
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and bonding in Au(I) chloride species: a critical examination of X-ray absorption spectroscopy (XAS) data†
Sin-Yuen
Chang
a,
Akihiro
Uehara‡
a,
Samuel G.
Booth
a,
Konstantin
Ignatyev
b,
J. Frederick W.
Mosselmans
b,
Robert A. W.
Dryfe
a and
Sven L. M.
Schroeder§
*ab
aSchool of Chemical Engineering and Analytical Science, School of Chemistry, The University of Manchester, Manchester M13 9PL, UK. E-mail: s.l.m.schroeder@leeds.ac.uk
bDiamond Light Source Ltd, Didcot, Oxfordshire OX11 0DE, UK
First published on 16th December 2014
Abstract
Au(I) chloride species are important reactants and intermediates in various processes across the chemical sciences and engineering. Structure and bonding in Au(I) species are often characterized by X-ray absorption spectroscopy (XAS), including measurements under reaction conditions. Previously reported XA spectra for Au(I) chloride species have varied significantly, likely as a result of radiation damage and/or partial disproportionation of [AuCl2]− ions, which are metastable under ambient conditions. By monitoring the decomposition of tetrabutylammonium dichloroaurate(I), TBA[AuCl2], in 1,2-dichlorobenzene we have obtained a reliable X-ray absorption spectrum of [AuCl2]− ions by combining the calculation of difference spectra with an extended X-ray absorption fine-structure (EXAFS) determination of the solution composition. The results show that the X-ray absorption near-edge structure (XANES) of [AuCl2]− is characterized by a weak Au 2p3/2 → 5d (‘white line’) transition, which agrees well with the spectrum predicted by electronic structure calculations using the FEFF8 code. Compared to [AuCl4]−, the determined [AuCl2]− spectrum has several distinctive features of diagnostic analytical value. A more detailed densities of states (DOS) analysis of the electronic structure suggests that the weak white line arises from a hybrid Au 6s/5d DOS band that is partially occupied, up to the level of the highest occupied molecular orbital (HOMO). Correlation of Cl coordination numbers determined from the EXAFS with the intensity of the white line in the XANES indicates that the decomposition is a primarily radiation-induced oxidation to Au(III) species with an average formula of [AuCl3OH]−.
1. Introduction
Au(I) chloride species are important reactants and intermediates in heterogeneous and homogeneous Au catalysis,1,2 electrochemical processes,3 nanoparticle synthesis,4–6 geochemical Au speciation,7–9 and gold mining.10 X-ray absorption spectroscopy (XAS) measurements can provide incisive information on the chemical state of Au in solid Au(I) chloride and its associated solution species.8 The X-ray absorption fine-structure (XAFS) in these spectra provides information on the electronic properties of the Au centers through the X-ray absorption near-edge structure (XANES) and on molecular structure, such as Au–Cl coordination numbers and bond lengths, through the extended X-ray absorption fine structure (EXAFS).11,12In the context of Au speciation analysis from L3-edge XANES, the spectral feature receiving most attention is the so-called ‘white line’ resonance, which is at a photon energy of approximately 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 921 eV for Au(I) chloride(Fig. 1). The white line arises from the excitation of core Au 2p3/2 electrons to unoccupied Au 5d or 6s states. Compared to metallic Au, Au(III) compounds tend to have lower 5d and 6s occupancies and therefore exhibit prominent white lines,13–18 while Au(I) compounds tend to have weaker white line absorptions.14,17 White line intensity analysis can therefore be used to identify Au oxidation states14 although there are cases in which such correlations need to be applied with care.19,20 For example, in the context of heterogeneous catalysis, Au chloride species are common precursors in catalyst preparation21 and reliable identification of Au species is important for generating the deep mechanistic understanding required for catalyst design. Certainty about the spectroscopic signature of Au(I) chloride is required to reach reliable conclusions.
921 eV for Au(I) chloride(Fig. 1). The white line arises from the excitation of core Au 2p3/2 electrons to unoccupied Au 5d or 6s states. Compared to metallic Au, Au(III) compounds tend to have lower 5d and 6s occupancies and therefore exhibit prominent white lines,13–18 while Au(I) compounds tend to have weaker white line absorptions.14,17 White line intensity analysis can therefore be used to identify Au oxidation states14 although there are cases in which such correlations need to be applied with care.19,20 For example, in the context of heterogeneous catalysis, Au chloride species are common precursors in catalyst preparation21 and reliable identification of Au species is important for generating the deep mechanistic understanding required for catalyst design. Certainty about the spectroscopic signature of Au(I) chloride is required to reach reliable conclusions.
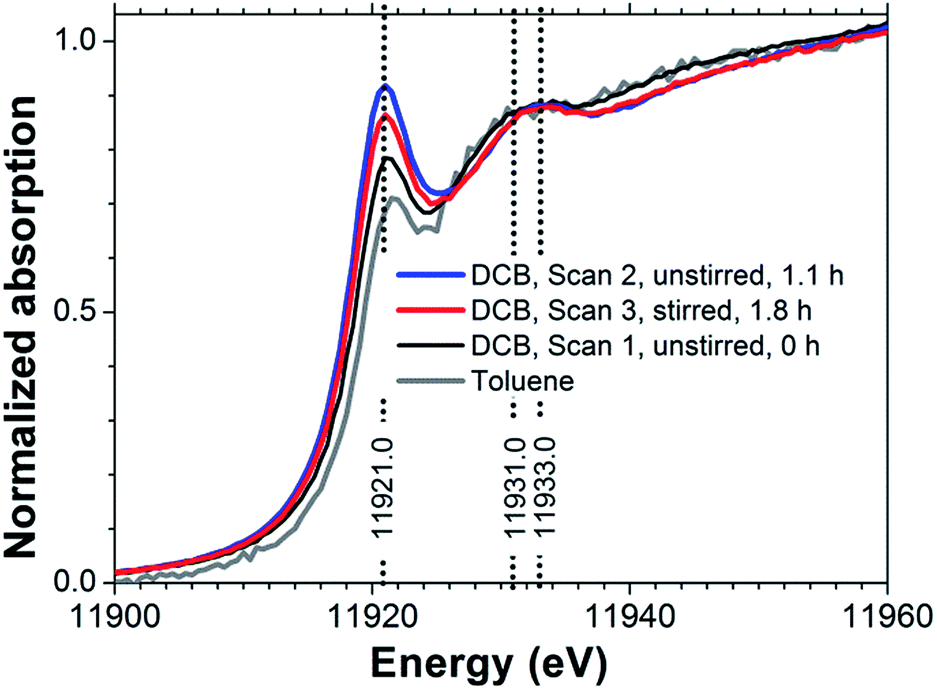 | ||
| Fig. 1 Au L3-edge XANES spectra of 6 mM TBA[AuCl2] in DCB (sample #1 in Table 1) and 1 mM TBA[AuCl2] in toluene (sample #2). | ||
However, previously published Au L3-edge XANES spectra of Au(I) chloride species6,8,13–16 have varied considerably. Quite prominent white lines have been reported in some cases,17,22 which suggest a superposition of absorption from significant concomitant Au(III) concentrations, either due to contamination or from reaction products formed spontaneously in solution. Even the published spectrum of [AuCl2]− obtained under hydrothermal conditions, which strongly favor Au(I), has significant intensity in the white line region.8 Generally, atmospheric pressure, non-acidic pH and absence of chloride ions favor Au(III), while hydrothermal and acidic chloride-rich aqueous electrolytes stabilize Au(I).8 The variations between published spectra are likely due to (i) the sensitivity of cationic Au species to radiation-induced redox transformations,23 and (ii) the metastability of Au(I) chloride under ambient conditions, especially in aqueous solution.9,17
Examining the dichloroaurate(I) ion as its tetrabutylammonium salt, TBA[AuCl2] in the non-aqueous solvents 1,2-dichlorobenzene (DCB), toluene and 1,2-dichloroethane (DCE) we also observed evidence for Au(III) formation as a clearly enhanced white line feature (Fig. 1). This feature was weaker in toluene than in DCB solution, suggesting higher stability of [AuCl2]− in toluene, but the low TBA[AuCl2] solubility of approximately 1 mM in toluene did not allow us to collect EXAFS data with sufficient signal-to-noise quality for a more detailed analysis. For DCE, the spectrum of 5 mM TBA[AuCl2] (not shown in Fig. 1) indicated metallic Au only, suggesting rapid reduction from Au(I) to Au(0). However, in DCB solutions containing 6 mM TBA[AuCl2], the spectral changes between each measurement were slow enough to enable monitoring of the reaction progress by XAS. In the following we will use this time-dependence of the Au L3-edge spectrum to advantage, to reliably isolate the [AuCl2]− XANES through a combined EXAFS and XANES analysis. The analysis will show that TBA[AuCl2] appears to be oxidized through the influence of radiation on the DCB solution, forming Au(III) species with an average composition of [AuCl3OH]−. We will compare the determined [AuCl2]− spectrum with the XANES predicted by electronic structure calculations and with the spectrum of aqueous [AuCl2]−.
2. Experimental
Table 1 summarizes the samples examined by XAS measurements in this study. For samples #1–3, tetrabutylammonium dichloroaurate (TCI Europe N. V.), (TBA)[AuCl2], was directly dissolved in the solvents (1,2-DCB, ≥99% Sigma Aldrich; toluene, ≥99.5% Sigma Aldrich; 1,2-DCE, ≥99% Sigma Aldrich) without further purification. Only the spectra arising from sample #1 will be analyzed in detail in this contribution.| # | Chemical | Solvent | Condition | Conc. (mM) | Modea |
|---|---|---|---|---|---|
| a ‘Fluor’ denotes fluorescence-yield detection, ‘trans’ denotes transmission detection. | |||||
| 1 | TBA[AuCl2] | DCB | Ambient | 6 | Fluor |
| 2 | TBA[AuCl2] | Toluene | Ambient | 1 | Fluor |
| 3 | TBA[AuCl2] | DCE | Ambient | 5 | Fluor |
| 4 | H[AuCl2] | Water | Ambient | 15 | Fluor, trans |
| 5 | TBA[AuCl2] | Solid | Ambient | — | Fluor |
| 6 | [AuCl2]− | Water | Hydrothermal | See ref. 8 | Fluor |
| 7 | TOA[AuCl4] | DCB | Ambient | 5 | Fluor |
To obtain a spectrum of aqueous [AuCl2]− (sample #4), it was extracted into water from a DCE solution through the DCE/water interface, similar to a previously described phase transfer process.24 A 20 mM (TBA)[AuCl2] solution in DCE was brought in contact with an aqueous solution containing 15 mM HAuCl4 and 0.1 M HCl. [AuCl2]− exchanges with [AuCl4]− in this system because of the hydrophilic nature of [AuCl2]−. Ion exchange resulted in an aqueous solution of H[AuCl2] of approximately 15 mM.
Tetraoctylammonium tetrachloroaurate, (TOA)[AuCl4], crystals (sample #7) were prepared by mixing equimolar quantities of HAuCl4·3H2O (≥99.99%, Alfa Aesar) and TOACl (≥97%, Aldrich) which were then dissolved in a minimal amount of methanol. Undissolved impurities were removed by filtration. The TOA[AuCl4] salt produced was purified further by repeating the recrystallization procedure. The purified TOA[AuCl4] crystals were then dissolved in DCB just prior to XAS measurements.
The Au L3 absorption edges of samples #1–3 and #7 were measured at the undulator beamline I18 of Diamond Light Source, UK.25 During measurements, the sample solutions were placed in capped Eppendorf centrifugation tubes. The synchrotron electron storage ring was operating at 3 GeV, 300 mA. A double Si(111) crystal monochromator giving an energy resolution of 1.4 × 10−4 dE/E was used. Kirkpatrick-Baez (KB) mirrors were used to focus the beam and to reject higher harmonics. The photon fluxes on the samples were between 0.9 × 1011 and 4.9 × 1011 s−1 at 11919 eV. Spectra were acquired in fluorescence-yield mode, with an Ortec multi-element solid-state Ge detector. The beam size during the experiments was approximately 45 (horizontal) μm × 50 (vertical) μm. The data acquisition time for each spectrum was around 30 min. The spectra of samples #4 and #5 were measured at the BL27B beamline of the Photon Factory at the High Energy Accelerator Research Organization (KEK), Japan. The spectrum of sample #6 (aqueous [AuCl2]−) has previously been reported.8 It was measured under hydrothermal conditions at 250 °C, 600 bar, starting from an aqueous solution containing 0.032 M HAuCl4, 0.54 M NaCl and 0.53 M HCl.
The Demeter software package (version 0.9.18) was used for XAFS data analysis.26 The data were first calibrated using gold foil and the edge-step heights normalized to 1. For the EXAFS analysis, a Hanning-type Fourier transform window was used. Fitting was done in R-space using k1-weighting. The EXAFS scattering paths were generated from the published crystal structure of AuCl.27 The error values reported for fitted parameters are the diagonals of the covariance matrix multiplied by the square-root of the reduced chi-square. The EXAFS ![[scr R, script letter R]](https://www.rsc.org/images/entities/char_e531.gif) -factor indicates the closeness-of-fit.12 Only 3 variables out of the 15.1 independent points were used. The fitted k- and R-ranges were 3–11 Å−1 and 1.0–4.0 Å respectively.
-factor indicates the closeness-of-fit.12 Only 3 variables out of the 15.1 independent points were used. The fitted k- and R-ranges were 3–11 Å−1 and 1.0–4.0 Å respectively.
FEFF8.2 (ref. 28) was used to simulate the XANES of monomer [AuCl2]−, with Au–Cl distance at 2.27 Å. XANES, FMS and SCF cards were used – FMS for full-multiple scattering XANES calculation; SCF to enable self-consistent field iterations. The ION card was not used.
3. Results
3.1 EXAFS analysis and the Au(III) species
Without exposure to the X-ray beam, [AuCl2]− in DCE is stable, as known from electrochemical experiments.29 Using cyclic voltammetry we likewise established the stability of [AuCl2]− in DCB solution (Fig. S1†). We can therefore conclude that the observed changes in Fig. 1 were indeed radiation-induced. We further established that radiation converted Au(I) to Au(III) in DCB by recording the spectrum of an unstirred solution, followed by a spectrum at the same position under stirring. As can be seen in Fig. 1, the white line intensity decreased upon stirring, indicating that Au(III) concentrations accumulated in the region of the unstirred solution exposed to the X-ray beam. Aqueous [AuCl2]− is known to undergo spontaneous disproportionation to [AuCl4]− and Au metal.9 However, for the radiation-induced oxidation observed in DCB, neither visual inspection of the sample cells nor EXAFS analysis (vide infra) provided any evidence for the presence of metallic Au. We return to the possible mechanism of the radiation-induced Au(III) formation in DCB further below, after establishing the likely composition of the Au(III) species from a combined EXAFS and XANES analysis.Fig. 2 displays EXAFS data for TOA[AuCl4] and TBA[AuCl2] in DCB. The spectra of both solutions have very similar features in both k- and R-spaces, except for the fact that the amplitude of the TBA[AuCl2] EXAFS is smaller because of its lower Cl coordination number. This similarity of the EXAFS functions is expected, as the length of the Au–Cl bond in [AuCl2]− is almost identical to that in [AuCl4]−.8 Consequently the scattering paths involving Cl in the linear Au(I) complex [AuCl2]− and in square planar Au(III) complexes such as [AuCl4]− and [AuCl3OH]− are practically identical.
 | ||
| Fig. 2 EXAFS of 6 mM TBA[AuCl2] in DCB (sample #1 in Table 1) contrasted with 5 mM TOA[AuCl4] in DCB (sample #7). The multiple scattering paths are also shown on the right. | ||
Four scattering paths were used for the EXAFS analysis of the three scans of TBA[AuCl2] in DCB (Fig. 1): the Au–Cl and Au–O single scattering (SS) paths as well as the linear multiple scattering (MS) paths Au–Cl– –Cl–Au and Au–Cl–Au–Cl–Au (Fig. 2). Such linear MS paths are commonly used in EXAFS models for linear and square planar Au(III) compounds.8,30 The parameters for the EXAFS model are summarized in Table 2. The amplitude factor S02 for all the scattering paths was fixed at 0.82 and the Debye Waller (DW) factor for Cl, σCl2 was fixed at 0.002 Å2. These values were obtained from the fitting of the TOA[AuCl4] reference solution in DCB (sample #7, Table 1) and are in close agreement with values previously reported for [AuCl4]−.8,30,31 We will in the following assume that identical DW factors apply to Au–Cl bonds in Au(I) and Au(III) complexes, which, strictly speaking, introduces uncertainty because the DW factor is dependent on the amplitude of the Au–Cl vibrations. However, given the similar Au–Cl bond lengths in [AuCl2]− and [AuCl4]−, it appears unlikely that the differences introduce a major error. No metallic Au–Au scattering was evident from the EXAFS analysis, suggesting the absence of dimers, Au(0), or other self-associated species. When carrying out the EXAFS fitting with k1-weighting we noted that the fit quality improved upon the inclusion of an Au–O scattering path, with σO2 and RO fixed at 0.002 Å2 and 1.96 Å, which are values comparable to those previously found for hydrolyzed Au(III) species in water,18,30,31 suggesting the presence of hydroxo ligands. Higher k-weightings were less sensitive to the contribution of this weak Au–O scattering, which is perhaps expected because Au–O scattering is strongest at low k-values. The initial E0 value was chosen within the post-white line and pre-EXAFS region at 11926 eV to minimize the shift in energy, ΔE in the EXAFS fitting.32
| Parametersa | Au–O (SS) | Au–Cl (SS) | Au–Cl (MS) |
|---|---|---|---|
|
a
S
0
2 is the amplitude reduction factor; σ2 is the Debye Waller factor; R is the Au–scatterer distance or the half-path length; xAu(III) is the mole fraction of Au(III) species; N is the coordination number or path degeneracy; ΔE is the edge energy shift; the initial choice for E0 was 11926 eV.
|
|||
| S 0 2 | 0.82 | 0.82 | 0.82 |
| σ 2/Å2 | 0.002 | 0.002 | 2 × 0.002 |
| R/Å | 1.96 | R Cl | 2RCl |
| N | x Au(III) | N Cl | N Cl |
| E 0/eV | ΔE | ΔE | ΔE |
As can be seen in Table 3, for all of the TBA[AuCl2] solutions in DCB the Cl coordination number (NCl) was significantly higher than the value of 2 expected for [AuCl2]−, confirming the presence of significant concentrations of Au(III) species with a higher Cl coordination number, such as [AuCl4]− or [AuCl3OH]−. A linear best fit to the correlation between white line intensity and NCl (Fig. 3) predicts a white line intensity of 1.6 at NCl = 4, the expected coordination number of [AuCl4]−. This intensity is much higher than the experimentally observed value of 1.1 we obtained for a solution of TOA[AuCl4] in DCB (Fig. 4). Much better agreement with experiment is achieved by extrapolation to NCl = 3, where the predicted white line intensity is 1.1. This value is in very good agreement with the white line intensity of 1.2 previously reported for [AuCl3OH]−.31 This suggests that [AuCl3OH]− is the dominant Au(III) species formed in DCB, which also explains our observation of significant Au–O scattering in the EXAFS.
| Parameters | Scan 1 | Scan 2 | Scan 3 |
|---|---|---|---|
| R(Au–Cl)/Å | 2.266 (6) | 2.28 (1) | 2.272 (8) |
| N Cl | 2.39 (5) | 2.63 (10) | 2.47 (8) |
| x Au(III)/% | 39 (5) | 63 (10) | 47 (8) |
| ΔE/eV | 0.1 (7) | 1.1 (10) | 0.09 (92) |
| -factor |
0.012 | 0.032 | 0.024 |
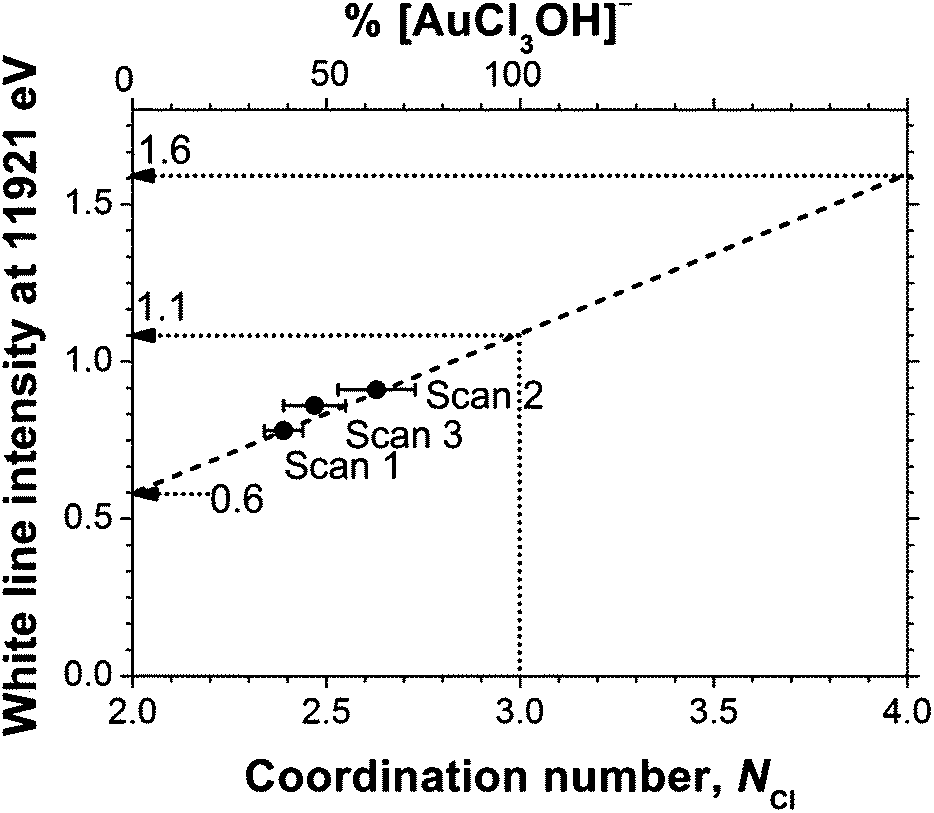 | ||
| Fig. 3 The white line absorption of 6 mM TBA[AuCl2] in DCB (Fig. 1) at 11921 eV as a function of NCl. NCl was quantified using EXAFS analysis, see Table 3. | ||
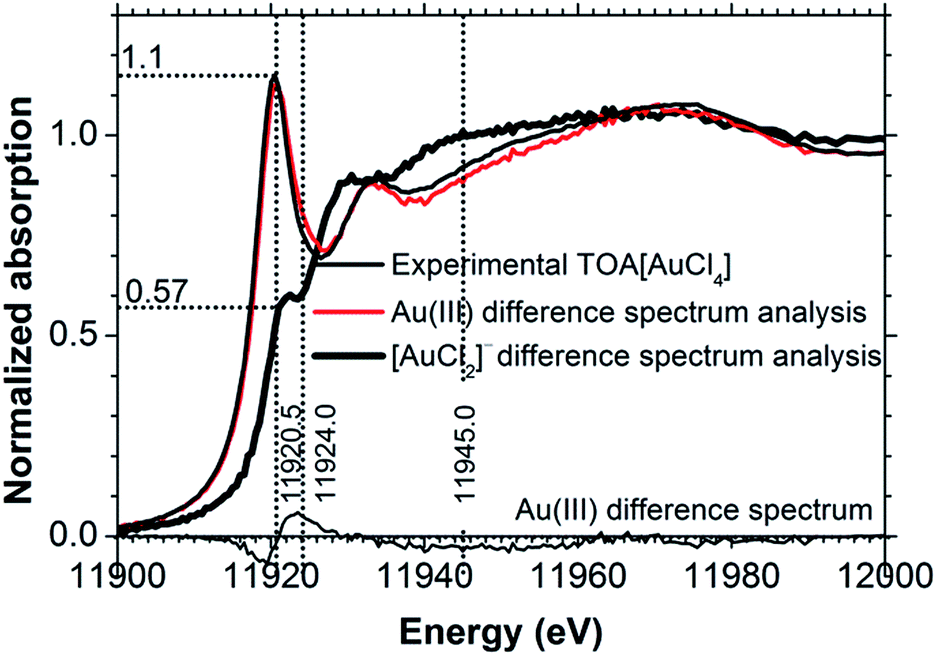 | ||
| Fig. 4 [AuCl2]− and Au(III) spectra determined by XANES difference spectrum analysis contrasted with the experimental spectrum of 5 mM TOA[AuCl4] in DCB. | ||
Assuming that the DCB solution contains a mixture of [AuCl2]− and [AuCl3OH]− we can now determine the mole fraction of [AuCl3(OH)]− from the EXAFS-derived NCl values, as xAu(III) = NCl − 2. This analysis yielded [AuCl3(OH)]− contents of 39%, 63% and 47% for the three solutions (Fig. 3, Table 3).
3.2 Determination of [AuCl2]− XANES by difference spectrum analysis
As a complementary and independent means for determining the mole fractions x of Au(I) and Au(III) in DCB solutions, a difference spectrum analysis of the XANES spectra was carried out. For this we assumed that the experimental spectra, Si,exp can be expressed as a sum of the spectra from [AuCl2]−, S[AuCl2]−, and an arbitrary Au(III) compound, SAu(III):| Si,exp = (1 − xAu(III),i)S[AuCl2]− + xAu(III),iSAu(III) | (1) |
We can then determine the spectrum of pure [AuCl2]−, S[AuCl2]−,det from two experimental spectra (scan 1 and 2) by eliminating SAu(III) from eqn (1), yielding eqn (2).
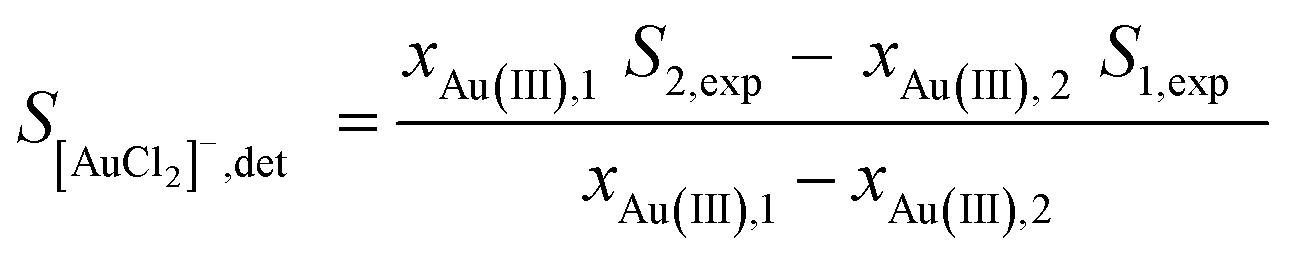 | (2) |
One such spectrum is shown in Fig. 4, labelled ‘[AuCl2]− difference spectrum analysis’. With knowledge of S[AuCl2]−,det, we can now also determine the corresponding Au(III) spectrum, SAu(III),det, shown in Fig. 4, labelled ‘Au(III) difference spectrum analysis’.
We used a least-square method to calculate xAu(III),1 and xAu(III),2. The difference between the determined Au(III) spectrum, SAu(III),det and the experimentally obtained Au(III) spectrum, SAu(III),exp was minimized by changing xAu(III),1 and xAu(III),2 using the Solver in Microsoft Excel. We used the spectrum of TOA[AuCl4] in DCB as SAu(III),exp. This analysis indicated xAu(III) values of 0.39 and 0.64 for scans 1 and 2 respectively. These values are in very good agreement with those obtained from the EXAFS fitting analysis in Section 3.1, where xAu(III) was found to be 0.39 and 0.63. Comparison of the determined Au(III) XANES spectrum with that of the [AuCl4]− standard (Fig. 4) reveals small differences; there is a weak shoulder feature around 11924 eV, while the shoulder at 11945 eV is somewhat lower in intensity. These are precisely the characteristics of the spectrum of partially hydrolyzed Au(III) chloride with a stoichiometry close to [AuCl3OH]−,31 confirming the conclusion drawn from the EXAFS analysis in Section 3.1.
A FEFF8 simulation of the [AuCl2]− L3-edge reproduces all salient features in the experimental XANES spectrum (Fig. 5), including the peaks at 11922 and 11930 eV. Analysis of the l-projected unoccupied density-of-states (DOS) plots shows that the weak absorption in the white line region arises from a transition to a hybrid partially unoccupied Au 6s/5d band intersected by what FEFF8 identifies as the ‘Fermi energy’, but should perhaps more correctly be referred to as the highest occupied molecular orbital (HOMO) for a molecular species. The well-known 6s/5d hybridization arises from the relativistic effect on the 6s state.33,34 There is also significant p-DOS in this energy range, but its influence on the XANES spectrum is not evident because only transitions with Δl = ±1, i.e. p3/2 → d or p3/2 → s, are dipole-allowed. Examination of the DOS in Fig. 5 further indicates that the resonance at 11930 eV, which is in the region of the onset of EXAFS backscattering, arises from a transition to unoccupied states with Cl 3d–Au 5d hybridization.30,35
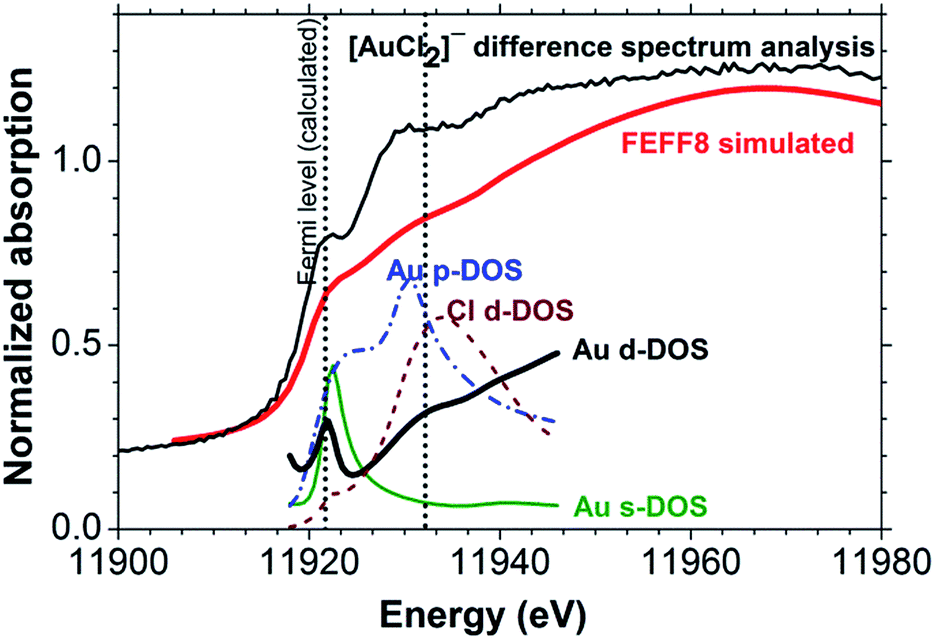 | ||
| Fig. 5 FEFF8 simulated Au(I) chloride and the projected density-of-states compared with [AuCl2]− determined using XANES difference spectrum analysis. | ||
4. Discussion
In the previous section, we have shown that TBA[AuCl2] in DCB was oxidized to [AuCl3OH]− under X-ray irradiation, and that metallic Au was not detected in the solution. Under ambient conditions,9 TBA[AuCl2] in DCB was stable in the absence of radiation (Section 3.1), in marked contrast to aqueous [AuCl2]− solutions, which undergo disproportionation to [AuCl4]− and metallic Au. These observations point towards a reaction induced by X-ray photons, converting TBA[AuCl2] in DCB to [AuCl3OH]−. In the absence of additional mechanistic information about the process it is difficult to identify the radiation-induced process with certainty, but it appears likely that radicals formed by irradiation of DCB cause the observed transformation. For example, a reaction involving Cl˙ radicals and water dissolved in DCB from the laboratory ambient could lead to an overall reaction according to eqn (3). But clearly more work is required to unequivocally confirm this or any alternative mechanism.| [AuCl2]− + 2Cl˙ + H2O → [AuCl3OH]− + HCl | (3) |
To examine the effect of spontaneous disproportionation of aqueous [AuCl2]− under ambient conditions we also measured its spectrum (sample #4 in Table 1, the group of spectra in the lower part of Fig. 6). We obtained both the transmission and the fluorescence-yield XANES spectra. Comparison of these data with the spectrum obtained in DCB indicated that partial disproportionation to Au(0) and Au(III) had taken place.9 This was evident from the fluorescence-yield XANES spectrum, which has a somewhat stronger white line than the transmission spectrum, as a result of metallic gold deposition on the walls of the sample cell during the measurement. Fluorescence-yield detection is much more sensitive to such thin wall deposits36 than transmission detection, which probes the bulk composition of the solution. In line with this, subsequent scans of the same sample (not shown) yielded fluorescence-yield spectra that evolved to mainly metallic features; meanwhile the transmission spectra acquired Au(III) and Au(0) features, supporting the fact that disproportionation of Au(I) to Au(III) and Au(0) had taken place in the aqueous solution during the measurements.
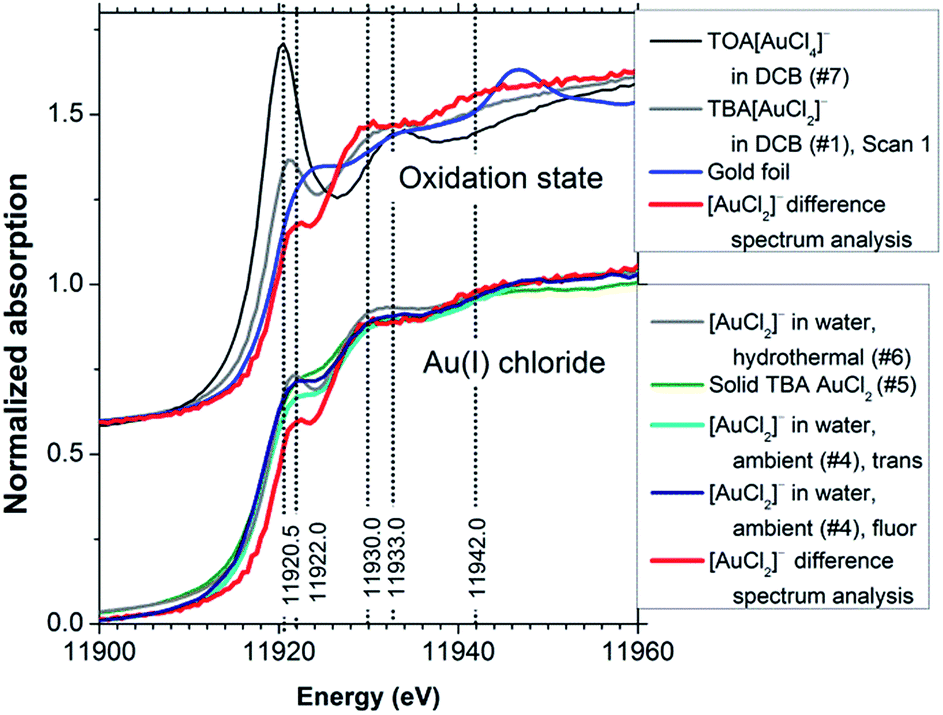 | ||
| Fig. 6 [AuCl2]− determined using XANES difference spectrum analysis compared with compounds of different oxidation states (top) and with the other Au(I) chloride spectra (bottom). Sample numbers are shown in brackets, refer to Table 1. ‘Trans’ denotes transmission detection, ‘fluor’ denotes fluorescence-yield detection. | ||
There are several distinctive features in the determined [AuCl2]− XANES spectrum that allow the identification of Au(I) chloride species. The upper group of spectra in Fig. 6 compares the determined [AuCl2]− spectrum with the spectrum of TOA[AuCl4] and with the original spectrum of the TBA[AuCl2] solution in DCB. It can be seen that the white line maximum for [AuCl2]− is shifted to higher photon energy by approximately 1.5 eV relative to the value of 11920.5 eV observed for [AuCl4]−. A broad shoulder is evident in the photon energy range around 11942 eV. However, the resonance at ∼11930 eV is most distinctive for [AuCl2]−, as it occurs several eV below the corresponding feature in the [AuCl4]− spectrum. We note that this transition is evident in a set of recently published time-resolved XANES data for the process of X-ray induced Au nanoparticle formation from [AuCl4]− in ionic liquids, where it is most clearly visible after approximately 4 h.37 Its presence suggests the formation of an [AuCl2]− intermediate in the process, possibly supporting the AuCl2-dimer model put forward in the study, which was derived from an EXAFS analysis. Judging from the XANES obtained in our work it would appear that a model involving Au nanoparticles, [AuCl2]− and [AuCl4]− may provide an alternative explanation.
The [AuCl2]− spectrum determined from XANES difference spectrum analysis has a weak white line, similar to that in some previously reported data for [AuCl2]− species – including measurements under ambient and hydrothermal conditions (600 bar, 250 °C)8 as well as a spectrum of solid TBA[AuCl2] (Fig. 6, lower set of spectra). The features at 11930 and 11942 eV are very similar in these Au(I) chloride XANES spectra. The agreement between these spectra in different environments indicates that the internal electronic structure of the ion is not strongly influenced by the solvent and cation. Somewhat stronger white lines in other reported data17,22 may be due to either residual Au(III) content or weak solvent interactions.
We note particularly that compared to Au foil (Fig. 6, upper set of spectra) the absorption in the white line region in the [AuCl2]− spectrum is actually weaker. This indicates that the relationship between oxidation state and white line intensity may be more complex than sometimes assumed.14 Indeed, the results of the FEFF8 DOS calculation (Fig. 5) suggest a high sensitivity to the exact position of the ‘Fermi energy’ (HOMO), which determines Au 6s/5d band occupancy, relative to the contributions from Au 5d, 6s and 6p states. This reflects the well-known dependence of chemical bonding on ligand species, coordination number and geometric arrangement around the X-ray absorbing central atom.38,39 More systematic studies are needed to elucidate these complex relationships further.
5. Conclusions
By combined difference spectrum and EXAFS analysis we have determined the XANES spectrum of [AuCl2]− from several consecutive XAS scans of TBA[AuCl2] in DCB. The [AuCl2]− spectrum obtained by our analysis can serve as a reliable reference for XANES studies requiring the identification and quantification of [AuCl2]− content. Radiation-induced oxidation of TBA[AuCl2] appears to result in [AuCl3OH]− as shown in the correlation between the EXAFS-derived Cl coordination numbers and the white line intensities in the spectra. Difference spectrum analysis of the XANES arrives at the same quantitative conclusions and likewise allowed us to eliminate the influence of the Au(III) species on the spectrum. Compared with [AuCl4]− the determined [AuCl2]− spectrum has several distinctive features with diagnostic value: a small white line at 11922 eV, a second peak at 11930 eV and a broad shoulder at 11942 eV, which are also evident in some previously reported Au(I) chloride spectra. The energetic positions of these features are quantitatively reproduced by FEFF8 calculations of the [AuCl2]− XANES. It appears that the radiation-induced oxidation and partial hydrolysis of [AuCl2]− to [AuCl3OH]− most likely follows a mechanism involving radicals formed in the DCB solvent matrix as well as water dissolved in the solvent.
Acknowledgements
We thank Diamond Light Source for the award of beamtime on I18 under proposal number SP-8861 and Dr Gleb Pokrovski for sharing his Au chloride data. RAWD and SLMS gratefully acknowledge financial support from the EPSRC through an EPSRC-NSF “Materials World Network” grant (EP/H047786/1). SYC thanks The University of Manchester, Mr and Mrs Clews for a President PhD scholarship. AU acknowledges Dr Y. Okamoto, Japan Atomic Energy Agency, Dr T. Fujii, Kyoto University Research Reactor Institute for the XAFS measurements at KEK.Notes and references
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- J. Huang and M. Haruta, in Bridging Heterogeneous and Homogeneous Catalysis: Concepts, Strategies and Applications, ed. C. Li and Y. Liu, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- L. Aldous, D. S. Silvester, C. Villagran, W. R. Pitner, R. G. Compton, M. Cristina Lagunas and C. Hardacre, New J. Chem., 2006, 30, 1576–1583 RSC.
- P. J. G. Goulet and R. B. Lennox, J. Am. Chem. Soc., 2010, 132, 9582–9584 CrossRef CAS PubMed.
- S. K. Meena and M. Sulpizi, Langmuir, 2013, 29, 14954–14961 CrossRef CAS PubMed.
- M. Harada and H. Einaga, Langmuir, 2007, 23, 6536–6543 CrossRef CAS PubMed.
- F. Gibert, M. L. Pascal and M. Pichavant, Geochim. Cosmochim. Acta, 1998, 62, 2931–2947 CrossRef CAS.
- G. S. Pokrovski, B. R. Tagirov, J. Schott, E. F. Bazarkina, J.-L. Hazemann and O. Proux, Chem. Geol., 2009, 259, 17–29 CrossRef CAS PubMed.
- C. H. Gammons, Y. Yu and A. E. Williams-Jones, Geochim. Cosmochim. Acta, 1997, 61, 1971–1983 CrossRef CAS.
- Ö. E. Kuzugüdenli and Ç. Kantar, Erciyes Üniversitesi Fen Bilimleri Dergisi, 1999, 15, 119–127 Search PubMed.
- G. Bunker, Introduction to XAFS: A Practical Guide to X-ray Absorption Fine Structure Spectroscopy, Cambridge University Press, New York, 2010 Search PubMed.
- S. Calvin, XAFS for Everyone, CRC Press, Boca Raton, USA, 2013 Search PubMed.
- G. Meitzner, G. H. Via, F. W. Lytle and J. H. Sinfelt, J. Phys. Chem., 1992, 96, 4960–4964 CrossRef CAS.
- A. Pantelouris, G. Küper, J. Hormes, C. Feldmann and M. Jansen, J. Am. Chem. Soc., 1995, 117, 11749–11753 CrossRef CAS.
- N. Weiher, E. A. Willneff, C. Figulla-Kroschel, M. Jansen and S. L. M. Schroeder, Solid State Commun., 2003, 125, 317–322 CrossRef CAS.
- J. A. van Bokhoven, C. Louis, J. T. Miller, M. Tromp, O. V. Safonova and P. Glatzel, Angew. Chem., Int. Ed., 2006, 45, 4651–4654 CrossRef CAS PubMed.
- M. F. Lengke, B. Ravel, M. E. Fleet, G. Wanger, R. A. Gordon and G. Southam, Can. J. Chem., 2007, 85, 651–659 CrossRef CAS.
- F. Farges, J. A. Sharps and G. E. Brown Jr, Geochim. Cosmochim. Acta, 1993, 57, 1243–1252 CrossRef CAS.
- M. C. Kung, R. J. Davis and H. H. Kung, J. Phys. Chem. C, 2007, 111, 11767–11775 CAS.
- N. Weiher, E. Bus, L. Delannoy, C. Louis, D. E. Ramaker, J. T. Miller and J. A. van Bokhoven, J. Catal., 2006, 240, 100–107 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- J. Gaudet, K. K. Bando, Z. Song, T. Fujitani, W. Zhang, D. S. Su and S. T. Oyama, J. Catal., 2011, 280, 40–49 CrossRef CAS PubMed.
- Q. Ma, R. Divan, D. C. Mancini and D. T. Keane, J. Phys. Chem. A, 2008, 112, 4568–4572 CrossRef CAS PubMed.
- Y. Gründer, H. L. T. Ho, J. F. W. Mosselmans, S. L. M. Schroeder and R. A. W. Dryfe, Phys. Chem. Chem. Phys., 2011, 13, 15681–15689 RSC.
- J. F. W. Mosselmans, P. D. Quinn, A. J. Dent, S. A. Cavill, S. D. Moreno, A. Peach, P. J. Leicester, S. J. Keylock, S. R. Gregory, K. D. Atkinson and J. R. Rosell, J. Synchrotron Radiat., 2009, 16, 818–824 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. Straehle and K. P. Loercher, Z. Naturforsch., 1974, B29, 266–267 Search PubMed.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- A. Uehara, T. Hashimoto and R. A. W. Dryfe, Electrochim. Acta, 2014, 118, 26–32 CrossRef CAS PubMed.
- X. Chen, W. Chu, D. Chen, Z. Wu, A. Marcelli and Z. Wu, Chem. Geol., 2009, 268, 74–80 CrossRef CAS PubMed.
- Z. Song, J. P. L. Kenney, J. B. Fein and B. A. Bunker, Geochim. Cosmochim. Acta, 2012, 86, 103–117 CrossRef CAS PubMed.
- S. D. Kelly and B. Ravel, AIP Conf. Proc., 2007, 882, 132–134 CrossRef CAS PubMed.
- D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed.
- M. G. Blaber, M. J. Ford and M. B. Cortie, in Gold: Science and Applications, ed. C. Corti and R. Holliday, CRC Press, Boca Raton, 2010 Search PubMed.
- A. L. Ankudinov, J. J. Rehr and S. R. Bare, Chem. Phys. Lett., 2000, 316, 495–500 CrossRef CAS.
- M. Kasrai, W. N. Lennard, R. W. Brunner, G. M. Bancroft, J. A. Bardwell and K. H. Tan, Appl. Surf. Sci., 1996, 99, 303–312 CrossRef CAS.
- J. Ma, Y. Zou, Z. Jiang, W. Huang, J. Li, G. Wu, Y. Huang and H. Xu, Phys. Chem. Chem. Phys., 2013, 15, 11904–11908 RSC.
- M. Tromp, J. Moulin, G. Reid and J. Evans, X-Ray Absorption Fine Structure-XAFS13, Stanford, USA, July 2006, pp. 9–14 Search PubMed.
- A. Pantelouris, H. Modrow, M. Pantelouris, J. Hormes and D. Reinen, Chem. Phys., 2004, 300, 13–22 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13087a |
| ‡ Present address: Division of Nuclear Engineering Science, Research Reactor Institute, Kyoto University, Asashironishi, Kumatori, Osaka, 590-0494, Japan. |
| § Present address: School of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK. |
| This journal is © The Royal Society of Chemistry 2015 |