
Synthesis of poly(stearyl methacrylate-b-3-phenylpropyl methacrylate) nanoparticles in n-octane and associated thermoreversible polymorphism
Yiwen Pei†
*,
Odilia R. Sugita,
Luckshen Thurairajah and
Andrew B. Lowe†*
School of Chemical Engineering, UNSW Australia, Kensington, Sydney, NSW 2052, Australia. E-mail: andrew.b.lowe@curtin.edu.au; yiwen.pei@curtin.edu.au
First published on 27th January 2015
Abstract
Poly(stearyl methacrylate) (PSMA) homopolymers with average degrees of polymerization (![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) n) ranging from 18–30 have been prepared by homogeneous RAFT radical polymerization in toluene and subsequently employed as macro-chain transfer agents (CTAs) in non-polar RAFT dispersion formulations with 3-phenylpropyl methacrylate (PPMA) as the comonomer in n-octane at 70 °C. With PSMA18 or PSMA19 macro-CTAs in n-octane at 20 wt%, a series of PSMAx–PPPMAy block copolymers are readily accessible in situ that form the full range of common nanoparticle morphologies, with the complexity of the nano-objects increasing (spheres-to-worms-to-vesicles) with increasing n of the PPPMA block as clearly evidenced by transmission electron microscopy (TEM). An evaluation of the effect of total solids for the preparation of block copolymers of common composition indicated that polymerizations conducted at higher concentrations favoured the formation of nanoparticles with more complex morphologies. In the case of block copolymers prepared with a PSMA30 macro-CTA the only accessible morphology was spheres regardless of compositional asymmetry. However, the size of the spheres increased monotonically with increasing PPPMA block length. Formulations that yielded (essentially) pure worm phases, such as PSMA18-b-PPPMA71, formed physical gels at ambient temperature. Heating the physical gels to (or beyond) a critical temperature resulted in a macroscopic transformation to a free flowing solution. The fundamental reason for the transformation, as evidenced by TEM, was a morphological transition from worm to sphere nanoparticles facilitated, in part, by a change in solvation of the PPPMA core-forming block with increasing temperature. DLS analysis indicated that the morphology transitions were fully reversible.
n) ranging from 18–30 have been prepared by homogeneous RAFT radical polymerization in toluene and subsequently employed as macro-chain transfer agents (CTAs) in non-polar RAFT dispersion formulations with 3-phenylpropyl methacrylate (PPMA) as the comonomer in n-octane at 70 °C. With PSMA18 or PSMA19 macro-CTAs in n-octane at 20 wt%, a series of PSMAx–PPPMAy block copolymers are readily accessible in situ that form the full range of common nanoparticle morphologies, with the complexity of the nano-objects increasing (spheres-to-worms-to-vesicles) with increasing n of the PPPMA block as clearly evidenced by transmission electron microscopy (TEM). An evaluation of the effect of total solids for the preparation of block copolymers of common composition indicated that polymerizations conducted at higher concentrations favoured the formation of nanoparticles with more complex morphologies. In the case of block copolymers prepared with a PSMA30 macro-CTA the only accessible morphology was spheres regardless of compositional asymmetry. However, the size of the spheres increased monotonically with increasing PPPMA block length. Formulations that yielded (essentially) pure worm phases, such as PSMA18-b-PPPMA71, formed physical gels at ambient temperature. Heating the physical gels to (or beyond) a critical temperature resulted in a macroscopic transformation to a free flowing solution. The fundamental reason for the transformation, as evidenced by TEM, was a morphological transition from worm to sphere nanoparticles facilitated, in part, by a change in solvation of the PPPMA core-forming block with increasing temperature. DLS analysis indicated that the morphology transitions were fully reversible.
Introduction
When AB diblock copolymers are placed in a selective solvent (one that is a good solvent for one block but poor for the other) the copolymer will undergo self-directed assembly (provided the concentration of the copolymer is above its critical aggregation concentration) to give polymeric nanoparticles whose morphology is governed by a number of factors. By far the most common reported morphology is spheres although this is primarily a consequence of the compositional nature of the synthesized block copolymers as opposed to other possible contributing structural or chemical factors. Historically the preparation of such nanoparticles has been accomplished by initially synthesizing well-defined parent copolymers under homogeneous conditions, which, after characterization, are subjected to a processing step(s) to give the target nano-objects. While perfectly valid, arguably the biggest drawback of this well established approach is the generally low concentrations at which such nanoparticles are formed (≤1 wt% is common).Reversible addition–fragmentation chain transfer radical dispersion polymerization (herein abbreviated to RAFTDP) has recently attracted significant academic interest as a convenient approach for the in situ preparation of polymeric nanoparticles of varying morphologies via polymerization-induced self-assembly under generally facile conditions. Indeed, given its ease of execution and versatility, soft matter nanoparticles of increasingly more complex design and final structure/morphology are beginning to be reported in the literature, often with associated interesting properties.1–4
One key advantage of RAFTDP is that appropriate formulations yield nano-objects exhibiting impressive polymorphism at concentrations much higher than those obtainable by more traditional post-polymerization processing routes with formulations at ≥50 wt% solids having been reported.5–7 Currently, the majority of RAFTDP formulations that yield nanoparticles of differing morphologies, have focused on block copolymerizations conducted in polar media such as water and alcohols (including water/alcohol solvent mixtures) as exemplified by the work of Armes et al.,5,8–14 Pan and coworkers,15–17 our group18–20 and others.21–26 Surprisingly, however, there are comparatively few examples of such RAFTDP formulations in non-polar media,27–32 and these studies have been fairly limited in their scope at least with respect to monomer/comonomer pairings and solvent choice. For example, the synthesis and characterization of spherical nanoparticles, in all-acrylic formulations, have been reported by Charleux et al.27,31,32 in isododecane, while Fielding and co-workers,28 have described methacrylic formulations of poly(lauryl methacrylate-b-benzyl methacrylate) copolymers prepared in n-heptane at 90 °C. In this case, tuning the poly(lauryl methacrylate) average degree of polymerization (n) facilitated the preparation of spherical nanoparticles of varying size as well as allowing access to higher ordered nano-objects including worms and vesicles. We very recently reported similar behaviour in AB diblock copolymer nanoparticles formed from poly(stearyl methacrylate) (PSMA) with poly(3-phenylpropyl methacrylate) (PPPMA) prepared in n-tetradecane.30 The full common range of nanoparticles were prepared and thermoreversible gelation in worm nanoparticle formulations was also demonstrated. Inline with our previous observations,19 such degelation was accompanied by a fundamental worm-to-sphere (W–S) morphology transition and was driven, in part, by changes in the relative solvation of the core and coronal blocks with increasing temperature.
Our main interest in 3-phenylpropyl methacrylate (PPMA) as a comonomer in RAFTDP formulations is due to similar recent observations relating to thermoreversible polymorphism exhibited by examples of poly[2-(dimethylamino)ethyl methacrylate-b-PPMA] (PDMAEMAx-b-PPPMAy) copolymers prepared by RAFTDP in EtOH, and most recently for the above mentioned PSMA–PPPMA block copolymers prepared in n-tetradecane.19,30 For example, a PDMAEMA20-b-PPPMA47 copolymer forms a physical gel at room temperature but becomes a free flowing solution upon heating to a critical temperature (ca. 60–70 °C in this case). Macroscopic physical degelation was due to a W–S transition as determined via TEM analysis and confirmed by dynamic light scattering (DLS). This morphological transition was both rapid and completely reversible and has important implications in the analysis of nanoparticles prepared by RAFTDP since it suggests that the nanoparticle morphology formed at elevated temperature, at least in certain formulations, may not be the same as the nano-object morphology at ambient temperature. Such W–S transitions are partly facilitated by a basic change in the solvation of the core forming PPPMA block that increases chain mobility, a process that would have changed the relative balance of the core and coronal volumes and hence the packing parameter p.33 Additionally, we noted that the PPPMA block possesses a relatively low glass transition temperature (Tg) of 2 °C 19 (compare this with structurally similar species such as poly(benzyl methacrylate) – Tg: 54 °C and poly(2-phenyl ethyl)methacrylate – Tg: 42 °C).34–36 While, technically, a solid-state property, this sub-ambient value was hypothesized to be at least partly responsible for the observed fast morphological transition since it requires the core-forming blocks to be able to extricate themselves from the solvophobic environment to facilitate rearrangement. Such thermoreversible morphology transitions are rare phenomena and relatively few examples exist in the open literature. However, in the realm of RAFTDP and polymerization-induced self-assembly, other examples of temperature-induced degelation–gelation have been reported in alternative systems in which worm nanoparticles form physical gels. For example, Armes et al. has noted thermoreversible degelation in several systems including examples of poly(glycerol monomethacrylate-b-2-hydroxypropyl methacrylate), poly[(galactose methacrylate-co-glycerol monomethacrylate)-b-2-hydroxypropyl methacrylate]11 and in poly(lauryl methacrylate-b-benzyl methacrylate)29 copolymers. However, the majority of these examples exhibit opposite thermal behaviour with degelation occurring upon cooling. However, the fundamental macroscopic changes are due to identical W–S nanoparticle morphology transitions. Likewise we have reported that vesicle-to-sphere transitions can also occur in some examples of PDMAEMA–PPPMA block copolymers although we have not, as yet, examined this is detail.19
Given our recent disclosure describing the RAFTDP synthesis and thermoresponsive properties of PSMA–PPPMA block copolymer nanoparticles prepared in n-tetradecane30 we decided to evaluate the effect of n-alkane solvent on the general RAFTDP synthesis characteristics of such nanoparticles under polymerization-induced self-assembly conditions. This is particularly important since it has been reported previously that solvents from a homologous series can have a clear and direct impact on the basic properties of an otherwise identical RAFTDP formulation. As such, in order to obtain a genuine full understanding of the behaviour of a given macro-CTA/comonomer pairing it is important to evaluate the effect of polymerization media when formulations can be evaluated in an homologous series of solvents. Given this, herein we report a fundamental study examining the RAFTDP of PPMA in n-octane with PSMA macro-CTAs. We examine the effect of the average degree of polymerization (n) of the PPPMA block for a low, fixed n of PSMA macro-CTA; the effect of concentration, i.e. total solids content for a fixed copolymer composition; the effect of n of the PSMA block on nanoparticle morphology, and the temperature effects on nanoparticle morphology with an emphasis on macroscopic thermally induced reversible degelation.
Experimental
All reagents were purchased from the Aldrich Chemical Company at the highest available purity and used as received unless noted otherwise. 2,2′-Azobis(isobutyronitrile) (AIBN) was purified by recrystallization twice from methanol and then stored in a freezer until needed. Stearyl methacrylate (SMA) and 3-phenylpropyl methacrylate (PPMA) were purified by passage through a column of basic alumina (3 times) and then stored in the freezer prior to use. 2-Cyanopropan-2-yl benzodithioate (CPDB) was stored in a freezer until needed.RAFT homopolymerization of stearyl methacrylate
Below is a typical procedure for the RAFT homopolymerization of stearyl methacrylate (SMA).To a reaction vial equipped with a magnetic stir bar was added AIBN (3.87 × 10−2 g, 2.36 × 10−4 mol) and SMA (9.2 mL, 2.36 × 10−2 mol). In a separate vessel, CPDB (2.61 × 10−1 g, 1.18 × 10−3 mol) was dissolved in 7.6 mL of toluene. This solution was added to the AIBN and then the vial was placed in a sonicator for ca. 30 min to ensure complete dissolution of the AIBN. The vial was then sealed with a rubber septum and the solution purged with nitrogen gas while immersed in an ice bath. The vial was then placed in an oil bath preheated to 70 °C. Polymerization was allowed to proceed for 16 h after which it was halted by exposure to air while cooling in an ice water bath. The polySMA (PSMA) homopolymer was isolated by precipitation in an excess of MeOH. After filtration the homopolymer was redissolved in CHCl3 and re-precipitated in MeOH distributed in four centrifuge tubes. After removal of the supernatant, the samples were dried in vacuo overnight prior to NMR spectroscopic and SEC analyses. The final monomer conversion was determined by 1H NMR analysis by comparing the integrals of the PPPMA peaks (C6H5–) at 7.10–7.50 ppm to those of the PPMA monomer vinyl peaks (CH2) at 5.5 and 6.1 ppm.
RAFT dispersion polymerization of PPMA with PSMAx in n-octane
Below is a typical procedure for the RAFT dispersion polymerization (RAFTDP) of PPMA with a PSMA homopolymer macro-CTA at 20 wt% solids. The same general procedure was employed for all other RAFTDP syntheses.PPMA (3.04 × 10−1 g, 1.49 × 10−3 mol) and AIBN (1.48 × 10−3 g, 9.02 × 10−6 mol) were added to a glass vial of 20 mL capacity equipped with a magnetic stir bar. The vial was then placed in a sonicator bath for ca. 30 min to ensure complete dissolution of the AIBN in PPMA. To a second vial was added PSMA macro-CTA with an average degree of polymerization (n) of 19 (PSMA19) (0.30 g, 4.51 × 10−5 mol) and 3.445 mL of n-octane. This solution was combined with the PPMA/AIBN solution, the vial capped with a rubber septum, placed in an ice water bath and the solution purged with nitrogen. Subsequently, the vial was placed in a preheated oil bath set to 70 °C and polymerization allowed to proceed for 24–48 h. The polymerization was halted by exposure to air while being cooled in an ice water bath. Block copolymers were isolated by precipitation into a large excess of MeOH, followed by filtration and subsequent drying in vacuo at 40 °C prior to NMR spectroscopic and SEC analyses.
Nuclear magnetic resonance (NMR) spectroscopy
NMR spectra were recorded on a Bruker 300 MHz spectrometer. 1H NMR spectra were recorded in deuterated chloroform (CDCl3) and sixty-four scans were averaged per spectrum. Residual CHCl3 (δ = 7.26 ppm) was used as the internal reference signal. Temperature-dependent NMR characterization analyses were performed using a Bruker Avance III 500 spectrometer (1H, 500.13 MHz) equipped with a 31P-TBI probe utilizing 7500 Hz sweep width, 3.2 s acquisition time, and 2 s recycle delay. The pulse program employing solvent suppression at a 1H chemical shift was used to eliminate the solvent peak. Samples were heated from 25 °C to 90 °C and allowed to equilibrate at each temperature for at least 10 min prior to measurements. Variable temperature NMR studies were performed in fully deuterated n-octane (d18-n-octane).Size exclusion chromatography (SEC)
SEC analysis was conducted on a Shimadzu modular system consisting of four phenogel columns (106, 104, 103 and 102 Å pore size) and a differential refractive index detector. Tetrahydrofuran (THF) was used as the mobile phase at a temperature of 40 °C and a flow rate of 1.0 mL min−1. The system was calibrated with a series of narrow molecular weight distribution polystyrene standards with molecular weights ranging from 0.58–1820 kg mol−1. The chromatograms were analyzed with Cirrus SEC software version 3.0. (Co)Polymer samples were prepared at a concentration of 3–5 mg mL−1 and filtered through a 0.45 μm nylon filter prior to analysis.Dynamic light scattering (DLS)
Nanoparticle size distributions were determined on a Malvern Zetasizer Nano Series instrument (laser power = 4 mW, wavelength = 633 nm, detection angle = 173°). Samples for analysis were prepared by taking 50 μL of the block copolymer dispersion, diluted with 1.45 mL of n-octane and filtered twice through 0.45 μm nylon filters. For temperature-dependent DLS analysis, samples were heated up and allowed to equilibrate at each temperature for at least 10 min prior to measurements. To study the reversibility of the thermally induced W–S morphological transition, variable-temperature DLS measurements were performed utilizing the 30 wt% PSMA18-b-PPPMA71 dispersion in n-octane. DLS samples (1 wt%) were freshly prepared by diluting parent copolymer dispersion in cold or hot n-octane (at the same temperature as copolymer samples).Transmission electron microscopy (TEM)
TEM analyses were conducted on a JEOL 1400 transmission electron microscope operating at 100 kV. 10 μL of block copolymer solution was added slowly to 1.49 mL of n-tetradecane under stirring. The copolymer solution (0.07–0.27 w/w%) was dropped onto the top of a carbon-coated copper grid (ProSciTech) and allowed to dry for 1 min. Excess solution was wicked off using filter paper. Samples were then stained by exposure to RuO4 vapour for 15 min. at room temperature prior to analysis. As a heavy metal compound, RuO4 can interact with the core-forming PPPMA block to improve contrast. The RuO4 was prepared as follows: ruthenium(IV) oxide hydrate (0.06 g) was added to a solution of sodium periodate (0.4 g) in water (10 mL) with stirring at 0 °C for 4 h. A yellow solution of RuO4 was obtained at the end of reaction and was stored in the freezer prior to use. For TEM grid preparation at high temperature, all materials and equipment were placed in a hot oven at 90 °C for 10 min. The 30 wt% PSMA18-b-PPPMA71 copolymer sample was kept under continuous stirring at 90 °C for 10 min and then diluted to 0.2 wt% with warm n-tetradecane (at same temperature of copolymer sample) prior to TEM sample preparation. The TEM grids were then stained as described above.Results and discussion
The overall approach for the synthesis of the target AB diblock copolymer nanoparticles is shown in Scheme 1.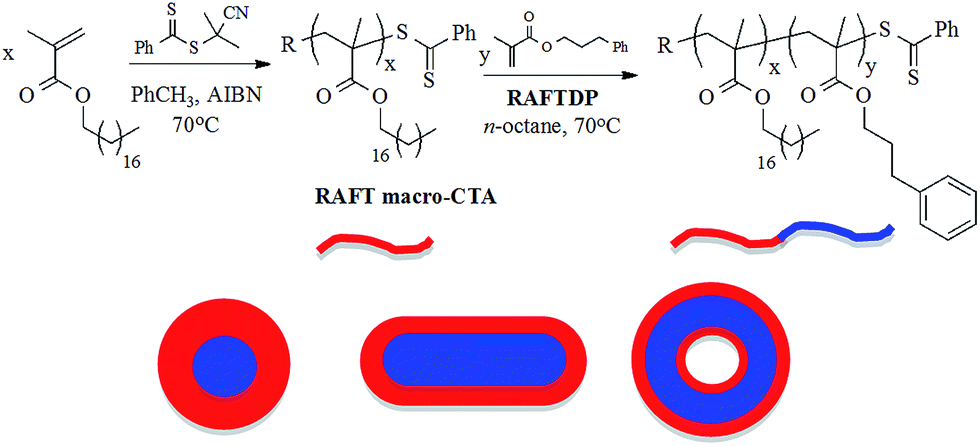 | ||
| Scheme 1 Outline for the general approach to soft matter nanoparticles based on poly(stearyl methacrylate-b-3-phenylpropyl methacrylate) copolymers prepared in n-octane at 70 °C for 24 h. | ||
In the first step, three poly(stearyl methacrylate) (PSMA) homopolymers with average degrees of polymerization, n, ranging from 18–30 were prepared by homogeneous RAFT radical polymerization in toluene at 70 °C. All polymerizations were taken to near quantitative conversion and yielded well-defined PSMA macro-CTAs with unimodal, near symmetric molecular weight distributions with measured dispersities (ĐM = ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w/n) ≤1.17. Table 1 gives a summary of the PSMA homopolymers prepared. The two PSMA homopolymers with the lowest n's were prepared, specifically, to allow for direct comparisons with PSMA–PPPMA AB diblock copolymers prepared by RAFTDP in n-tetradecane that we reported recently.30
w/n) ≤1.17. Table 1 gives a summary of the PSMA homopolymers prepared. The two PSMA homopolymers with the lowest n's were prepared, specifically, to allow for direct comparisons with PSMA–PPPMA AB diblock copolymers prepared by RAFTDP in n-tetradecane that we reported recently.30
n) and dispersities
| Macro-CTA | PPMA/CTA | Conv.a (%) | MWNMRb | n SECc |
ĐM |
|---|---|---|---|---|---|
| a As determined by 1H NMR spectroscopy.b As determined by 1H NMR spectroscopy and end-group analysis.c As determined in THF on a system calibrated with a series of narrow molecular weight distribution polystyrene standards. | |||||
| PSMA18 | 20 | 99 | 6500 | 6400 | 1.17 |
| PSMA19 | 20 | 98 | 6700 | 8000 | 1.11 |
| PSMA30 | 30 | 99 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
14300 |
1.13 |
RAFTDP of PPMA in n-octane at 20 wt% for a fixed PSMA ![[X with combining macron]](https://www.rsc.org/images/entities/h3_i_char_0058_0304.gif) n
n
With PSMA macro-CTAs available we first examined the RAFTDP of PPMA with the PSMA18 and PSMA19 macro-CTAs at 20 wt% total solids in n-octane for variable targeted n's of the PPPMA block. A summary of the block copolymers prepared is given in Table 2. Under the specified conditions, conversions of PPMA ranged from 73 to 91%. These conversions are slightly lower than typically observed in all-methacrylic formulations (Table 2 entries 1–9), including our complementary study detailing the synthesis of similar AB diblock copolymers in n-tetradecane, and may be the first indication of a solvent effect for otherwise essentially identical formulations.30 Extending the polymerization time to 48 h (entry 10) had minimal effect although did result in the highest observed conversion. Regardless of final PPMA conversions, the polymerizations yielded a series of asymmetric block copolymers in a controlled fashion with NMR and SEC-measured molecular weights increasing with increasing n of the PPPMA block as expected. Additionally, all block copolymers had narrow molecular weight distributions with SEC-measured ĐM values ≤1.23 and more typically ca. 1.14. These values are perfectly consistent with similar block copolymers prepared in n-tetradecane where measured ĐM's spanned a similar range (1.14–1.23).30 As a representative example, Fig. 1 shows the experimentally measured SEC traces for the PSMA19-b-PPPMAy series of block copolymers and digital pictures of examples of the resulting RAFTDP solutions. The PSMA18-b-PPPMAy series yielded essentially identical results.
| Entry | PPMA/macro-CTA/AIBN | Copolymer compositionc | PPMA conv.c | NMR MW | SEC nd |
SEC ĐM | DLS Dh (nm) | DLS PDI (μ2/Γ2) | TEM morp.e |
|---|---|---|---|---|---|---|---|---|---|
| a All polymerizations were performed in n-octane at 70 °C for 24 h.b All polymerizations were performed in n-octane at 70 °C for 48 h.c As determined by 1H NMR spectroscopy.d As measured in THF on a system calibrated with a series of narrow molecular weight distribution polystyrene standards.e S = spheres, W = worms, V = vesicle. | |||||||||
| 1a | 33/1/0.2 | PSMA19-b-PPPMA31 | 85% | 13000 |
9600 | 1.14 | 10.9 | 0.09 | S |
| 2a | 72/1/0.2 | PSMA19-b-PPPMA57 | 77% | 18200 |
12500 |
1.14 | 15.5 | 0.01 | S |
| 3a | 105/1/0.2 | PSMA19-b-PPPMA75 | 86% | 22000 |
14700 |
1.14 | 38.3 | 0.09 | S + W |
| 4a | 112/1/0.2 | PSMA19-b-PPPMA88 | 79% | 24600 |
17700 |
1.14 | 74.7 | 0.14 | S + W |
| 5a | 130/1/0.2 | PSMA19-b-PPPMA101 | 73% | 27200 |
20400 |
1.14 | 188.1 | 0.22 | W + V |
| 6a | 154/1/0.2 | PSMA19-b-PPPMA131 | 87% | 30800 |
24600 |
1.14 | 212.6 | 0.23 | V |
| 7a | 38/1/0.2 | PSMA18-b-PPPMA36 | 90% | 13700 |
9400 | 1.19 | 21.3 | 0.06 | S |
| 8a | 87/1/0.2 | PSMA18-b-PPPMA72 | 85% | 21000 |
13300 |
1.22 | 111.9 | 0.19 | S + W |
| 9a | 102/1/0.2 | PSMA18-b-PPPMA76 | 89% | 22000 |
13300 |
1.23 | 256.6 | 0.32 | S + W |
| 10b | 140/1/0.2 | PSMA18-b-PPPMA130 | 91% | 32900 |
19900 |
1.14 | 823.9 | 0.36 | V |
 | ||
| Fig. 1 Representative, normalized, SEC traces for the PSMA19-b-PPPMAy series of AB diblock copolymers prepared in n-octane and digital pictures of the RAFTDP solutions at 20 °C for an n of PPPMA of (1) 31, (2) 57, (3) 75, (4) 88, (5) 101 and (6) 131. | ||
The transition from optically clear, albeit coloured, solutions to those with a distinct milky appearance is qualitatively indicative of a step-wise change in nanoparticle morphology with increasing n of the PPPMA block. To evaluate the nanoparticle morphology, samples were extracted and imaged by TEM.
Initial attempts at imaging the nanoparticles by dilution of the n-octane solutions with additional n-octane and deposition on carbon-coated copper grids proved generally unsuccessful for reasons that are, at present, unclear. As noted above, we have recently reported the RAFTDP of PPMA with PSMA macro-CTAs in n-tetradecane.30 Dilution of such formulations with additional n-tetradecane followed by TEM analysis was successful so we opted to image the nanoparticles detailed herein after dilution with this higher alkane.30 However, to check that this approach did not adversely impact the formed nanoparticle morphology we conducted a control experiment in which we imaged particles formed by PSMA19-b-PPPMA131 as noted above by dilution with n-tetradecane and the same nanoparticle solution diluted with n-octane but deposited on purpose-purchased hydrophobic octane-modified TEM grids (as opposed to the cheaper more commonly employed carbon-coated copper grids) Fig. 2.
 | ||
| Fig. 2 Representative TEM images of the PSMA19-b-PPPMA131 AB diblock copolymer nanoparticles after (1) dilution with n-tetradecane and deposition on carbon-coated copper grids and (2) after dilution with n-octane and deposition on hydrophobic octane modified grids. Both samples were stained by exposure to RuO4 vapor. | ||
The images in Fig. 2 both show the presence of vesicular species whose sizes are comparable regardless of the method of preparation and specifically the solvent utilized for sample dilution. Since this suggests that dilution with n-tetradecane prior to imaging has a minimal effect on the nanoparticle morphology formed in n-octane all further TEM studies were performed with n-tetradecane diluted solutions and imaging accomplished using copper-based TEM grids. We acknowledge that this is not an ideal solution but since we have employed this approach uniformly we can qualitatively draw useful conclusions and comparisons.
Fig. 3 shows representative TEM images obtained for examples of the PSMA19-b-PPPMAy block copolymer nanoparticles with increasing PPPMA block lengths (y).
 | ||
| Fig. 3 Representative TEM images of the nanoparticles formed by PSMA19-b-PPPMAy copolymers prepared in n-octane and diluted with n-tetradecane at 20 °C prior to imaging. Particles were stained by exposure to RuO4 vapor. | ||
For the block copolymers with the two lowest PPPMA n's (31 and 57) pure spherical nanoparticle morphologies were observed (Fig. 3A shows the TEM image of the nano-objects for the PSMA19-b-PPPMA57 copolymer). The estimated TEM size is 15–20 nm for this particular sample and agrees well with the hydrodynamic diameter (Dh) measured by DLS of 15.5 nm (with an associated DLS polydispersity of 0.01). Increasing the n of the PPPMA block to 75, and then 88, resulted in mixed morphologies with nanoparticles having spherical and worm-like structures, Fig. 3B. The diameter of the worms is approximately the same as the Dh of the spherical species and is consistent with worm formation via the 1D coalescence of the spherical particles. The Dh values for these mixed phases are given in Table 2 but should only be treated as ‘sphere-equivalent’ values given data treatment employing the Stokes–Einstein equation. A further increase in the PPPMA n to 101 results in another mixed phase, Fig. 3C, but in this instance consisting of a mixture of worms and species that could be associated with the early formation of vesicles although the former appears to be the major structural form. Finally, for the PSMA19-b-PPPMA131 sample a pure vesicle phase was observed, Fig. 3D, whose TEM sizes (approaching an average of ca. 150 nm) are consistent with the DLS measured size of 212.6 nm. Similar observations were made for the PSMA18-b-PPPMAy series of copolymers with the nanoparticle morphology progressing to more complex structures with increasing compositional asymmetry. These results clearly demonstrate that not only is the RAFTDP of PPMA in n-octane a viable approach to soft matter nanoparticles exhibiting the full common range of morphologies, their syntheses are complementary to the ethanolic and n-tetradecane formulations with PPMA as a comonomer reported previously.19,30 However, we do note that the critical compositions associated with morphology transitions appear to be shifted to higher values of the PPPMA block compared to essentially identical block copolymers prepared directly in n-tetradecane. For example, PSMA19-b-PPPMA87 and PSMA19-b-PPPMA98 prepared in n-tetradecane at 20 wt% form nanoparticles with worm and vesicular species for the first example and a pure vesicle phase in the case of the latter. In contrast, as noted above, the PSMA19-b-PPPMA88 copolymer forms a mixture of spherical and worm nanoobjects while the PSMA19-b-PPPMA101 sample consisted of worms and some vesicles. Since these samples were prepared with identical PSMA macro-CTAs under identical conditions (except reaction media) these differences can only be attributed to the use of different n-alkane solvents. This is also consistent with reports from Armes et al. who synthesized poly(lauryl methacrylate-b-benzyl methacrylate) copolymer nanoparticles via RAFTDP in n-heptane and n-dodecane.28,29
One key feature of RAFTDP formulations that result in differing nanoparticle morphologies is the large number of experimental variables that can have a direct impact on the resulting nano-object size and shape. While the solvophobic block length is generally acknowledged to be the primary structural feature determining nanoparticle morphology other factors such as concentration (total solids) and the solvophilic block length (for a fixed n of solvophobic block) can also play a key role.
Effect of total solids on nanoparticle morphology for a fixed target PSMA18-b-PPPMAy composition
Having shown that then of the PPPMA block for a low, fixed n of a PSMA macro-CTA, is a convenient approach for preparing a range of nanoparticles of increasingly complex morphology we next examined the effect of total solids on nanoparticle morphology for a fixed block copolymer composition. This approach is synthetically more challenging to conduct successfully since it requires the repeated preparation of compositionally identical block copolymer species under different experimental conditions. Four block copolymers were prepared at between 10 and 40 wt% total solids employing the PSMA18 macro-CTA for a typical target n of the PPPMA block of 70. Table 3 gives a summary of the block copolymers prepared while Fig. 4 shows the SEC traces of the PSMA18 macro-CTA as well as the four AB diblock copolymers prepared.
| Entry | Compositiona | Solids content | PPMA conv.a | NMR MWb | SEC nc |
ĐM | DLS Dh (nm) | DLS PDI (μ2/Γ2) | TEM morp.d |
|---|---|---|---|---|---|---|---|---|---|
| a As determined by 1H NMR spectroscopy.b As determined by 1H NMR spectroscopy and end-group analysis.c Measured on a system calibrated with a series of narrow molecular weight distribution polystyrene standards.d S = spheres, W = worms, V = vesicles. | |||||||||
| 1 | PSMA18-b-PPPMA73 | 10 wt% | 65% | 21200 |
20000 |
1.23 | 39.8 | 0.28 | S + W |
| 2 | PSMA18-b-PPPMA71 | 20 wt% | 91% | 20800 |
19100 |
1.27 | 65.0 | 0.14 | S + W |
| 3 | PSMA18-b-PPPMA71 | 30 wt% | 89% | 20800 |
19600 |
1.19 | 145.6 | 0.19 | W |
| 4 | PSMA18-b-PPPMA68 | 40 wt% | 95% | 20200 |
19800 |
1.18 | 84.9 | 0.12 | W |
 | ||
| Fig. 4 SEC traces of the PSMA18 macro-CTA and the four AB diblock copolymers with PPMA prepared at 10–40 wt% total solids in n-octane. | ||
Gratifyingly, we were able to prepare AB diblock copolymers with near identical average compositions at different total solids contents. 1H NMR spectroscopy, Table 3, as well as SEC confirmed this, Fig. 4, where we can clearly see that the chromatograms of each block copolymer are almost perfectly superimposed and NMR-measured compositions are similar. There is some evidence of low molecular weight impurity, presumably PSMA18 macro-CTA, in the 20 wt% formulation but otherwise the results are consistent with the targeted block copolymers.
A clear effect of the block copolymer concentration on final nanoparticle morphology was observed as judged by TEM analysis, Fig. 5. Under the most dilute conditions, at 10 wt%, a mixed morphology was observed that consisted of predominantly spheres along with short, oligomeric worms. At 20 wt% we see an increase in the concentration of the worm species although they are still oligomeric in nature and there are clearly still spherical nano-objects present. The Dh for the 20 wt% sample was significantly higher than for the comparable species prepared with the PSMA19 macro-CTA, Table 2 entry 3. However, it must be remembered that these values are for mixed phases consisting of spheres and worms and that a difference in the relative nanoparticle composition could account for the difference in the average Dh values. At 30 and 40 wt% we observe phases that consist predominantly of worms although the TEM images appear to show some evidence of coalescence, early stage vesicle formation, or possibly signs of film formation. However, the effect of concentration is evident and consistent with previous studies by us and others,18,19 and highlights the important role concentration can play in targeting and accessing specific nanoparticle morphologies.
 | ||
| Fig. 5 Representative TEM images of the nanoparticles formed by PSMA–PPPMA of average composition of ca. 18:70 as a function of increasing total solids. | ||
Effect of n of the PSMA macro-CTA on nanoparticle morphology
Having demonstrated that PSMA macro-CTAs with n's of 18 or 19 can be employed in the RAFTDP of PPMA in n-octane to give nanoparticles with the full range of commonly observed morphologies we next evaluated the effect of a PSMA macro-CTA with an n of 30 on the final nanoparticle morphology for 20 wt% solid formulations. While the effect of the solvophilic block n has not historically been considered to be as important a structural feature as the solvophobic block length it has been reported that there is commonly a critical n of the solvophilic, stabilizing block beyond which only spherical species can be accessed, even in block copolymers of extreme compositional asymmetry. Presumably, at some key n of the corona-forming block effective steric stabilization is attained precluding morphological transitions. This critical value is system specific and may also be dictated by both the volume fraction and n of the solvophilic block. For example, in n-heptane a poly(lauryl methacrylate) macro-CTA with an n of 37 when used to polymerize benzyl methacrylate at 90 °C yields a series of spherical nanoparticles of increasing hydrodynamic diameter even in block copolymers with benzyl methacrylate n's of 100–900.28 To check for possible similar behaviour in these particular non-polar RAFTDP formulations we prepared a series of four AB diblock copolymers in n-octane with a PSMA30 macro-CTA at 20 wt% with final n's of the core PPPMA block ranging from 72–289, Table 4. Polymerizations generally proceeded smoothly although PPMA conversions were again lower than observed in n-tetradecane and the ĐM values were slightly higher than other formulations especially for the most asymmetric block copolymer. Fig. 6 shows representative TEM images of the nanoparticles obtained.
| Entry | Compositiona | PPMA conv.a | NMR MWb | SEC nc |
ĐM | DLS Dh (nm) | DLS PDI (μ2/Γ2) | TEM morp.d |
|---|---|---|---|---|---|---|---|---|
| a As determined by 1H NMR spectroscopy.b As determined by 1H NMR spectroscopy and end-group analysis.c Measured on a system calibrated with a series of narrow molecular weight distribution polystyrene standards.d S = spheres. | ||||||||
| 1 | PSMA30-b-PPPMA72 | 85% | 25100 |
28100 |
1.25 | 41.5 | 0.09 | S |
| 2 | PSMA30-b-PPPMA139 | 73% | 38800 |
31100 |
1.33 | 65.3 | 0.14 | S |
| 3 | PSMA30-b-PPPMA226 | 71% | 63700 |
39400 |
1.14 | 68.0 | 0.02 | S |
| 4 | PSMA30-b-PPPMA289 | 93% | 69400 |
41800 |
1.43 | 70.4 | 0.02 | S |
 | ||
| Fig. 6 Representative TEM images of PSMA30-b-PPPMAy spherical nanoparticles formed at 20 wt% in n-octane at 70 °C with varying DP of the PPPMA block. | ||
In all instances only spherical nanoparticles were observed by TEM whose average size increased with increasing n of the PPPMA block. Interestingly, even for the shortest PPPMA block length (with an n of 72, i.e. PSMA30-b-PPPMA72) when compared to PSMA19-b-PPPMA75 (Table 1) under otherwise identical conditions we observed nanoparticles with higher ordered morphologies (S + W) for the latter species. This highlights the effect of the n of the solvophilic block length in terms of the ease of access to nanoparticle morphologies of differing shape and size and likewise reinforces the notion that morphology is not solely dictated by the n of the solvophobic block but rather there is a balancing act. Dynamic light scattering confirmed the increase in size of the spherical nanoparticles with increasing n of the PPPMA block, Fig. 7. Also, we note that the DLS data is entirely consistent with the TEM data and also inline with the report from Fielding et al.28 regarding DLS-measured Dh's for poly(lauryl methacrylate-b-benzyl methacrylate) spherical nano-objects prepared in n-heptane with compositions in the same range as ours. These results clearly show that the critical n for PSMA macro-CTAs, with respect to the ability to access multiple nanoparticle morphologies, lies somewhere between 19 and 30, and therefore we note that if the goal is to prepare nano-objects with variable morphologies then shorter PSMA macro-CTAs should clearly be employed in RAFTDP syntheses. In contrast, the use of a PSMA macro-CTA with a higher n will allow straightforward access to spherical species with different hydrodynamic diameters.
 | ||
| Fig. 7 Representative DLS-measured intensity-average size distributions for the spherical nanoparticles prepared with PSMA30 at 20 wt% in n-octane with PPMA as comonomer. | ||
Thermoreversible degelation and associated morphology transitions
The ability of soft matter nanoparticles to undergo temperature induced morphology transitions, via heating and or cooling, is known although there is comparatively little in the literature on the topic. For example, LaRue et al.37 reported a worm-to-sphere (W–S) transition in block copolymer nanoparticles formed by polystyrene-b-polyisoprene in n-heptane (a selective solvent for polyisoprene) upon heating from 25 to 35 °C. This process was completely reversible although the corresponding S–W transition observed upon cooling was very slow compared to the W–S morphological change. Similar thermally induced transitions have been observed by Abbas et al.38 in polystyrene-b-polydimethylsiloxane copolymers in various phthalates as solvents, while a thermally induced sphere-to-vesicle (S–V) transition was reported by Moughton and O'Reilly in block copolymers prepared by conventional RAFT radical polymerization.39 One feature in certain RAFTDP formulations that has emerged is the ability to induce macroscopic physical changes upon heating or cooling and most notably reversible gelation. Such macroscopic changes occur due to a fundamental change in the nanoparticle morphology. The ability of worm micelles, above a critical concentration, to form physical gels is well known for polymers as well as species formed from small molecule building blocks,40,41 and has been observed by us19 and others42 in specific RAFTDP formulations. For example, we have previously shown that the AB diblock copolymer poly[2-(dimethylamino)ethyl methacrylate20-b-PPMA47] (PDMAEMA20-b-PPPMA47) forms a physical gel at RT in EtOH at a concentration of 21 wt% due to the formation of a pure worm morphological phase and associated worm entanglements. Upon heating to 70 °C the sample undergoes a macroscopic transition from a solid physical gel to a free flowing solution. This change was due to a thermally induced morphology change from worm species at ambient temperature to a pure spherical phase at elevated temperature. This was a rapid and completely reversible process that was facilitated, in part presumably, by the low Tg of the PPPMA core-forming block as well as changes in relative block solvation. Interestingly, we have observed almost identical behaviour for select examples of the PSMA–PPPMA block copolymers in non-polar media. For example, the PSMA18-b-PPPMA71 block copolymer prepared at 30 wt% solids formed an essentially pure worm phase and existed as a physical gel at ambient temperature, Fig. 8. Heating this solution to 70 °C for 5 min resulted in macroscopic degelation and the formation of a free flowing solution. This macroscopic transition was completely reversible, and cooling back to ambient temperature resulted in reformation of the gelled state. Interestingly, this critical transition temperature is almost identical to that we reported previously for similar degelation behaviour with PDMAEMA20-b-PPPMA47 in EtOH,19 but somewhat lower than the ∼85 °C transitional temperature we reported for similar PSMA–PPPMA block copolymers prepared in n-tetradecane.30 | ||
| Fig. 8 Digital images demonstrating reversible, thermally induced degelation–gelation for the PSMA18-b-PPPMA71 copolymer at 30 wt% in n-octane and TEM images obtained for the PSMA18-b-PPPMA71 block copolymer nanoparticles prepared at 30 wt% in octane at 25 °C and after heating to 90 °C for 5 min. | ||
As in previous reports, we evaluated this macroscopic change employing a combination of 1H NMR spectroscopy, DLS and TEM.
TEM is the most convenient technique for demonstrating the assumed nanoparticle morphology transition. Fig. 8 shows representative TEM images obtained after the withdrawal of aliquots at room temperature and after heating. As noted at ambient temperature a predominant worm phase is observed with some evidence of possible film formation or early transitional processes to vesicular species. After heating at 90 °C for 5 min followed by dilution in hot n-tetradecane and staining, we observe nanoparticles with a now predominant spherical morphology. This is consistent with our previous work on thermally induced morphology changes and with the above observed macroscopic change, i.e. degelation.
The change in nanoparticle morphology is also evident in the DLS analysis. Fig. 9 shows the measured intensity average size distributions for the PSMA18-b-PPPMA71 copolymer nanoparticles at 25 °C and after heating to 70 °C for 5 min. and then 92 °C for an additional 5 min. At ambient temperature we observe a ‘sphere equivalent’ size of ca. 146 nm. After heating at 70 °C for 5 min. there is a pronounced drop in the Dh to 64 nm, and continues to decrease after heating at ca. 92 °C with a final measured Dh of 35 nm. This is entirely consistent with the TEM data with an estimated sphere size of ca. 30 nm. However, we do point out that while these thermally induced changes are rapid they are slower and require slightly higher temperatures than similar changes for other PPPMA-based block copolymer nanoparticles we have examined.
 | ||
| Fig. 9 DLS data for the PSMA18-b-PPPMA71 block copolymer, measured in n-octane as a functional of increasing temperature. | ||
The nanoparticle morphology change is facilitated not just by the low Tg of the PPPMA core-forming block but also by a change in the relative solvation (and hence interfacial surface energy and/or relative block volume fractions), mainly of, the PPPMA core block. To examine this in more detail we conducted a temperature dependent 1H NMR study of the PSMA18-b-PPPMA71 copolymer in fully deuterated n-octane. Fig. 10 shows key sections of the NMR spectra as a function of heating from ambient temperature to 90 °C. Specifically, we have highlighted the regions associated with the Ph groups of the PPPMA block (δ = 7.9–7.6 ppm), the benzylic CH2 groups also associated with the PPMA repeat units (δ = 3.5–3.15 ppm) and the methylene groups directly adjacent to the methacrylic ester groups which, in this instance, are associated with the PSMA and PPPMA repeat units (δ = 4.9–4.6 ppm).
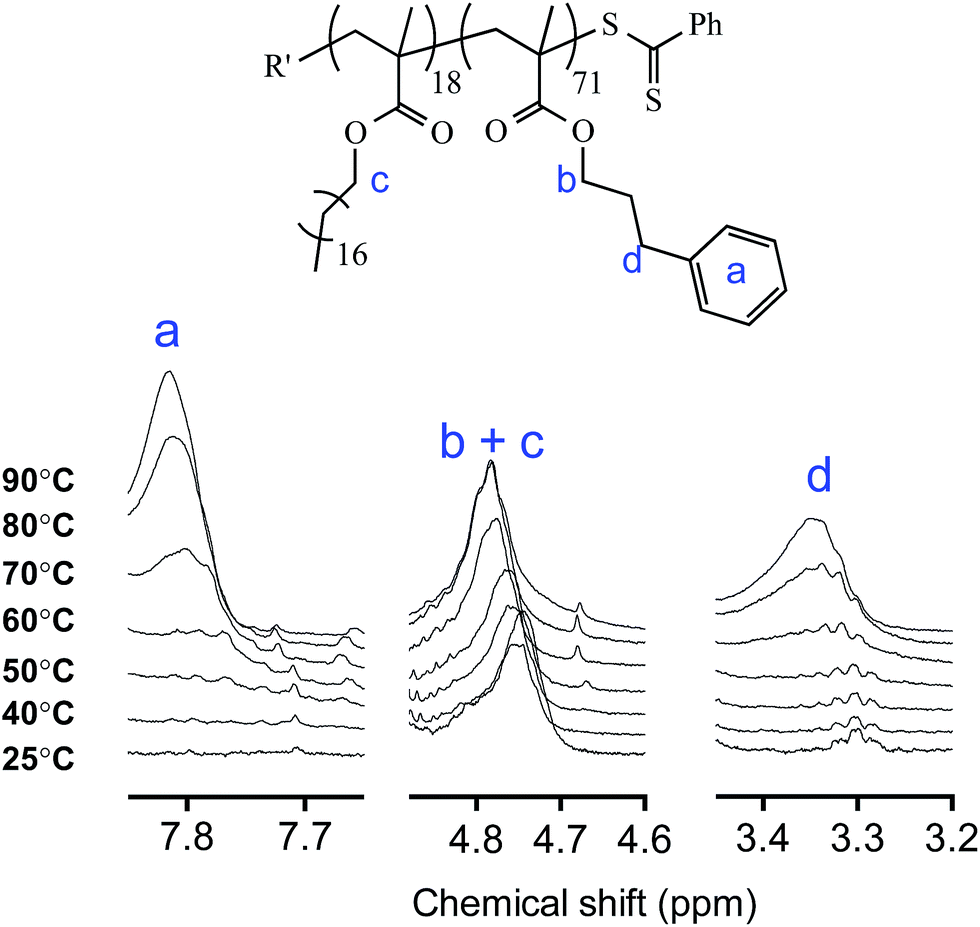 | ||
| Fig. 10 Sectional 1H NMR spectra recorded over the temperature range 25–90 °C in d18-n-octane for the PSMA18-b-PPPMA71 copolymer highlighting the change in solvation of key groups associated with the PPPMA block with increasing temperature. Measurements were made at a concentration of 0.5 wt%. | ||
At 25 °C we see no evidence of the Ph groups associated with the PPPMA block indicating these groups are not solvated under these conditions. Likewise, the signal associated with the benzylic hydrogens, while visible, has a significantly reduced intensity based on the composition. Indeed, this situation does not change even after heating to 60 °C. However, at 70 °C we begin to observe an increase in the intensity of both signals, a trend that continues with further heating to 80 and then 90 °C. This indicates that at these elevated temperatures the PPPMA side chains become increasingly solvated. This, in turn, increases core chain mobility that helps facilitate the observed nanoparticle morphology transitions and the degelation process. The side chains never become completely solvated (based on the measured integral values) since this would imply molecular dissolution and hence no nanoparticle formation. However, this does demonstrate that solvation, coupled with the low Tg of PPPMA, are the key features responsible for the fast and reversible nature of the observed morphology changes.
To demonstrate the reversibility of these thermally induced morphology transitions the PSMA18-b-PPPMA71 copolymer sample was subjected to variable temperature heating–cooling cycles in n-octane and monitored in real-time by DLS. Fig. 11 shows the systematic change in hydrodynamic diameter and DLS polydispersity over four heating–cooling cycles.
 | ||
| Fig. 11 Change in hydrodynamic diameter and DLS polydispersity for the nano-objects formed by PSMA18-b-PPPMA71 AB diblock copolymer during four heating–cooling cycles. Measurements were made in n-octane at a block copolymer concentration of 1 wt%. | ||
In all instances, when the temperature is at 25 °C nano-objects with intensity-average hydrodynamic diameters of ca. 130–150 nm were observed whose DLS polydispersities were ca. 0.2. This data is consistent with the DLS data shown in Fig. 9 when the nanoparticles exist in a predominantly worm state. Likewise, in all instances when the temperature of the solution was raised to 92 °C we see a large drop in the average hydrodynamic diameter to an average of ca. 35–45 nm with low associated polydispersities of 0.05. Again this is consistent with the data in Fig. 9 and likewise with the nanoparticles now existing in a spherical state.
Conclusions
Herein we have reported the use of poly(stearyl methacrylate) (PSMA) homopolymers as macro-CTAs in non-polar RAFT dispersion polymerization (RAFTDP) employing n-octane as the reaction medium and 3-phenylpropyl methacrylate (PPMA) as comonomer in a polymerization-induced self-assembly process. At 20 wt% solids and employing a PSMA homopolymer with a low average degree of polymerization (n) it is possible to access the full range of common nanoparticle morphologies with increasing complexity being attained with increasing n of the PPPMA block. Nanoparticle morphology can also be tuned by varying the total solids content although this is perhaps not as straightforward as simply varying the n of the PPPMA block for a fixed n of PSMA. In the case of a PSMA30 macro-CTA, RAFTDP of PPMA resulted in the formation of only spherical nanoparticles regardless of the extent of block copolymer asymmetry. This observation is consistent with previous observations where there often exists a critical solvophilic block length at and beyond which only spherical nanoparticles are accessible. Finally, block copolymers forming worm phases were able to undergo a thermally induced macroscopic transition from gels to free flowing solutions. This was shown to be due to a fundamental worm-to-sphere morphology change that was facilitated by both a change in the relative solvation of the core-forming PPPMA block and its associated low Tg. Real time DLS experiments confirmed that this process was fully reversible.
Acknowledgements
ABL thanks the Australian Research Council (ARC) for funding via the Future Fellowship program (FT110100046).References
- Q. Li, X. He, Y. Cui, P. Shi, S. Li and W. Zhang, Polym. Chem., 2014, 6, 70–78 RSC.
- F. Huo, S. Li, X. He, S. A. Shah, Q. Li and W. Zhang, Macromolecules, 2014, 47, 8262–8269 CrossRef CAS.
- M. Dan, F. Huo, X. Xiao, Y. Su and W. Zhang, Macromolecules, 2014, 47, 1360–1370 CrossRef CAS.
- J. R. Lovett, N. J. Warren, L. P. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2014, 53, 1–6 CrossRef.
- M. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- W.-M. Wan and C.-Y. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CrossRef CAS.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS PubMed.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 128–139 CrossRef CAS.
- W.-D. He, X.-L. Sun, W.-M. Wan and C.-Y. Pan, Macromolecules, 2011, 44, 3358–3365 CrossRef CAS.
- C.-Q. Huang and C.-Y. Pan, Polymer, 2010, 51, 5115–5121 CrossRef CAS.
- W. M. Wan, X. L. Sun and C. Y. Pan, Macromol. Rapid Commun., 2010, 31, 399–404 CrossRef CAS PubMed.
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC.
- Y. Pei, N. C. Dharsana, J. A. van Hensbergen, R. P. Burford, P. J. Roth and A. B. Lowe, Soft Matter, 2014, 10, 5787–5796 RSC.
- Y. Pei, N. C. Dharsana and A. B. Lowe, Aust. J. Chem., 2015 DOI:10.1071/ch14490.
- M. Zong, K. J. Thurecht and S. M. Howdle, Chem. Commun., 2008, 5942–5944 RSC.
- J. Jennings, M. Beija, J. T. Kennon, H. Willcock, R. K. O'Reilly, S. Rimmer and S. M. Howdle, Macromolecules, 2013, 46, 6843–6851 CrossRef CAS.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- Q. Li, X. He, Y. Cui, P. Shi, S. Li and W. Zhang, Polym. Chem., 2015, 6, 70–78 RSC.
- X. He, Q. Li, P. Shi, Y. Cui, S. Li and W. Zhang, Polym. Chem., 2014, 5, 7090–7099 RSC.
- L. Houillot, C. Bui, C. Farcet, C. Moire, J. A. Raust, H. Pasch, M. Save and B. Charleux, ACS Appl. Mater. Interfaces, 2010, 2, 434–442 CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- Y. Pei, L. Thurairajah, O. Sugita and A. B. Lowe, Macromolecules, 2015, 48, 236–244 CrossRef CAS.
- J. A. Raust, L. Houillot, M. Save, B. Charleux, C. Moire, C. Farcet and H. Pasch, Macromolecules, 2010, 43, 8755–8765 CrossRef CAS.
- L. Houillot, C. Bui, M. Save, B. Charleux, C. Farcet, C. Moire, J. A. Raust and I. Rodriguez, Macromolecules, 2007, 40, 6500–6509 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- T. K. Mandal and E. M. Woo, Macromol. Chem. Phys., 1999, 200, 1143–1149 CrossRef CAS.
- G. Sakellariou, A. Siakali-Kioulafa and N. Hadjichristidis, Int. J. Polym. Anal. Charact., 2003, 8, 269–277 CrossRef CAS.
- H. Lee, G. Tae and Y. H. Kim, Macromol. Res., 2008, 16, 614–619 CrossRef CAS.
- I. LaRue, M. Adam, M. Pitsikalis, N. Hadjichristdis, M. Rubenstein and S. S. Sheiko, Macromolecules, 2006, 39, 309–314 CrossRef CAS.
- S. Abbas, Z. Li, H. Hassan and T. P. Lodge, Macromolecules, 2007, 40, 4048–4052 CrossRef CAS.
- A. O. Moughton and R. K. O'Reilly, Chem. Commun., 2010, 46, 1091–1093 RSC.
- S. R. Raghavan and J. F. Douglas, Soft Matter, 2012, 8, 8539–8546 RSC.
- C. A. Dreiss, Soft Matter, 2007, 3, 956–970 RSC.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Doudlas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
Footnote |
| † Current address: Nanochemistry Research Institute & Department of Chemistry, Curtin University, Bentley Campus, Bentley, Perth, WA 6102, Australia. |
| This journal is © The Royal Society of Chemistry 2015 |