng, J. Mater. Sci., 2013, 48, 532–542 CrossRef CAS.
-
(a) M. D. McCluskey and S. J. Jokela, J. Appl. Phys., 2009, 106, 071101 CrossRef PubMed;
(b) B. Ludi and M. Niederberger, Dalton Trans., 2013, 12554–12568 RSC;
(c) J. Li, S. Ma, X. Liu, Z. Zhou and C. Q. Sun, Chem. Rev., 2012, 112, 2833–2852 CrossRef CAS PubMed.
-
(a) X. Wang, J. Song and Z. L. Wang, J. Mater. Chem., 2007, 17, 711–720 RSC;
(b) C. Klingshirn, J. Fallert, H. Zhou, J. Sartor, C. Thiele, F. M. Fliag, D. Schneider and H. Kalt, Phys. Status Solidi B, 2010, 247, 1424–1447 CrossRef CAS.
-
(a) U. Ozgur, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Dogan, V. Avrutin, S. J. Cho and H. Morkoc, J. Appl. Phys., 2005, 98, 041301 CrossRef PubMed;
(b) B. Weintraub, Z. Zhou, Y. Li and Y. Deng, Nanoscale, 2010, 2, 1573–1587 RSC.
-
(a) A. B. Djurisic, X. Chen, Y. H. Leung and A. M. C. Ng, J. Mater. Chem., 2012, 22, 6526–6535 RSC;
(b) S. Dutta, S. Chattopadhyay, A. Sarkar, M. Chakrabarti, D. Sanyal and D. Jana, Prog. Mater. Sci., 2009, 54, 89–136 CrossRef CAS PubMed.
-
(a) A. D. Paola, E. G. Lopez, G. Marci and L. Palmisano, J. Hazard. Mater., 2012, 211–212, 3–29 CrossRef PubMed;
(b) I. Udom, M. K. Ram, E. K. Stefanakos, A. F. Hepp and D. Y. Goswami, Mater. Sci. Semicond. Process., 2013, 16, 2070–2083 CrossRef CAS PubMed;
(c) Y. Zhang, M. K. Ram, E. K. Stefanakos and D. Y. Goswami, J. Nanomater., 2012, 624520 Search PubMed.
-
(a) S. M. Lam, J. C. Sin, A. Z. Abdullah and A. R. Mohamed, Desalin. Water Treat., 2012, 41, 131–169 CrossRef CAS;
(b) S. H. S. Chan, T. Y. Wu, J. C. Juan and C. Y. Teh, J. Chem. Technol. Biotechnol., 2011, 86, 1130–1158 CrossRef CAS;
(c) R. Kumar, G. Kumar and A. Umar, Nanosci. Nanotechnol. Lett., 2014, 6, 631–650 CrossRef PubMed.
-
(a) M. D. H. Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC;
(b) M. Montazer and M. M. Amiri, J. Phys. Chem. B, 2014, 118, 1453–1470 CrossRef CAS PubMed.
-
(a) S. Rehman, R. Ullah, A. M. Butt and N. D. Gohar, J. Hazard. Mater., 2009, 170, 560–569 CrossRef CAS PubMed;
(b) A. K. Radzimska and T. Jesionowski, Materials, 2014, 7, 2833–2881 CrossRef PubMed.
-
(a) M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS;
(b) A. Ajmal, I. Majeed, R. N. Malik, H. Idriss and M. A. Nadeem, RSC Adv., 2014, 4, 37003–37026 RSC.
-
(a) J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed;
(b) D. Friedmann, C. Mendive and D. Bahnemann, Appl. Catal., B, 2010, 99, 398–406 CrossRef CAS PubMed.
- Y. Li, W. Xie, X. Hu, G. Shen, X. Zhou, Y. Xiang, X. Zhao and P. Fang, Langmuir, 2010, 26, 591–597 CrossRef CAS PubMed.
-
(a) Z. Li, Y. Fang, X. Zhan and S. Xu, J. Alloys Compd., 2013, 564, 138–142 CrossRef CAS PubMed;
(b) D. Chatterjee and S. Dasgupta, J. Photochem. Photobiol., C, 2005, 6, 186–205 CrossRef CAS PubMed.
-
(a) R. Vinu, S. Polisetti and G. Madras, Chem. Eng. J., 2010, 165, 784–797 CrossRef CAS PubMed;
(b) Y. C. Hsiao, T. F. Wu, Y. S. Wang, C. C. Hu and C. Huang, Appl. Catal., B, 2014, 148–149, 250–257 CrossRef CAS PubMed.
- X. Li, Y. Cheng, S. Kang and J. Mu, Appl. Surf. Sci., 2010, 256, 6705–6709 CrossRef CAS PubMed.
- Y. Li, X. Zhou, X. Hu, X. Zhao and P. Fang, J. Phys. Chem. C, 2009, 113, 16188–16192 CAS.
- R. Comparelli, E. Fanizza, M. L. Curri, P. D. Cozzoli, G. Mascolo and A. Agostiano, Appl. Catal., B, 2005, 60, 1–11 CrossRef CAS PubMed.
- H. Wang and C. Xie, J. Phys. Chem. Solids, 2008, 69, 2440–2444 CrossRef CAS PubMed.
- D. S. Bohle and C. J. Spina, J. Am. Chem. Soc., 2009, 131, 4397–4404 CrossRef CAS PubMed.
- J. Becker, K. R. Raghupathi, J. St. Pierre, D. Zhao and R. T. Koodali, J. Phys. Chem. C, 2011, 115, 13844–13850 CAS.
- H. Wang, C. Xie, W. Zhang, S. Cai, Z. Yang and Y. Gui, J. Hazard. Mater., 2007, 141, 645–652 CrossRef CAS PubMed.
- A. C. Dodd, A. J. McKinley, M. Saunders and T. Tsuzuki, J. Nanopart. Res., 2006, 8, 43–51 CrossRef CAS.
-
(a) L. G. Devi and S. G. Kumar, Appl. Surf. Sci., 2012, 261, 137–146 CrossRef CAS PubMed;
(b) L. G. Devi and S. G. Kumar, Appl. Surf. Sci., 2011, 257, 2779–2790 CrossRef CAS PubMed;
(c) L. G. Devi, N. Kottam and S. G. Kumar, J. Phys. Chem. C, 2009, 113, 15593–15601 CrossRef CAS;
(d) L. G. Devi, N. Kottam, B. N. Murthy and S. G. Kumar, J. Mol. Catal. A: Chem., 2010, 328, 44–52 CrossRef CAS PubMed.
- P. S. Casey, C. J. Rossouw, S. Boskovic, K. A. Lawrence and T. W. Turney, Superlattices Microstruct., 2006, 39, 97–106 CrossRef CAS PubMed.
- T. R. Giraldi, G. V. F. Santos, V. R. de Mendonca, C. Ribeiro and I. T. Weber, Mater. Chem. Phys., 2012, 136, 505–511 CrossRef CAS PubMed.
- S. Sakthivel, B. Neppolian, M. V. Shankar, B. Arabindoo, M. Palanichamy and V. Murugesan, Sol. Energy Mater. Sol. Cells, 2003, 77, 65–82 CrossRef CAS.
- Y. Huang, Y. Wei, J. Wu, C. Guo, M. Wang, S. Yin and T. Sato, Appl. Catal., B, 2012, 123–124, 9–17 CrossRef CAS PubMed.
- S. K. Pardeshi and A. B. Patil, Sol. Energy, 2008, 82, 700–705 CrossRef CAS PubMed.
-
(a) D. W. Bahnemann, C. K. Kormann and M. R. Hoffmann, J. Phys. Chem., 1987, 91, 3789–3798 CrossRef CAS;
(b) G. A. Parks, Chem. Rev., 1965, 65, 177–198 CrossRef CAS;
(c) H. Yin and P. S. Casey, RSC Adv., 2014, 4, 26149–26157 RSC.
- A. N. Rao, B. Sivasankar and V. Sadasivam, J. Hazard. Mater., 2009, 166, 1357–1361 CrossRef PubMed.
- E. Evgenidou, I. Konstantinou, K. Fytianos, I. Poulios and T. Albanis, Catal. Today, 2007, 124, 156–162 CrossRef CAS PubMed.
-
(a) L. G. Devi, K. S. A. Raju, S. G. Kumar and K. E. Rajashekhar, J. Taiwan Inst. Chem. Eng., 2011, 42, 341–349 CrossRef CAS PubMed;
(b) X. Wang, L. Wang, J. Li, J. Qiu, C. Cai and H. Zhang, Sep. Purif. Technol., 2014, 122, 41–46 CrossRef CAS PubMed;
(c) N. Sobana and M. Swaminathan, Sep. Purif. Technol., 2007, 56, 101–107 CrossRef CAS PubMed.
- G. K. Prasad, P. V. R. K. Ramacharyulu, B. Singh, K. Batra, A. R. Srivastava, K. Ganesan and R. Vijayaraghavan, J. Mol. Catal. A: Chem., 2011, 349, 55–62 CrossRef CAS PubMed.
- C. Lu, Y. Wu, F. Mai, W. Chung, C. Wu, W. Lin and C. Chen, J. Mol. Catal. A: Chem., 2009, 310, 159–165 CrossRef CAS PubMed.
- C. Chen, J. Mol. Catal. A: Chem., 2007, 264, 82–92 CrossRef CAS PubMed.
- D. Mijin, M. Savic, P. Snezana, A. Smiljanic, O. Glavaski, M. Jovanovic and S. Petrovic, Desalination, 2009, 249, 286–292 CrossRef CAS PubMed.
- C. Karunakaran, S. Senthilvelan, S. Karuthapandain and K. Balaraman, Catal. Commun., 2004, 5, 283–290 CrossRef CAS PubMed.
- M. Iranifam, M. Zarei and A. R. Khataee, J. Electroanal. Chem., 2011, 659, 107–112 CrossRef CAS PubMed.
- J. Wang, P. Liu, X. Fu, Z. Li, W. Han and X. Wang, Langmuir, 2009, 25, 1218–1223 CrossRef CAS PubMed.
- J. Wang, P. Liu, S. Wang, W. Han, X. Wang and X. Fu, J. Mol. Catal. A: Chem., 2007, 273, 21–25 CrossRef CAS PubMed.
- P. Liu, J. Bandara, Y. Lin, D. Elgin, L. F. Allard and Y. P. Sun, Langmuir, 2002, 18, 10398–10401 CrossRef CAS.
- S. Wang, P. Liu, X. Wang and X. Fu, Langmuir, 2005, 21, 11969–11973 CrossRef CAS PubMed.
- P. Pathak, M. J. Meziani, Y. Li, L. T. Cureton and Y. P. Sun, Chem. Commun., 2004, 1234–1235 RSC.
- H. Zhang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2009, 113, 4605–4611 CAS.
- Z. Pei, L. Ding, M. Lu, Z. Fan, S. Weng, J. Hu and P. Liu, J. Phys. Chem. C, 2014, 118, 9570–9577 CAS.
- J. Wang, Z. Wang, B. Huang, Y. Ma, Y. Liu, X. Qin, X. Zhang and Y. Dai, ACS Appl. Mater. Interfaces, 2012, 4, 4024–4030 CAS.
- R. W. Matthews and S. R. McEvoy, J. Photochem. Photobiol., A, 1992, 66, 355–366 CrossRef CAS.
- A. M. Ali, E. A. C. Emanuelsson and D. A. Patterson, Appl. Catal., B, 2011, 106, 323–336 CrossRef CAS PubMed.
- R. A. Reichle, K. G. McCurdy and L. G. Hepler, Can. J. Chem., 1975, 53, 3841–3845 CrossRef CAS.
- A. Moezzi, M. Cortie and A. McDonagh, Dalton Trans., 2011, 4871–4878 RSC.
-
(a) S. Yamabi and H. Imai, J. Mater. Chem., 2002, 12, 3773–3778 RSC;
(b) A. Moezzi, M. B. Cortie and A. M. McDonagh, Dalton Trans., 2013, 14432–14437 RSC;
(c) W. J. Li, E. W. Shi, W. Z. Zhong and Z. W. Yin, J. Cryst. Growth, 1999, 203, 186–196 CrossRef CAS.
- S. G. Kumar and K. S. R. K. Rao, Nanoscale, 2014, 6, 11574–11632 RSC.
- N. C. S. Selvam, J. J. Vijaya and L. J. Kennedy, Ind. Eng. Chem. Res., 2012, 51, 16333–16345 CrossRef.
-
(a) J. H. Qian, L. W. Zhu and R. Guo, J. Chin. Chem. Soc., 2005, 52, 1245–1252 CAS;
(b) N. C. S. Selvam, J. J. Vijaya and L. J. Kennedy, J. Colloid Interface Sci., 2013, 407, 215–224 CrossRef PubMed.
- J. Y. Dong, Y. J. Hsu, D. S. H. Wong and S. Y. Lu, J. Phys. Chem. C, 2010, 114, 8867–8872 CAS.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- S. S. Shinde, P. S. Shinde, C. H. Bhosale and K. Y. Rajpure, J. Photochem. Photobiol., B, 2011, 104, 425–433 CrossRef CAS PubMed.
- P. R. Potti and V. C. Srivastava, Ind. Eng. Chem. Res., 2012, 51, 7948–7956 CrossRef CAS.
- C. C. Hwang and T. Y. Wu, Mater. Sci. Eng., B, 2004, 111, 197–206 CrossRef PubMed.
- K. M. Fang, Z. Z. Wang, M. Zhang, A. J. Wang, Z. Y. Meng and J. J. Feng, J. Colloid Interface Sci., 2013, 402, 68–74 CrossRef CAS PubMed.
- H. Liu, J. Yang, J. Liang, Y. Huang and C. Tang, J. Am. Ceram. Soc., 2008, 91, 1287–1291 CrossRef CAS PubMed.
- M. Y. Guo, A. M. C. Ng, F. Liu, A. B. Djurisic, W. K. Chan, H. Su and K. S. Wong, J. Phys. Chem. C, 2011, 115, 11095–11101 CAS.
- Q. Wan, T. H. Wang and J. C. Zhao, Appl. Phys. Lett., 2005, 87, 083105 CrossRef PubMed.
- Y. Wang, X. Li, G. Lu, X. Quan and G. Chen, J. Phys. Chem. C, 2008, 112, 7332–7336 CAS.
- E. S. Jang, J. H. Won, S. J. Hwang and J. H. Choy, Adv. Mater., 2006, 18, 3309–3312 CrossRef CAS.
-
(a) X. Zou, H. Fan, Y. Tian and S. Yan, CrystEngComm, 2014, 16, 1149–1156 RSC;
(b) P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539–14548 CrossRef CAS PubMed.
- Y. Liu, J. Shi, Q. Peng and Y. Li, J. Mater. Chem., 2012, 22, 6539–6541 RSC.
- B. Liu, J. Xu, S. Ran, Z. Wang, D. Chen and G. Shen, CrystEngComm, 2012, 14, 4582–4588 RSC.
- Z. Peng, Y. Jiang, Y. Song, C. Wang and H. Zhang, Chem. Mater., 2008, 20, 3153–3162 CrossRef CAS.
-
(a) A. Subramania, G. V. Kumar, A. R. S. Priya and T. Vasudevan, Nanotechnology, 2007, 18, 225601 CrossRef;
(b) L. H. Zhao and S. Q. Sun, CrystEngComm, 2011, 13, 1864–1869 RSC.
- J. Yu and X. Yu, Environ. Sci. Technol., 2008, 42, 4902–4907 CrossRef CAS.
-
(a) H. Yu, J. Yu, S. Liu and S. Mann, Chem. Mater., 2007, 19, 4327–4334 CrossRef CAS;
(b) C. Zhu, B. Lu, Q. Su, E. Xie and W. Lan, Nanoscale, 2012, 4, 3060–3064 RSC;
(c) J. Yu and J. Zhang, Dalton Trans., 2010, 5860–5867 RSC;
(d) H. Li, Z. Bian, J. Zhu, D. Zhang, G. Li, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 8406–8407 CrossRef CAS PubMed.
- E. Yassitepe, H. C. Yatmaz, C. Ozturk, K. Ozturk and C. Duran, J. Photochem. Photobiol., A, 2008, 198, 1–6 CrossRef CAS PubMed.
-
(a) A. V. Dijken, A. H. Janssen, M. H. P. Smitsmans, D. Vanmaekelbergh and A. Meijerink, Chem. Mater., 1998, 10, 3513–3522 CrossRef;
(b) R. I. Bickley and F. S. Stone, J. Catal., 1973, 31, 389–397 CrossRef CAS.
- Q. Xie, J. Li, Q. Tian and R. Shi, J. Mater. Chem., 2012, 22, 13541–13547 RSC.
-
(a) H. C. Zeng, Curr. Nanosci., 2007, 3, 177–181 CrossRef CAS;
(b) X. Chen, X. Jing, J. Wang, J. Liu, D. Song and L. Liu, CrystEngComm, 2013, 15, 7243–7249 RSC;
(c) W. Ostwald, Z. Phys. Chem., 1897, 22, 289–330 CAS;
(d) W. Ostwald, Z. Phys. Chem., 1900, 34, 495–503 Search PubMed.
- F. Lu, W. Cai and Y. Zhang, Adv. Funct. Mater., 2008, 18, 1047–1056 CrossRef CAS.
- C. Ye, Y. Bando, G. Shen and D. Golberg, J. Phys. Chem. B, 2006, 110, 15146–15151 CrossRef CAS PubMed.
- J. Gupta, K. C. Barick and D. Bahadur, J. Alloys Compd., 2011, 509, 6725–6730 CrossRef CAS PubMed.
- I. Altln, I. Polat, E. Bacakslz and M. Sokmen, Appl. Surf. Sci., 2012, 258, 4861–4865 CrossRef PubMed.
- J. Xie, H. Wang, M. Duan and L. Zhang, Appl. Surf. Sci., 2011, 257, 6358–6363 CrossRef CAS PubMed.
- D. Chen, D. Yang, Q. Wang and Z. Jiang, Ind. Eng. Chem. Res., 2006, 45, 4110–4116 CrossRef CAS.
- H. Wang, S. Dong, Y. Chang, X. Zhou and X. Hu, Appl. Surf. Sci., 2012, 258, 4288–4293 CrossRef CAS PubMed.
- L. Sun, R. Shao, Z. Chen, L. Tang, Y. Dai and J. Ding, Appl. Surf. Sci., 2012, 258, 5455–5461 CrossRef CAS PubMed.
- A. Mclaren, T. V. Solis, G. Li and S. C. Tsang, J. Am. Chem. Soc., 2009, 131, 12540–12541 CrossRef CAS PubMed.
- L. Xu, Y. L. Hu, C. Pelligra, C. H. Chen, L. Jin, H. Huang, S. Sithambaram, M. Aindow, R. Joesten and S. L. Suib, Chem. Mater., 2009, 21, 2875–2885 CrossRef CAS.
-
(a) M. Wang, Y. Zhang, Y. Zhou, F. Yang, E. J. Kim, S. H. Hahn and S. G. Seong, CrystEngComm, 2013, 15, 754–763 RSC;
(b) S. Das and S. Ghosh, Dalton Trans., 2013, 42, 1645–1656 RSC;
(c) S. Das, K. Dutta and A. Pramanik, CrystEngComm, 2013, 15, 6349–6358 RSC;
(d) M. Raula, M. Biswas and T. K. Mandal, RSC Adv., 2014, 4, 5055–5064 RSC.
-
(a) W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef;
(b) C. M. Teh and A. R. Mohamed, J. Alloys Compd., 2011, 509, 1648–1660 CrossRef CAS PubMed;
(c) L. G. Devi, N. Kottam, S. G. Kumar and K. S. A. Raju, Catal. Lett., 2009, 131, 612–617 CrossRef;
(d) J. Choi, H. Park and M. R. Hoffmann, J. Phys. Chem. C, 2010, 114, 783–792 CrossRef CAS.
- H. Benhebal, M. Chaib, C. Malengreaux, S. D. Lambert, A. Leonard, M. Crine and B. Heinrichs, J. Taiwan Inst. Chem. Eng., 2014, 45, 249–253 CrossRef CAS PubMed.
- V. Etacheri, R. Roshan and V. Kumar, ACS Appl. Mater. Interfaces, 2012, 4, 2717–2725 CAS.
-
(a) B. E. Sernelius, K. F. Berggren, Z. C. Jin, I. Hamberg and C. G. Granqvist, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 10244 CrossRef CAS;
(b) Z. Li, W. Shen, S. Xue and X. Zu, Colloids Surf., A, 2008, 320, 156–160 CrossRef CAS PubMed.
- J. Z. Kong, A. D. Li, X. Y. Li, H. F. Zhai, W. Q. Zhang, Y. P. Gong, H. Li and D. Wu, J. Solid State Chem., 2010, 183, 1359–1364 CrossRef CAS PubMed.
- J. Z. Kong, A. D. Li, H. F. Zhai, Y. P. Gong, H. Li and D. Wu, J. Solid State Chem., 2009, 182, 2061–2067 CrossRef CAS PubMed.
-
(a) A. Janotti and C. G. V. de Walle, Appl. Phys. Lett., 2005, 87, 122102 CrossRef PubMed;
(b) A. Janotti and C. G. V. de Walle, J. Cryst. Growth, 2006, 287, 58–65 CrossRef CAS PubMed.
- C. Wu, L. Shen, Y. C. Zhang and Q. Huang, Mater. Lett., 2011, 65, 1794–1796 CrossRef CAS PubMed.
- C. Xu, L. Cao, G. Su, W. Liu, X. Qu and Y. Yu, J. Alloys Compd., 2010, 497, 373–376 CrossRef CAS PubMed.
- K. Jayanthi, S. Chawla, A. G. Joshi, Z. H. Khan and R. K. Kotnala, J. Phys. Chem. C, 2010, 114, 18429–18434 CAS.
- R. Ullah and J. Dutta, J. Hazard. Mater., 2008, 156, 194–200 CrossRef CAS PubMed.
- F. Tuomisto, K. Sarrinen, D. C. Look and G. C. Farlow, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085206 CrossRef.
- M. A. Mahmood, S. Baruah and J. Dutta, Mater. Chem. Phys., 2011, 130, 531–535 CrossRef CAS PubMed.
- L. X. Zhang, P. Liu and Z. X. Su, Mater. Res. Bull., 2006, 41, 1631–1637 CrossRef CAS PubMed.
- A. Dodd, A. McKinley, T. Tsuzuki and M. Saunders, Mater. Chem. Phys., 2009, 114, 382–386 CrossRef CAS PubMed.
- K. C. Barick, S. Singh, M. Aslam and D. Bahadur, Microporous Mesoporous Mater., 2010, 134, 195–202 CrossRef CAS PubMed.
- B. M. Rajbongshi and S. K. Samdarshi, Appl. Catal., B, 2014, 144, 435–441 CrossRef CAS PubMed.
- N. V. Kaneva, D. T. Dimitrov and C. D. Dushkin, Appl. Surf. Sci., 2011, 257, 8113–8120 CrossRef CAS PubMed.
- K. Sato and H. K. Yoshida, Semicond. Sci. Technol., 2002, 17, 367–376 CrossRef CAS.
- J. Zhao, L. Wang, X. Yan, Y. Yang, Y. Lei, J. Zhou, Y. Huang, Y. Gu and Y. Zhang, Mater. Res. Bull., 2011, 46, 1207–1210 CrossRef CAS PubMed.
- J. B. Zhong, J. Z. Li, X. Y. He, J. Zeng, Y. Lu, W. Hu and K. Lin, Curr. Appl. Phys., 2012, 12, 998–1001 CrossRef PubMed.
-
(a) L. G. Devi, N. Kottam, S. G. Kumar and K. E. Rajashekhar, Cent. Eur. J. Chem., 2009, 8, 142–148 CrossRef;
(b) K. E. Karakitsou and X. E. Verykios, J. Phys. Chem., 1993, 97, 1184–1189 CrossRef CAS;
(c) A. W. Xu, Y. Gao and H. Q. Liu, J. Catal., 2002, 207, 151–157 CrossRef CAS.
- M. Fu, Y. Li, S. Wu, P. Lu, J. Liu and F. Dong, Appl. Surf. Sci., 2011, 258, 1587–1591 CrossRef CAS PubMed.
- H. Ma, L. Yue, C. Yu, X. Dong, X. Zhang, M. Xue, X. Zhang and Y. Fu, J. Mater. Chem., 2012, 22, 23780–23788 RSC.
-
(a) B. Donkova, D. Dimitrov, M. Kostadinov, E. Mitkova and D. Mehandjiev, Mater. Chem. Phys., 2010, 123, 563–568 CrossRef CAS PubMed;
(b) Y. Kanai, Jpn. J. Appl. Phys., 1991, 30, 703–707 CrossRef CAS.
-
(a) X. Zhang, M. Zhou and L. Lei, Carbon, 2005, 43, 1700–1708 CrossRef CAS PubMed;
(b) G. L. Puma, A. Bono, D. Krishnaiah and J. G. Collin, J. Hazard. Mater., 2008, 157, 209–219 CrossRef PubMed;
(c) A. E. Eliyas, L. Ljutzkanov, I. D. Stambolova, V. N. Blaskov, S. V. Vassilev, E. N. R. Velkova and D. R. Mehandjiev, Cent. Eur. J. Chem., 2013, 11, 464–470 CrossRef CAS PubMed.
- N. M. Jacob, G. Madras, N. Kottam and T. Thomas, Ind. Eng. Chem. Res., 2014, 53, 5895–5904 CrossRef.
- O. Yayapao, T. Thongtem, A. Phuruangrat and S. Thongtem, Mater. Lett., 2013, 90, 83–86 CrossRef CAS PubMed.
- M. Khatamian, A. A. Khandar, B. Divband, M. Haghighi and S. Ebrahimiasl, J. Mol. Catal. A: Chem., 2012, 365, 120–127 CrossRef CAS PubMed.
- S. Suwanboon, P. Amornpitoksuk, A. Sukolrat and N. Muensit, Ceram. Int., 2013, 39, 2811–2819 CrossRef CAS PubMed.
- S. Anandan, A. Vinu, T. Mori, N. Gokulakrishnan, P. Srinivasu, V. Murugesan and K. Ariga, Catal. Commun., 2007, 8, 1377–1382 CrossRef CAS PubMed.
- S. Anandan, A. Vinu, K. L. P. S. Lovely, N. Gokulakrishnan, P. Srinivasu, T. Mori, V. Murugesan, V. Sivamurugan and K. Ariga, J. Mol. Catal. A: Chem., 2007, 266, 149–157 CrossRef CAS PubMed.
- M. Rezaei and A. H. Yangjeh, Appl. Surf. Sci., 2013, 265, 591–596 CrossRef CAS PubMed.
- B. Subash, B. Krishnakumar, R. Velmurugan, M. Swaminathan and M. Shanthi, Catal. Sci. Technol., 2012, 2, 2319–2326 CAS.
-
(a) M. V. Dozzi and E. Selli, J. Photochem. Photobiol., C, 2013, 14, 13–28 CrossRef CAS PubMed;
(b) J. Zhang, Y. Wu, M. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715–726 RSC;
(c) L. G. Devi and R. Kavitha, Appl. Catal., B, 2013, 140–141, 559–587 CrossRef CAS PubMed;
(d) L. G. Devi and R. Kavitha, RSC Adv., 2014, 4, 28265–28299 RSC;
(e) R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS PubMed.
- S. Sun, X. Chang, X. Li and Z. Li, Ceram. Int., 2013, 39, 5197–5203 CrossRef CAS PubMed.
- Y. Qiu, M. Yang, H. Fan, Y. Xu, Y. Shao, X. Yang and S. Yang, Mater. Lett., 2013, 99, 105–107 CrossRef CAS PubMed.
- C. Shifu, Z. Wei, Z. Sujuan and L. Wei, Chem. Eng. J., 2009, 148, 263–269 CrossRef CAS PubMed.
-
(a) K. Chen, G. H. Fan, Y. Zhang and S. F. Ding, Acta Phys.-Chim. Sin., 2008, 24, 61–66 CAS;
(b) J. Li, R. Kykyneshi, J. Tate and A. W. Sleight, Solid State Sci., 2007, 9, 613–618 CrossRef CAS PubMed.
- D. Zhang, J. Gong, J. Ma, G. Han and Z. Tong, Dalton Trans., 2013, 16556–16561 RSC.
- Z. Yu, L. C. Yin, Y. Xie, G. Liu, X. Ma and H. M. Cheng, J. Colloid Interface Sci., 2013, 400, 18–23 CrossRef CAS PubMed.
- X. Zong, C. Sun, H. Yu, Z. G. Chen, Z. Xing, D. Ye, G. Q. Lu, X. Li and L. Wang, J. Phys. Chem. C, 2013, 117, 4937–4942 CAS.
- P. Gu, X. Wang, T. Li and H. Meng, Mater. Res. Bull., 2013, 48, 4699–4703 CrossRef CAS PubMed.
- H. Qin, W. Li, Y. Xia and T. He, ACS Appl. Mater. Interfaces, 2011, 3, 3152–3156 CAS.
- D. Li and H. Haneda, J. Photochem. Photobiol., A, 2003, 155, 171–178 CrossRef CAS.
- D. Li and H. Haneda, J. Photochem. Photobiol., A, 2003, 160, 203–212 CrossRef CAS.
- D. Li, H. Haneda, N. Ohashi, S. Hishita and Y. Yoshikawa, Catal. Today, 2004, 93–95, 895–901 CrossRef CAS PubMed.
- D. Li and H. Haneda, Chemosphere, 2004, 54, 1099–1110 CrossRef CAS PubMed.
- K. Tennakone, O. A. Iieperuma, J. M. S. Bandara and W. C. B. Kiridena, Semicond. Sci. Technol., 1992, 7, 423–424 CrossRef CAS.
- J. S. Lee, O. S. Kwon and J. Jang, J. Mater. Chem., 2012, 22, 14565–14572 RSC.
-
(a) H. Yoo, C. Bae, Y. Yang, S. Lee, M. Kim, H. Kim, Y. Kim and H. Shin, Nano Lett., 2014, 14, 4413–4417 CrossRef CAS PubMed;
(b) Y. Yang, M. Nogami, J. Shi, H. Chen, Y. Liu and S. Qian, J. Mater. Chem., 2003, 13, 3026–3032 RSC;
(c) N. Zhang, S. Liu and Y. J. Xu, Nanoscale, 2012, 4, 2227–2238 RSC;
(d) A. Ayati, A. Ahmadpour, F. F. Bamoharram, B. Tanhaei, M. Manttari and M. Sillanpaa, Chemosphere, 2014, 107, 163–174 CrossRef CAS PubMed;
(e) R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed;
(f) A. Primo, A. Corma and H. Garcia, Phys. Chem. Chem. Phys., 2011, 13, 886–910 RSC;
(g) A. Bumajdad and M. Madkour, Phys. Chem. Chem. Phys., 2014, 16, 7146–7158 RSC;
(h) M. Jakob, H. Levanon and P. V. Kamat, Nano Lett., 2003, 3, 353–358 CrossRef CAS;
(i) H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
-
(a) H. Zhu, X. Chen, Z. Zheng, X. Ke, E. Jaatinen, J. Zhao, C. Guo, T. Xie and D. Wang, Chem. Commun., 2009, 7524–7526 RSC;
(b) S. Sarina, E. R. Waclawik and H. Zhu, Green Chem., 2013, 15, 1814–1833 RSC;
(c) S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed;
(d) X. Zhou, G. Liu, J. Yu and W. Fan, J. Mater. Chem., 2012, 22, 21337–21354 RSC.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- Y. Zheng, C. Chen, Y. Zhan, X. Lin, Q. Zheng, K. Wei and J. Zhu, J. Phys. Chem. C, 2008, 112, 10773–10777 CAS.
- M. M. Montero, A. Borras, Z. Saghi, P. R. Gomez, J. R. S. Valencia, J. C. Gonzalez, A. Barranco, P. Midgley, J. Cotrino and A. R. G. Elipe, J. Mater. Chem., 2012, 22, 1341–1346 RSC.
- Q. Deng, X. Duan, D. H. L. Ng, H. Tang, Y. Yang, M. Kong, Z. Wu, W. Cai and G. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6030–6037 CAS.
- Y. Zheng, L. Zheng, Y. Zhan, X. Lin, Q. Zheng and K. Wei, Inorg. Chem., 2007, 46, 6980–6986 CrossRef CAS PubMed.
- C. Ren, B. Yang, M. Wu, J. Xu, Z. Fu, Y. Lv, T. Guo, Y. Zhao and C. Zhu, J. Hazard. Mater., 2010, 182, 123–129 CrossRef CAS PubMed.
- C. Tian, W. Li, K. Pan, Q. Zhang, G. Tian, W. Zhou and H. Fu, J. Solid State Chem., 2010, 183, 2720–2725 CrossRef CAS PubMed.
- W. Xie, Y. Li, W. Sun, J. Huang, H. Xie and X. Zhao, J. Photochem. Photobiol., A, 2010, 216, 149–155 CrossRef CAS PubMed.
-
(a) A. Sclafani and J. M. Hermann, J. Photochem. Photobiol., A, 1998, 113, 181–188 CrossRef CAS;
(b) C. Yu, K. Yang, Y. Xie, Q. Fan, J. C. Yu, Q. Shu and C. Wang, Nanoscale, 2013, 5, 2142–2151 RSC;
(c) B. Chai, X. Wang, S. Cheng, H. Zhou and F. Zhang, Ceram. Int., 2014, 40, 429–435 CrossRef CAS PubMed.
- Y. Zhang and J. Mu, J. Colloid Interface Sci., 2007, 309, 478–484 CrossRef CAS PubMed.
- R. Georgekutty, M. K. Serry and S. C. Pillai, J. Phys. Chem. C, 2008, 112, 13563–13570 CAS.
- Q. Simon, D. Barreca, D. Bekermann, A. Gasparotto, C. Maccato, E. Comini, V. Gombac, P. Fornasiero, O. I. Lebedev, S. Turner, A. Devi, R. A. Fischer and G. V. Tendeloo, Int. J. Hydrogen Energy, 2011, 36, 15527–15537 CrossRef CAS PubMed.
-
(a) K. Lalitha, J. K. Reddy, M. V. P. Sharma, V. D. Kumari and M. Subrahmanyam, Int. J. Hydrogen Energy, 2010, 35, 3991–4001 CrossRef CAS PubMed;
(b) S. Ma, J. Xue, Y. Zhou and Z. Zhang, J. Mater. Chem. A, 2014, 2, 7272–7280 RSC;
(c) S. M. Lam, J. C. Sin, A. Z. Abdullah and A. R. Mohamed, Chem. Pap., 2013, 67, 1277–1284 CrossRef CAS;
(d) M. Wu, J. Yan, M. Zhao and Q. Jiang, ChemPlusChem, 2012, 77, 931–935 CrossRef CAS.
- H. R. Liu, G. X. Shao, J. F. Zhao, Z. X. Zhang, Y. Zhang, J. Liang, X. G. Liu, H. S. Jia and B. S. Xu, J. Phys. Chem. C, 2012, 116, 16182–16190 CAS.
- C. Gu, C. Cheng, H. Huang, T. Wong, N. Wang and T. Y. Zhang, Cryst. Growth Des., 2009, 9, 3278–3285 CAS.
- J. Liqiang, W. Baiqi, X. Baifu, L. Shudan, S. Keying, C. Weimin and F. Honggang, J. Solid State Chem., 2004, 177, 4221–4227 CrossRef PubMed.
-
(a) B. Tian, J. Zhang, T. Tong and F. Chen, Appl. Catal., B, 2008, 79, 394–401 CrossRef CAS PubMed;
(b) M. Sadeghi, W. Liu, T. G. Zhang, P. Stavropoulos and B. Levy, J. Phys. Chem., 1996, 100, 19466–19474 CrossRef CAS.
- H. Zeng, P. Liu, W. Cai, S. Yang and X. Xu, J. Phys. Chem. C, 2008, 112, 19620–19624 CAS.
- P. Li, Z. Wei, T. Wu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2011, 133, 5660–5663 CrossRef CAS PubMed.
-
(a) Y. Chen, M. Kim, G. Lian, M. B. Johnson and X. Peng, J. Am. Chem. Soc., 2005, 127, 13331–13337 CrossRef CAS PubMed;
(b) F. Li, Y. Ding, P. Gao, X. Xin and Z. L. Wang, Angew. Chem., Int. Ed., 2004, 43, 5238–5242 CrossRef CAS PubMed;
(c) Z. L. Wang, X. Y. Kong and J. M. Zuo, Phys. Rev. Lett., 2003, 91, 185502 CrossRef CAS.
- M. Ahmad, S. Yingying, A. Nisar, H. Sun, W. Shen, M. Wei and J. Zhu, J. Mater. Chem., 2011, 21, 7723–7729 RSC.
- E. C. H. Sykes, F. J. Williams, M. S. Tikhov and R. M. Lambert, J. Phys. Chem. B, 2002, 106, 5390–5394 CrossRef CAS.
- Q. Wang, B. Geng and S. Wang, Environ. Sci. Technol., 2009, 43, 8968–8973 CrossRef CAS PubMed.
- N. Udawatte, M. Lee, J. Kim and D. Lee, ACS Appl. Mater. Interfaces, 2011, 3, 4531–4538 CAS.
-
(a) M. Lee, P. Amaratunga, J. Kim and D. Lee, J. Phys. Chem. C, 2010, 114, 18366–18371 CrossRef CAS;
(b) P. Amaratunga, M. Lee, J. Kim and D. Lee, Can. J. Chem., 2011, 89, 1001–1009 CrossRef CAS.
- J. Lee, H. S. Shim, M. Lee, J. K. Song and D. Lee, J. Phys. Chem. Lett., 2011, 2, 2840–2845 CrossRef CAS.
- P. Pawinrat, O. Mekasuwandumrong and J. Panpranot, Catal. Commun., 2009, 10, 1380–1385 CrossRef CAS PubMed.
- A. Wood, M. Giersig and P. Mulvaney, J. Phys. Chem. B, 2001, 105, 8810–8815 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2003, 107, 7479–7485 CrossRef CAS.
-
(a) A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS;
(b) M. Ashokkumar, Int. J. Hydrogen Energy, 1998, 23, 427–438 CrossRef CAS.
- J. J. Wu and C. H. Tseng, Appl. Catal., B, 2006, 66, 51–57 CrossRef CAS PubMed.
- W. I. Park, G. C. Yi, J. W. Kim and S. M. Park, Appl. Phys. Lett., 2003, 82, 4358–4360 CrossRef CAS PubMed.
- S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS.
-
(a) B. Divband, M. Khatamian, G. R. K. Eslamian and M. Darbandi, Appl. Surf. Sci., 2013, 284, 80–86 CrossRef CAS PubMed;
(b) D. Lin, H. Wu, R. Zhang and W. Pan, Chem. Mater., 2009, 21, 3479–3484 CrossRef CAS;
(c) Z. Zhang, H. Liu, H. Zhang, H. Dong, X. Liu, H. Jia and B. Xu, Superlattices Microstruct., 2014, 65, 134–145 CrossRef CAS PubMed;
(d) C. Tian, Q. Zhang, B. Jiang, G. Tian and H. Fu, J. Alloys Compd., 2011, 509, 6935–6941 CrossRef CAS PubMed.
-
(a) R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS;
(b) P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed;
(c) S. G. Kumar and K. S. R. K. Rao, Energy Environ. Sci., 2014, 7, 45–102 RSC;
(d) H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
-
(a) H. Ma, J. Han, Y. Fu, Y. Song, C. Yu and X. Dong, Appl. Catal., B, 2011, 102, 417–423 CrossRef CAS PubMed;
(b) H. G. Kim, P. H. Borse, W. Choi and J. S. Lee, Angew. Chem., Int. Ed., 2005, 44, 4585–4589 CrossRef CAS PubMed;
(c) X. Wu, S. Yin, Q. Dong, B. Liu, Y. Wang, T. Sekino, S. W. Lee and T. Sato, Sci. Rep., 2013, 3, 2918 Search PubMed;
(d) Y. Chen, J. C. Crittenden, S. Hackney, L. Sutter and D. W. Hand, Environ. Sci. Technol., 2005, 39, 1201–1208 CrossRef CAS;
(e) S. Ida, A. Takashiba, S. Koga, H. Hagiwara and T. Ishihara, J. Am. Chem. Soc., 2014, 136, 1872–1878 CrossRef CAS PubMed.
- D. Chen, H. Zhang, S. Hu and J. Li, J. Phys. Chem. C, 2008, 112, 117–122 CAS.
-
(a) M. Law, L. E. Greene, A. Radenovic, T. Kuykendall, J. Liphardt and P. Yang, J. Phys. Chem. B, 2006, 110, 22652–22663 CrossRef CAS PubMed;
(b) J. Tian, Z. Zhao, A. Kumar, R. I. Boughton and H. Liu, Chem. Soc. Rev., 2014, 43, 6920–6937 RSC;
(c) B. Weng, S. Liu, Z. R. Tang and Y. J. Xu, RSC Adv., 2014, 4, 12685–12700 RSC.
- Q. Zhang, W. Fan and L. Gao, Appl. Catal., B, 2007, 76, 168–173 CrossRef CAS PubMed.
- J. Tian, L. Chen, Y. Yin, X. Wang, J. Dai, Z. Zhu, X. Liu and P. Wu, Surf. Coat. Technol., 2009, 204, 205–214 CrossRef CAS PubMed.
- J. Tian, J. Wang, J. Dai, X. Wang and Y. Yin, Surf. Coat. Technol., 2009, 204, 723–730 CrossRef CAS PubMed.
- J. Mani, H. Sakeek, S. Habouti, M. Dietze and M. E. Souni, Catal. Sci. Technol., 2012, 2, 379–385 CAS.
-
(a) K. Mallick and M. S. Scurrell, Appl. Catal., A, 2003, 253, 527–536 CrossRef CAS;
(b) C. H. Wu, Chemosphere, 2004, 57, 601–608 CrossRef CAS PubMed.
- S. Liao, H. Donggen, D. Yu, Y. Su and G. Yuan, J. Photochem. Photobiol., A, 2004, 168, 7–13 CrossRef CAS PubMed.
- M. Konyar, H. C. Yatmaz and K. Ozturk, Appl. Surf. Sci., 2012, 258, 7440–7447 CrossRef CAS PubMed.
- G. Yang, Z. Yan and T. Xiao, Appl. Surf. Sci., 2012, 258, 8704–8712 CrossRef CAS PubMed.
- C. Wang, B. Q. Xu, X. Wang and J. Zhao, J. Solid State Chem., 2005, 178, 3500–3506 CrossRef CAS PubMed.
- Y. Wei, Y. Huang, J. Wu, M. Wang, C. Guo, Q. Dong, S. Yin and T. Sato, J. Hazard. Mater., 2013, 248–249, 202–210 CrossRef CAS PubMed.
-
(a) J. Yang and J. H. Swisher, Mater. Charact., 1996, 37, 153–159 CrossRef CAS;
(b) C. L. Wang, W. S. Hwang, H. L. Chu, C. S. Hsi, H. H. Ko, K. M. Chang, X. Zhao, M. C. Wang and W. L. Li, Ceram. Int., 2014, 40, 7407–7415 CrossRef CAS PubMed;
(c) Y. L. Chai, Y. S. Chang, G. J. Chen and Y. J. Hsiao, Mater. Res. Bull., 2008, 43, 1066–1073 CrossRef CAS PubMed.
- Z. Zhang, C. Shao, X. Li, L. Zhang, H. Xue, C. Wang and Y. Liu, J. Phys. Chem. C, 2010, 114, 7920–7925 CAS.
- L. Zheng, Y. Zheng, C. Chen, Y. Zhan, X. Lin, Q. Zheng, K. Wei and J. Zhu, Inorg. Chem., 2009, 48, 1819–1825 CrossRef CAS PubMed.
- M. T. Uddin, Y. Nicolas, C. Olivier, T. Toupance, L. Servant, M. M. Muller, H. J. Kleebe, J. Ziegler and W. Jaegermann, Inorg. Chem., 2012, 51, 7764–7773 CrossRef CAS PubMed.
- D. Y. T. Martinez, R. C. Perez, G. T. Delgado and O. Z. Angel, J. Photochem. Photobiol., A, 2012, 235, 49–55 CrossRef PubMed.
- C. Wang, X. Wang, B. Q. Xu, J. Zhao, B. Mai, P. Peng, G. Sheng and J. Fu, J. Photochem. Photobiol., A, 2004, 168, 47–52 CrossRef CAS PubMed.
-
(a) T. Hashemi, H. M. Al-Allak, J. Illingsworth, A. W. Brinkman and J. Woods, J. Mater. Sci. Lett., 1990, 9, 776–778 CrossRef CAS;
(b) F. Belliard, P. A. Connor and J. T. S. Irvine, Solid State Ionics, 2000, 135, 163–167 CrossRef CAS;
(c) J. Fang, A. Huang, P. Zhu, N. Xu, J. Xie, J. Chi, S. Feng, R. Xu and M. Wu, Mater. Res. Bull., 2001, 36, 1391–1397 CrossRef CAS.
- X. Huang, L. Shang, S. Chen, J. Xia, X. Qi, X. Wang, T. Zhang and X. M. Meng, Nanoscale, 2013, 5, 3828–3833 RSC.
- K. Tennakone and J. Bandara, Appl. Catal., A, 2001, 208, 335–341 CrossRef CAS.
-
(a) Q. Kuang, Z. Y. Jiang, Z. X. Xie, S. C. Lin, Z. W. Lin, S. Y. Xie, R. B. Huang and L. S. Zheng, J. Am. Chem. Soc., 2005, 127, 11777–11784 CrossRef CAS PubMed;
(b) M. Davis, W. M. Hikal, C. Gumeci and L. J. H. Weeks, Catal. Sci. Technol., 2012, 2, 922–924 RSC;
(c) M. Li, Y. Hu, S. Xie, Y. Huang, Y. Tong and X. Lu, Chem. Commun., 2014, 4341–4343 RSC;
(d) S. Balachandran, K. Selvam, B. Babu and M. Swaminathan, Dalton Trans., 2013, 16365–16374 RSC;
(e) H. Wang, S. Baek, J. Lee and S. Lim, Chem. Eng. J., 2009, 146, 355–361 CrossRef CAS PubMed.
- Z. L. Liu, J. C. Deng, J. J. Deng and F. F. Li, Mater. Sci. Eng., B, 2008, 150, 99–104 CrossRef CAS PubMed.
- R. Saravanan, S. Karthikeyan, V. K. Gupta, G. Sekaran, V. Narayanan and A. Stephen, Mater. Sci. Eng., C, 2013, 33, 91–98 CrossRef CAS PubMed.
- C. C. Hsu and N. L. Wu, J. Photochem. Photobiol., A, 2005, 172, 269–274 CrossRef CAS PubMed.
- C. Shifu, Z. Wei, L. Wei, Z. Huaye and Y. Xiaoling, Chem. Eng. J., 2009, 155, 466–473 CrossRef PubMed.
-
(a) C. Shifu, C. Lei, G. Shen and C. Gengyu, Mater. Chem. Phys., 2006, 98, 116–120 CrossRef PubMed;
(b) C. Shifu, Z. Wei, L. Wei, Z. Huaye, Y. Xiaoling and C. Yinghao, J. Hazard. Mater., 2009, 172, 1415–1423 CrossRef PubMed.
- N. V. Kaneva and C. D. Dushkin, Colloids Surf., A, 2011, 382, 211–218 CrossRef CAS PubMed.
- X. Guo, H. Zhu and Q. Li, Appl. Catal., B, 2014, 160–161, 408–414 CrossRef CAS PubMed.
- W. Liu, M. Wang, C. Xu, S. Chen and X. Fu, Mater. Res. Bull., 2013, 48, 106–113 CrossRef CAS PubMed.
-
(a) Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. S. Williams, H. Yang, J. Cao, W. Luo, Z. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed;
(b) Y. Bi, S. Ouyang, J. Cao and J. Ye, Phys. Chem. Chem. Phys., 2011, 13, 10071–10075 RSC.
- Z. Zhan, Y. Wang, Z. Lin, J. Zhang and F. Huang, Chem. Commun., 2011, 4517–4519 RSC.
- J. M. Khoshman and M. E. Kordesch, Thin Solid Films, 2007, 515, 7393–7399 CrossRef CAS PubMed.
- F. Zhou, X. Li, J. Shu and J. Wang, J. Photochem. Photobiol., A, 2011, 219, 132–138 CrossRef CAS PubMed.
-
(a) L. Gai, L. Ma, H. Jiang, Y. Ma, Y. Tian and H. Liu, CrystEngComm, 2012, 14, 7479–7486 RSC;
(b) Z. Wang, B. Huang, Y. Dai, X. Qin, X. Zhang, P. Wang, H. Liu and J. Yu, J. Phys. Chem. C, 2009, 113, 4612–4617 CrossRef CAS.
- W. Wu, S. Zhang, X. Xiao, J. Zhou, F. Ren, L. Sun and C. Jiang, ACS Appl. Mater. Interfaces, 2012, 4, 3602–3609 CAS.
- Z. Zhang, C. Shao, X. Li, C. Wang, M. Zhang and Y. Liu, ACS Appl. Mater. Interfaces, 2010, 2, 2915–2923 CAS.
-
(a) H. Ohta, M. Hirano, K. Nakahara, H. Maruta, T. Tanabe, M. Kamiya, T. Kamiya and H. Hosono, Appl. Phys. Lett., 2003, 83, 1029–1031 CrossRef CAS PubMed;
(b) M. J. Ma, B. Lu, T. T. Zhou, Z. Z. Ye, J. G. Lu and X. H. Pan, J. Appl. Phys., 2013, 113, 163704 CrossRef PubMed;
(c) J. Wang, C. Lee, Y. Chen, C. Chen, Y. Chen, C. Lin and Y. Chen, Appl. Phys. Lett., 2009, 95, 131117 CrossRef PubMed.
- B. Li and Y. Wang, J. Phys. Chem. Solids, 2011, 72, 1165–1169 CrossRef CAS PubMed.
- X. Cao, P. Chen and Y. Guo, J. Phys. Chem. C, 2008, 112, 20560–20566 CAS.
-
(a) Y. Tak, S. J. Hong, J. S. Lee and K. Yong, J. Mater. Chem., 2009, 19, 5945–5951 RSC;
(b) Y. Choi, M. Beak and K. Yong, Nanoscale, 2014, 6, 8914–8918 RSC.
- F. Xu, Y. Yuan, H. Han, D. Wu, Z. Gao and K. Jiang, CrystEngComm, 2012, 14, 3615–3622 RSC.
- P. Kundu, P. A. Deshpande, G. Madras and N. Ravishankar, J. Mater. Chem., 2011, 21, 4209–4216 RSC.
- M. Villani, D. Calestani, L. Lazzarini, L. Zanotti, R. Mosca and A. Zappettini, J. Mater. Chem., 2012, 22, 5694–5699 RSC.
- H. M. Pathan and C. D. Lokhande, Bull. Mater. Sci., 2004, 27, 85–111 CrossRef CAS.
- S. Khanchandani, S. Kundu, A. Patra and A. K. Ganguli, J. Phys. Chem. C, 2012, 116, 23653–23662 CAS.
- S. Hotchandani and P. V. Kamat, J. Phys. Chem., 1992, 96, 6834–6839 CrossRef CAS.
- J. Nayak, S. N. Sahu, J. Kasuya and S. Nozaki, Appl. Surf. Sci., 2008, 254, 7215–7218 CrossRef CAS PubMed.
- C. Li, T. Ahmed, M. Ma, T. Edvinsson and J. Zhu, Appl. Catal., B, 2013, 138–139, 175–183 CrossRef CAS PubMed.
- H. X. Sang, X. T. Wang, C. C. Fan and F. Wang, Int. J. Hydrogen Energy, 2012, 37, 1348–1355 CrossRef CAS PubMed.
- M. Basu, N. Garg and A. K. Ganguli, J. Mater. Chem. A, 2014, 2, 7517–7525 CAS.
-
(a) P. Zhang, C. Shao, Z. Zhang, M. Zhang, J. Mu, Z. Guo and Y. Liu, Nanoscale, 2011, 3, 2943–2949 RSC;
(b) P. Zhang, C. Shao, Z. Zhang, M. Zhang, J. Mu, Z. Guo, Y. Sun and Y. Liu, J. Mater. Chem., 2011, 21, 17746–17753 RSC;
(c) V. Kiran and S. Sampath, Nanoscale, 2013, 5, 10646–10652 RSC.
- X. Zhou, Y. Li, T. Peng, W. Xie and X. Zhao, Mater. Lett., 2009, 63, 1747–1749 CrossRef CAS PubMed.
- S. Liu, C. Li, J. Yu and Q. Xiang, CrystEngComm, 2011, 13, 2533–2541 RSC.
- S. Cho, J. W. Jang, J. S. Lee and K. H. Lee, CrystEngComm, 2010, 12, 3929–3935 RSC.
- S. Cho, H. Jeong, D. H. Park, S. H. Jung, H. J. Kim and K. H. Lee, CrystEngComm, 2010, 12, 968–976 RSC.
- S. T. Kochuveedu, Y. H. Jang, Y. J. Jang and D. H. Kim, J. Mater. Chem. A, 2013, 1, 898–905 CAS.
- M. Zhou, X. Gao, Y. Hu, J. Chen and X. Hu, Appl. Catal., B, 2013, 138–139, 1–8 CrossRef CAS PubMed.
- F. Waltz, G. Wißmann, J. Lippke, A. M. Schneider, H. C. Schwarz, A. Feldhoff, S. Eiden and P. Behrens, Cryst. Growth Des., 2012, 12, 3066–3075 CAS.
- J. Mu, C. Shao, Z. Guo, Z. Zhang, M. Zhang, P. Zhang, B. Chen and Y. Liu, ACS Appl. Mater. Interfaces, 2011, 3, 590–596 CAS.
-
(a) L. Zhang, D. Austin, V. I. Merkulov, A. Meleshko, K. L. Klein, M. A. Guillorn, D. H. Lowndes and M. L. Simpson, Appl. Phys. Lett., 2004, 84, 3972–3974 CrossRef CAS PubMed;
(b) H. E. Unalan, D. Wei, K. Suzuki, S. Dalal, P. Hiralal, H. Matsumoto, S. Imaizumi, M. Minagawa, A. Tanioka, A. J. Flewitt, W. I. Milne and G. A. Amaratunga, Appl. Phys. Lett., 2008, 93, 133116 CrossRef PubMed.
- N. Sobana and M. Swamtinathan, Sol. Energy Mater. Sol. Cells, 2007, 91, 727–734 CrossRef CAS PubMed.
- Y. Guo, H. Wang, C. He, L. Qiu and X. Cao, Langmuir, 2009, 25, 4678–4684 CrossRef CAS PubMed.
- S. Ma, J. Xue, Y. Zhou, Z. Zhang and X. Wu, CrystEngComm, 2014, 16, 4478–4484 RSC.
- H. F. Yu and H. Y. Chou, Powder Technol., 2013, 233, 201–207 CrossRef CAS PubMed.
- S. Lian, H. Huang, J. Zhang, Z. Kang and Y. Liu, Solid State Commun., 2013, 155, 53–56 CrossRef CAS PubMed.
- L. Zhang, H. Cheng, R. Zong and Y. Zhu, J. Phys. Chem. C, 2009, 113, 2368–2374 Search PubMed.
-
(a) G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS;
(b) W. J. D. Beenken, Chem. Phys., 2009, 357, 144–150 CrossRef CAS PubMed;
(c) L. Zhao, X. Chen, X. Wang, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 3317–3321 CrossRef CAS PubMed.
-
(a) S. Chowdhury and R. Balasubramanian, Appl. Catal., B, 2014, 160–161, 307–324 CrossRef CAS PubMed;
(b) N. Zhang, Y. Zhang and Y. J. Xu, Nanoscale, 2012, 4, 5792–5813 RSC;
(c) D. Chen, H. Zhang, Y. Liu and J. Li, Energy Environ. Sci., 2013, 6, 1362–1387 RSC;
(d) X. An and J. C. Yu, RSC Adv., 2011, 1, 1426–1434 RSC;
(e) Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC;
(f) R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef CAS PubMed.
- Q. Zhang, C. Tian, A. Wu, T. Tan, L. Sun, L. Wang and H. Fu, J. Mater. Chem., 2012, 22, 11778–11784 RSC.
- Y. Yang, L. Ren, C. Zhang, S. Huang and T. Liu, ACS Appl. Mater. Interfaces, 2011, 3, 2779–2785 CAS.
- B. Li, T. Liu, Y. Wang and Z. Wang, J. Colloid Interface Sci., 2012, 377, 114–121 CrossRef CAS PubMed.
- B. Li and H. Cao, J. Mater. Chem., 2011, 21, 3346–3349 RSC.
-
(a) Z. Xiong, L. L. Zhang, J. Ma and X. S. Zhao, Chem. Commun., 2010, 6099–6101 RSC;
(b) N. Yang, J. Zhai, D. Wang, Y. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS PubMed;
(c) T. Lv, L. Pan, X. Liu and Z. Sun, Catal. Sci. Technol., 2012, 2, 2297–2301 RSC.
- P. V. Kamat, Chem. Rev., 1993, 93, 267–300 CrossRef CAS.
- X. Liu, L. Pan, T. Lv, T. Lu, G. Zhu, Z. Sun and C. Sun, Catal. Sci. Technol., 2011, 1, 1189–1193 CAS.
-
(a) Y. B. Tang, C. S. Lee, J. Xu, Z. T. Liu, Z. H. Chen, Z. He, Y. L. Cao, G. Yuan, H. Song, L. Chen, L. Luo, H. M. Cheng, W. J. Zhang, I. Bello and S. T. Lee, ACS Nano, 2010, 4, 3482–3488 CrossRef CAS PubMed;
(b) Z. Chen, N. Zhang and Y. J. Xu, CrystEngComm, 2013, 15, 3022–3030 RSC;
(c) D. Fu, G. Han, F. Yang, T. Zhang, Y. Chang and F. Liu, Appl. Surf. Sci., 2013, 283, 654–659 CrossRef CAS PubMed.
- D. Fu, G. Han, Y. Chang and J. Dong, Mater. Chem. Phys., 2012, 132, 673–681 CrossRef CAS PubMed.
- T. Xu, L. Zhang, H. Cheng and Y. Zhu, Appl. Catal., B, 2011, 101, 382–387 CrossRef CAS PubMed.
- S. Gayathri, P. Jayabal, M. Kottaisamy and V. Ramakrishnan, J. Appl. Phys., 2014, 115, 173504 CrossRef PubMed.
- Y. Liu, Y. Hu, M. Zhou, H. Qian and X. Hu, Appl. Catal., B, 2012, 125, 425–431 CrossRef CAS PubMed.
- X. Liu, L. Pan, Q. Zhao, T. Lv, G. Zhu, T. Chen, T. Lu, Z. Sun and C. Sun, Chem. Eng. J., 2012, 183, 238–243 CrossRef CAS PubMed.
- J. Wang, T. Tsuzuki, B. Tang, X. Hou, L. Sun and X. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 3084–3090 CAS.
-
(a) S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS;
(b) F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171–15174 RSC;
(c) F. Dong, Z. Zhao, T. Xiong, Z. Ni, W. Zhang, Y. Sun and W. K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CrossRef CAS PubMed;
(d) Y. Cao, Z. Zhang, J. Long, J. Liang, H. Lin, H. Lin and X. Wang, J. Mater. Chem. A, 2014, 2, 17797–17807 RSC.
-
(a) F. Dong, Y. Sun, L. Wu, M. Fu and Z. Wu, Catal. Sci. Technol., 2012, 2, 1332–1335 RSC;
(b) W. Zhang, Y. Sun, F. Dong, W. Zhang, S. Duan and Q. Zhang, Dalton Trans., 2014, 12026–12036 RSC;
(c) Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015 10.1039/c4nr03008g;
(d) A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC;
(e) Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- Y. Wang, R. Shi, J. Lin and Y. Zhu, Energy Environ. Sci., 2011, 4, 2922–2929 CAS.
- S. C. Yan, S. B. Lv, Z. S. Li and Z. G. Zou, Dalton Trans., 2010, 1488–1491 RSC.
- J. X. Sun, Y. P. Yuan, L. G. Qiu, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, Dalton Trans., 2012, 6756–6763 RSC.
- W. Liu, M. Wang, C. Xu, S. Chen and X. Fu, J. Mol. Catal. A: Chem., 2013, 368–369, 9–15 CrossRef CAS PubMed.
- W. Liu, M. Wang, C. Xu and S. Chen, Chem. Eng. J., 2012, 209, 386–393 CrossRef CAS PubMed.
- D. Chen, K. Wang, D. Xiang, R. Zong, W. Yao and Y. Zhu, Appl. Catal., B, 2014, 147, 554–561 CrossRef CAS PubMed.
- D. Chen, K. Wang, T. Ren, H. Ding and Y. Zhu, Dalton Trans., 2014, 13105–13114 RSC.
- Y. P. Zhu, M. Li, Y. L. Liu, T. Z. Ren and Z. Y. Yuan, J. Phys. Chem. C, 2014, 118, 10963–10971 CAS.
- L. Jiang, W. Zhang, Y. Yu and J. Wang, Electrochem. Commun., 2011, 13, 627–630 CrossRef CAS PubMed.
- W. Zhang, L. Jiang and J. Ye, J. Phys. Chem. C, 2009, 113, 16247–16253 CAS.
- T. A. Saleh, M. A. Gondal and Q. A. Drmosh, Nanotechnology, 2010, 21, 495705 CrossRef PubMed.
- X. Liu, L. Pan, T. Lv, Z. Sun and C. Sun, J. Mol. Catal. A: Chem., 2012, 363–364, 417–422 CrossRef CAS PubMed.
-
(a) K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233–2239 CrossRef CAS;
(b) G. Zhu, L. Pan, T. Lu, T. Xu and Z. Sun, J. Mater. Chem., 2011, 21, 14869–14875 RSC;
(c) H. Chu, L. Wei, R. Cui, J. Wang and Y. Li, Coord. Chem. Rev., 2010, 254, 1117–1134 CrossRef CAS PubMed;
(d) Y. Yu, J. C. Yu, J. Yu, Y. Kwok, Y. Che, J. Zhao, L. Ding, W. Ge and P. Wong, Appl. Catal., A, 2005, 289, 186–196 CrossRef CAS PubMed.
-
(a) O. Akhavan, R. Azimirad, S. Safa and M. M. Larijani, J. Mater. Chem., 2010, 20, 7386–7392 RSC;
(b) I. Shakir, J. H. Choi, M. Shahid, Z. Ali and D. J. Kang, J. Mater. Chem., 2012, 22, 20549–20553 RSC;
(c) L. Tian, L. Ye, J. Liu and L. Zan, Catal. Commun., 2012, 17, 99–103 CrossRef CAS PubMed.
- M. Samadi, H. A. Shivaee, M. Zanetti, A. Pourjavadi and A. Moshfegh, J. Mol. Catal. A: Chem., 2012, 359, 42–48 CrossRef CAS PubMed.
- C. Yu, Y. Wang, Y. Liu, C. Guo and Y. Hu, Mater. Lett., 2013, 100, 278–281 CrossRef CAS PubMed.
- R. M. Mohamed and M. A. Salam, Mater. Res. Bull., 2014, 50, 85–90 CrossRef CAS PubMed.
- H. Fu, T. Xu, S. Zhu and Y. Zhu, Environ. Sci. Technol., 2008, 42, 8064–8069 CrossRef CAS.
- P. V. Kamat, J. Am. Chem. Soc., 1991, 113, 9705–9707 CrossRef CAS.
- T. Hasobe, S. Hattori, P. V. Kamat and S. Fukuzumi, Tetrahedron, 2006, 62, 1937–1946 CrossRef CAS PubMed.
- H. Yu, H. Zhang, H. Huang, Y. Liu, H. Li, H. Ming and Z. Kang, New J. Chem., 2012, 36, 1031–1035 RSC.
-
(a) H. Zhang, H. Ming, S. Lian, H. Huang, H. Li, L. Zhang, Y. Liu, Z. Kang and S. T. Lee, Dalton Trans., 2011, 10822–10825 RSC;
(b) H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S. T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed;
(c) X. Yu, J. Liu, Y. Yu, S. Zuo and B. Li, Carbon, 2014, 68, 718–724 CrossRef CAS PubMed;
(d) H. Wang, Z. Wei, H. Matsui and S. Zhou, J. Mater. Chem. A, 2014, 2, 15740–15745 RSC;
(e) C. Han, M. Q. Yang, B. Weng and Y. J. Xu, Phys. Chem. Chem. Phys., 2014, 16, 16891–16903 RSC.
-
(a) W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946–2008 RSC;
(b) G. Liu, H. G. Yang, J. Pan, Y. Q. Yang, G. Q. Lu and H. M. Cheng, Chem. Rev., 2014, 114, 9559–9612 CrossRef CAS PubMed;
(c) Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2008, 48, 60–103 CrossRef PubMed;
(d) W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, ChemSusChem., 2014, 7, 690–719 CrossRef CAS PubMed;
(e) W. Q. Fang, X. Q. Gong and H. G Yang, J. Phys. Chem. Lett., 2011, 2, 725–734 CrossRef CAS;
(f) E. Grabowska, M. Diak, M. Marchelek and A. Zaleska, Appl. Catal., B, 2014, 156–157, 213–235 CrossRef CAS PubMed.
- L. Zhang, L. Yin, C. Wang, N. Lun and Y. Qi, ACS Appl. Mater. Interfaces, 2010, 2, 1769–1773 CAS.
- S. He, S. Zhang, J. Lu, Y. Zhao, J. Ma, M. Wei, D. G. Evans and X. Duan, Chem. Commun., 2011, 10797–10799 RSC.
- H. Lu, S. Wang, L. Zhao, J. Li, B. Dong and Z. Xu, J. Mater. Chem., 2011, 21, 4228–4234 RSC.
-
(a) X. Wang, J. C. Yu, C. Ho, Y. Hou and X. Fu, Langmuir, 2005, 21, 2552–2559 CrossRef CAS PubMed;
(b) L. Zhang and J. C. Yu, Chem. Commun., 2003, 2078–2079 RSC.
- J. Chang and E. R. Waclawik, CrystEngComm, 2012, 14, 4041–4048 RSC.
- J. Yang, J. Wang, X. Li, J. Lang, F. Liu, L. Yang, H. Zhai, M. Gao and X. Zhao, J. Alloys Compd., 2012, 528, 28–33 CrossRef CAS PubMed.
- L. Jia, W. Cai, H. Wang and H. Zeng, Cryst. Growth Des., 2008, 8, 4367–4371 CAS.
- R. Boppella, K. Anjaneyulu, P. Basak and S. V. Manorama, J. Phys. Chem. C, 2013, 117, 4597–4605 CAS.
- L. Zhang, H. Yang, J. Ma, L. Li, X. Wang, L. Zhang, S. Tian and X. Wang, Appl. Phys. A, 2010, 100, 1061–1067 CrossRef CAS.
- D. Li, V. Balek, N. Ohashi, T. Mitsuhashi, S. Hishita and H. Haneda, J. Colloid Interface Sci., 2005, 289, 472–478 CrossRef CAS PubMed.
- Y. Yamaguchi, M. Yamazaki, S. Yoshihara and T. Shirakashi, J. Electroanal. Chem., 1998, 442, 1–3 CrossRef CAS.
- D. Li, H. Haneda, N. Ohashi, N. Saito and S. Hishita, Thin Solid Films, 2005, 486, 20–23 CrossRef CAS PubMed.
- S. Cho, J. W. Jang, J. S. Lee and K. H. Lee, Langmuir, 2010, 26, 14255–14262 CrossRef CAS.
- A. Wander and N. M. Harrison, Surf. Sci., 2000, 468, L851–L855 CrossRef CAS.
- J. H. Zeng, B. B. Jin and Y. F. Wang, Chem. Phys. Lett., 2009, 472, 90–95 CrossRef CAS PubMed.
- M. Farbod and E. Jafarpoor, Mater. Lett., 2012, 85, 47–49 CrossRef CAS PubMed.
- X. Wang, L. Yin, G. Liu, L. Wang, R. Saito, G. Q. Lu and H. M. Cheng, Energy Environ. Sci., 2011, 4, 3976–3979 CAS.
- U. Rau and M. Schmidt, Thin Solid Films, 2001, 387, 141–146 CrossRef CAS.
- Z. B. Yu, Y. P. Xie, G. Liu, G. Q. Lu, X. L. Ma and H. M. Cheng, J. Mater. Chem. A, 2013, 1, 2773–2776 CAS.
- M. Huang, Y. Yan, W. Feng, S. Weng, Z. Zheng, X. Fu and P. Liu, Cryst. Growth Des., 2014, 14, 2179–2186 CAS.
- G. R. Li, T. Hu, G. L. Pan, T. Y. Yan, X. P. Gao and H. Y. Zhu, J. Phys. Chem. C, 2008, 112, 11859–11864 CAS.
- G. Tang, S. Tian, Z. Zhou, Y. Wen, A. Pang, Y. Zhang, D. Zeng, H. Li, B. Shan and C. Xie, J. Phys. Chem. C, 2014, 118, 11833–11841 CAS.
- Y. Chen, H. Zhao, B. Liu and H. Yang, Appl. Catal., B, 2015, 163, 189–197 CrossRef CAS PubMed.
- X. G. Han, H. Z. He, Q. Kuang, X. Zhou, X. H. Zhang, T. Xu, Z. X. Xie and L. S. Zheng, J. Phys. Chem. C, 2009, 113, 584–589 CAS.
Footnote |
| † (i) the excitation source for all those references/literature dealing with UV light photocatalysis is not mentioned in this review article, for clarity; (ii) the term UV/visible light indicates the photocatalytic activity dealt with both UV and visible light illumination; (iii) UV-vis implies that the excitation source used has an emission wavelength in both the UV and visible region. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 



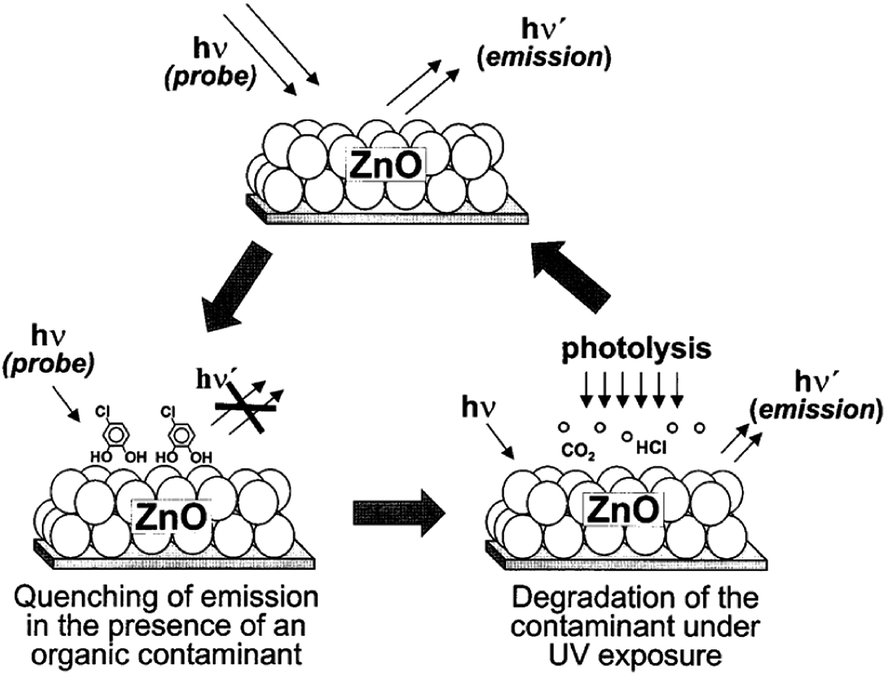
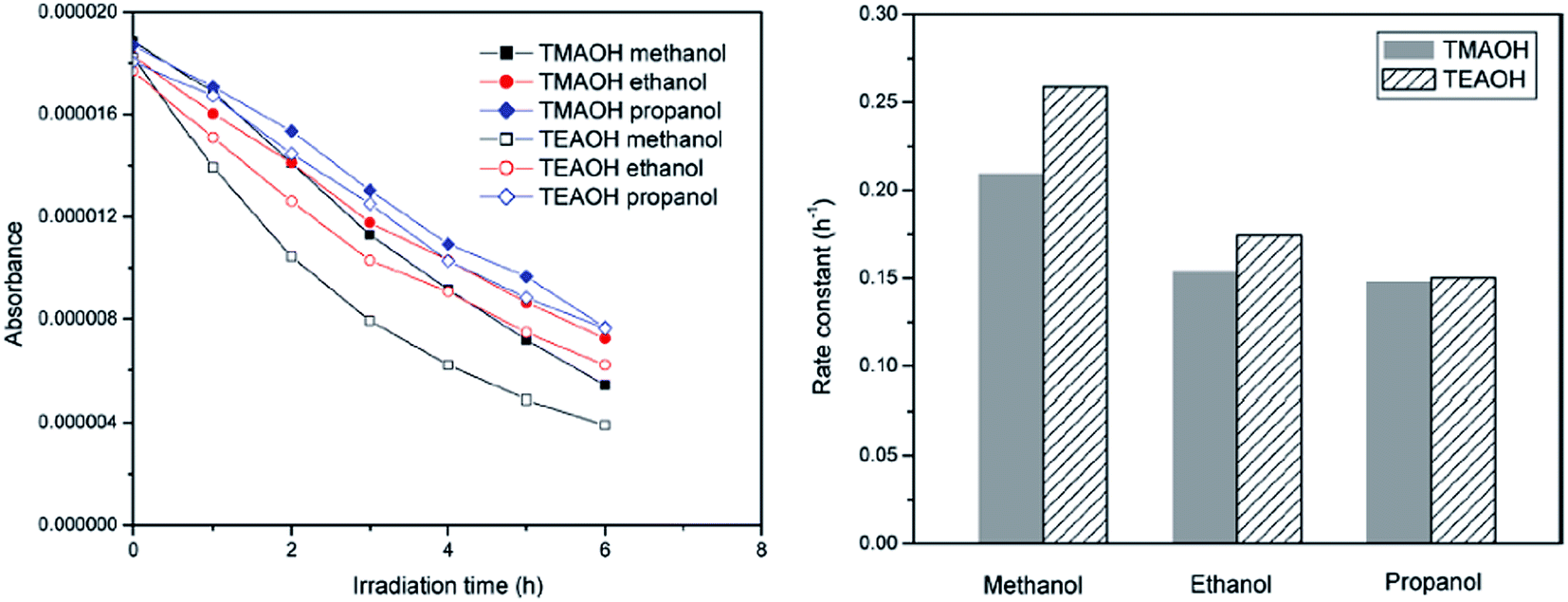
 and
and  ) and Vo (
) and Vo ( and
and  ) improves the charge transport and shifts the Fermi level towards the CB, which facilitates charge separation at the semiconductor–electrolyte interface and ultimately supplements the photocatalytic efficiency. The PANI behaves like a p-type semiconductor, which is an excellent hole transporting material and has become a better choice for preparing organic–inorganic hybrid photocatalysts. The oxygen deficient ZnO obtained from the calcination of ZnO2 was found to be efficient for the decomposition of DCP under visible light.88 The oxygen defects are introduced during the decomposition of O22− and were tailored by heat treatment at various temperatures. At high calcination temperatures (800 °C, 2 h), the pale yellow colour of oxygen-deficient ZnO was transformed to a white color. However, annealing under an argon atmosphere preserves the visible light absorption edge, as the oxygen vacancies are retained under an oxygen free condition. The impurity states associated with these high oxygen vacancies become more delocalized and overlap with the VB edge, and this raises the position of VB, making the ZnO more efficient for visible light absorption. The photocatalytic activity of oxygen-deficient ZnO gradually decreased with increases in the calcination temperature from 400 to 800 °C, which is in agreement with the concentration of oxygen vacancies. However, very low oxygen vacancies did not exert any impact on the optical properties. In contrast, oxygen deficiency was not observed for ZnO prepared by the calcination of Zn(OH)2.88 Thus, the electronic and optical properties of ZnO are strongly dependent on the nature and concentration of the defects generated during the crystallization process. For ZnO-based photocatalysis, excitation at high energy wavelength (254 nm) triggers both photolysis and photocatalysis;89 however, low energy (340 nm) induces only photocatalysis, due to its close association with the bandgap absorption. This indicates an increase in the surface or near surface reactions with the latter excitation source, and that the former operates by a MVK type mechanism, in which the oxygen from the crystal lattice is removed and used in the oxidation reaction. On the basis of a series of experiments, Ali et al.90 reported that a different degradation mechanism of MB operated for different ZnO films at different excitation wavelengths, with competition between the two distinctive mechanisms: conventional redox radicals and lattice oxygen driven oxidation. Surface photocatalyzed radical formation was prominent for highly aligned and more crystalline morphologies of ZnO with plentiful oxygen. However, the lattice oxygen mediated photodegradation was significant for less aligned and more amorphous morphologies with more defects. The high energy associated with 254 nm allows the activation energy barrier to overcome for lattice oxygen abstraction; furthermore, a high number of lattice defects lower the overall activation energy. However, the MVK type mechanism deactivates the catalyst surface, thereby inhibiting the redox reactions.90 Despite the debate concerning the exact role of defects in photocatalysis, it is generally accepted that the surface defects (states) act as shallow trapping sites, whereas the localized bulk defects promote the recombination process. The defects in mediated photocatalysis are vital for realizing the photocatalysis pathways, as these defects not only modulate the photoactivity, but are also inherent in pristine metal oxides.
) improves the charge transport and shifts the Fermi level towards the CB, which facilitates charge separation at the semiconductor–electrolyte interface and ultimately supplements the photocatalytic efficiency. The PANI behaves like a p-type semiconductor, which is an excellent hole transporting material and has become a better choice for preparing organic–inorganic hybrid photocatalysts. The oxygen deficient ZnO obtained from the calcination of ZnO2 was found to be efficient for the decomposition of DCP under visible light.88 The oxygen defects are introduced during the decomposition of O22− and were tailored by heat treatment at various temperatures. At high calcination temperatures (800 °C, 2 h), the pale yellow colour of oxygen-deficient ZnO was transformed to a white color. However, annealing under an argon atmosphere preserves the visible light absorption edge, as the oxygen vacancies are retained under an oxygen free condition. The impurity states associated with these high oxygen vacancies become more delocalized and overlap with the VB edge, and this raises the position of VB, making the ZnO more efficient for visible light absorption. The photocatalytic activity of oxygen-deficient ZnO gradually decreased with increases in the calcination temperature from 400 to 800 °C, which is in agreement with the concentration of oxygen vacancies. However, very low oxygen vacancies did not exert any impact on the optical properties. In contrast, oxygen deficiency was not observed for ZnO prepared by the calcination of Zn(OH)2.88 Thus, the electronic and optical properties of ZnO are strongly dependent on the nature and concentration of the defects generated during the crystallization process. For ZnO-based photocatalysis, excitation at high energy wavelength (254 nm) triggers both photolysis and photocatalysis;89 however, low energy (340 nm) induces only photocatalysis, due to its close association with the bandgap absorption. This indicates an increase in the surface or near surface reactions with the latter excitation source, and that the former operates by a MVK type mechanism, in which the oxygen from the crystal lattice is removed and used in the oxidation reaction. On the basis of a series of experiments, Ali et al.90 reported that a different degradation mechanism of MB operated for different ZnO films at different excitation wavelengths, with competition between the two distinctive mechanisms: conventional redox radicals and lattice oxygen driven oxidation. Surface photocatalyzed radical formation was prominent for highly aligned and more crystalline morphologies of ZnO with plentiful oxygen. However, the lattice oxygen mediated photodegradation was significant for less aligned and more amorphous morphologies with more defects. The high energy associated with 254 nm allows the activation energy barrier to overcome for lattice oxygen abstraction; furthermore, a high number of lattice defects lower the overall activation energy. However, the MVK type mechanism deactivates the catalyst surface, thereby inhibiting the redox reactions.90 Despite the debate concerning the exact role of defects in photocatalysis, it is generally accepted that the surface defects (states) act as shallow trapping sites, whereas the localized bulk defects promote the recombination process. The defects in mediated photocatalysis are vital for realizing the photocatalysis pathways, as these defects not only modulate the photoactivity, but are also inherent in pristine metal oxides.