
H2CO3 → CO2 + H2O decomposition in the presence of H2O, HCOOH, CH3COOH, H2SO4 and HO2 radical: instability of the gas-phase H2CO3 molecule in the troposphere and lower stratosphere†
Abstract
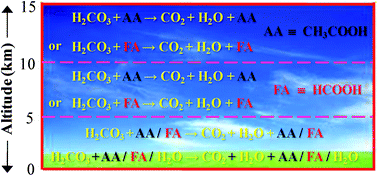
To understand the stability of the gas-phase carbonic acid (H2CO3) molecule, especially in the Earth's troposphere and lower stratosphere, here we report high level quantum chemistry calculations investigating the energetics for the H2CO3 → CO2 + H2O decomposition reaction via its shortest route in the presence of one to three water (H2O) molecules as well as in the presence of formic acid (FA), acetic acid (AA), sulfuric acid (SA) and hydroperoxide (HO2) radical. The calculations have been performed at the MP2/aug-cc-pVDZ, MP2/aug-cc-pVTZ, MP2/6-311++G(3df,3pd) and CCSD(T)/aug-cc-pVTZ levels of theory. The comparison of the reaction rates including tunneling corrections according to the unsymmetrical Eckart potential barriers suggests that at 0 km altitude in the clean environment of the Earth's atmosphere, the gaseous H2CO3 molecule becomes an unstable species in the presence of the H2O monomer, dimer, FA and AA. This follows as the FA- and AA-assisted H2CO3 → CO2 + H2O decomposition reactions are effectively near-barrierless processes and the reaction rates for the H2O monomer-, dimer-, FA- and AA-assisted H2CO3 decomposition reactions are comparable within a factor of ∼15. Similarly, at 0 km altitude in a polluted environment and also in the 5 to 15 km altitude range, only the FA- or AA-assisted H2CO3 decomposition is the dominant reaction pathway, especially, among all the pathways that have been considered here. It is seen from the CCSD(T)/aug-cc-pVTZ level prediction results that at altitudes of 5, 10, and 15 km in the Earth's atmosphere, the reaction rates for the FA-assisted H2CO3 decomposition depending upon the average concentrations of FA are respectively ∼102, 105 and 106 times higher than the reaction rates associated with the water monomer-assisted H2CO3 decomposition. Moreover, it is thought that the catalytic efficiencies of FA, AA and SA upon the H2CO3 → CO2 + H2O decomposition reaction are similar to each other, but nevertheless, SA, because of its low concentration, does not play a significant role in the H2CO3 → CO2 + H2O decomposition reaction, especially in the 0 to 15 km altitude range of the Earth's atmosphere.
 Please wait while we load your content...
Please wait while we load your content...

