
Feasible nonaqueous route to synthesize a high-voltage spinel cathode material for lithium ion batteries†
Yi-Chun Jin and
Jenq-Gong Duh*
Department of Materials Science and Engineering, National Tsing Hua University, Hsinchu, 30013, Taiwan. E-mail: jgd@mx.nthu.edu.tw; s9931512@m99.nthu.edu.tw; Tel: +886-3-5712686
First published on 10th December 2014
Abstract
High voltage spinels are of great interest as positive electrode materials for lithium-ion batteries in recent years due to the increasing market of electric vehicles. In this study, a new organic solvent system is explored for preparing this composite material via a co-precipitation process. The use of a functional polymer surfactant significantly affects both the structure and stoichiometry of the spinel compounds in the non-aqueous batch reaction. It was revealed that the residual halides and impurities can be completely eliminated at the precursor stage. A good cycling stability was demonstrated at room temperature and a greatly improved rate capability was successfully derived from this novel system as compared with the traditional aqueous system. The superior rate performance is mainly attributed to the appreciable formation of trivalent Mn, correlating with the change of Ni/Mn ordering and crystallographic phase transformation in the detailed structural analysis.
Introduction
Facing the current challenges of worldwide energy shortage and ever-increasing environmental pollution, the lithium ion battery is expected to play a crucial role as a large-scale electrical storage device for use in electric vehicles and the modern electrical grid.1,2 In this content, the increasing demand of electrode materials with high power density has attracted much investigation recently.3 Among the various cathode candidates, nickel substituted spinel (LiNi0.5Mn1.5O4) has received extensive attention due to its high redox plateau around 4.7 V during Li-intercalation.4,5 The ultimate working potential elevates the overall energy output and increases the possibility of high power delivery.A variety of methods have been examined to prepare the cathode compound, such as solid-state reaction,6 sol–gel,7 emulsion drying,8 hydrothermal,9 molten salt,10 combustion11 and spray pyrolysis.12 Comparing with a wide variety methods, co-precipitation process is known as a preferable process for synthesizing multicomponent materials.13,14 In most cases, the precursor route of co-precipitation process is under an aqueous system.15,16 The low boiling point of water limits the reaction kinetic of metathesis below 100 °C. In addition, the hydrophilic interaction between metal salt and precipitator usually causes localized aggregation after water vaporizations, leading to nonstoichiometric products.17 Moreover, some inorganic precipitant are used in aqueous system to obtain the hydroxide precipitation, followed by repeated filtering to get rid of residual alkaline or halogen ions.18 Apart from aqueous chemistry, few methodologies have been reported for non-aqueous synthesis of multicomponent metal oxides, still less studies with deeper discussion on polymeric surfactants are used or not.
In a solvent-controlled synthetic approach, the organic solvent acts as both reactant and control agent for particle growth.19,20 The reactivity of metal oxide precursors in organic solvent is greatly decreased under the exclusion of water,21 therefore, makes it easier to control the particle sizes. The role of organic species in co-precipitation approaches is rather complex, however, it can be organized into several steps:22 (1) metathesis reaction of metal salts and reagents, (2) condensation of carboxylate groups, (3) formation of nanometric framework and (4) elimination of alkyl halide.
DMAc (dimethylacetamide) is a dipolar, aprotic solvent with great solving power for high molecular-weight polymers and good miscibility with a wide range of organic and inorganic compounds.23 The polar nature of DMAc enables it to act as a combined solvent in catalyst reactions.24 Furthermore, its boiling point (166 °C) allows reactions to be carried out at much higher temperatures without the need to operate under pressure. Polyamic acid is an intermediate product for manufacture of polyimide.25 It preserves strong hydrogen bonds and good solubility in organic solvent.
In this study, DMAc solvent is first employed in co-precipitation process to fabricate spinel compounds. The pre-mediate of polyamic acid was found necessary for synthesizing pure-phase spinel compound in the non-aqueous system. As applied precipitator, oxalic acid is a strong ligand for transition metal ion. It allows to acidizes ionic halides into volatile hydroxyl gases easily.26 Since both the surfactant and the precipitator can be easily removed through heating process, the repetitive filtration step can be eliminated. All experimental results were compared to the sample fabricated in an aqueous system, regarding morphology, crystal structure, composition and electrochemical performances.
Experimental
Synthetic procedures
Dehydrate metal chlorides (LiCl, MnCl2, and NiCl2) were used as cationic sources with a stoichiometric ratio (Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni:Mn = 2:1:3). Initially, the dark blue solution was obtained as precursor powder dissolved in a DMAc solvent. After ultrasonic vibration, the solution colour completely transferred to a transparent green. Afterward, appropriate amount of polyamic acid (M.W = 10000–50000) was added into the previous solution with continuously stirring for 1 h to assure the homogeneous reaction. Then, oxalic acid was slowly dripped into the former solution route while the temperature was kept around 150 °C. The solution eventually turned into a jelly-like substance with pale green colour. The viscous mixture was vacuum dried at 250 °C for 3 h to yield the polymeric matrix. Finally, the dried compound underwent two-step calcinations in ambient atmosphere (first step: 400 °C for 3 h, second step: 700 °C for 10 h). A prolong annealing time is sufficient for crystallization of spinel framework.27 As-fabricated samples with/without the pre-mediation of polymer are denoted as NAQ-P/NAQ, respectively. All the aforementioned process was identically repeated in the aqueous system as for comparison, whereas the resulting sample is denoted as “AQ”. Unlike DMAc system, the temperature of aqueous mixing has to be limited below 100 °C for preventing water dissipations.
:Ni:Mn = 2:1:3). Initially, the dark blue solution was obtained as precursor powder dissolved in a DMAc solvent. After ultrasonic vibration, the solution colour completely transferred to a transparent green. Afterward, appropriate amount of polyamic acid (M.W = 10000–50000) was added into the previous solution with continuously stirring for 1 h to assure the homogeneous reaction. Then, oxalic acid was slowly dripped into the former solution route while the temperature was kept around 150 °C. The solution eventually turned into a jelly-like substance with pale green colour. The viscous mixture was vacuum dried at 250 °C for 3 h to yield the polymeric matrix. Finally, the dried compound underwent two-step calcinations in ambient atmosphere (first step: 400 °C for 3 h, second step: 700 °C for 10 h). A prolong annealing time is sufficient for crystallization of spinel framework.27 As-fabricated samples with/without the pre-mediation of polymer are denoted as NAQ-P/NAQ, respectively. All the aforementioned process was identically repeated in the aqueous system as for comparison, whereas the resulting sample is denoted as “AQ”. Unlike DMAc system, the temperature of aqueous mixing has to be limited below 100 °C for preventing water dissipations.
Materials characterization
Powder X-ray diffraction (Rigaku-6000, Cu-Kα radiation) was recorded by an automated diffractometer with step size and exposure time set as 0.02° and 4 s, separately. The sample morphology was observed by In-lens thermal field emission scanning electron microscope (JSM-7600, JOEL) with high probe current energy dispersive X-ray spectrometer. Thermogravimetric analyzer (TGA-7, Perkin Elmer) was performed under air flow of 20 mL min−1 with a heating rate of 10 °C min−1. Fourier transform infrared spectroscopy analysis (Perkin-Elmer, Horiba F730) was carried out in a wavelength range from 400 to 700 cm−1 with a spectral resolution of 4 cm−1. The sample pellet was prepared by diluting the prepared powder with ten times KBr in volume. X-ray photoelectron analysis was carried out by Ulvac-PHI (PHI 1600) electron spectrometer at a base pressure in the analysis chamber of 5 × 10−10 Pa (during the measurement 1.5 × 10−9 Pa) with Mg Kα X-ray source (excitation energy hν = 1253.6 eV). The energy scale and shifts is corrected to the C 1s peak maximum at 285 eV for electrostatic charging. The instrumental resolution measured as the full width at a half-maximum (FWHM) is 1.88 eV. The integrated fitting of the recorded XPS spectra was performed, using a symmetrical Gaussian–Lorentzian curve after a Shirley-type subtraction of the background by Xpspeak41 software.Electrochemical measurement
80 wt% as-prepared materials were mixed with 13 wt% carbon black and 7 wt% polyvinylidenefluoride (PVDF) in N-methylpyrrolidinone (NMP) for fabrication of cathode electrodes. The slurry underwent 2 hour ball-milling, and was then casted onto an Al-foil afterward. The loading of as-fabricated cathode electrodes was in a range of 2.2 to 2.5 mg cm−2 for both AQ and NAQ-P. The 2032 type coin cell was assembled in an argon-filled glove box with a controlled atmosphere, whereas both H2O and O2 were under 0.1 ppm. For the half-cell test, lithium metal as negative electrodes and Celgard 2400 membrane as separators in an electrolyte mixture. The 1 M LiPF6 was dissolved in 1:2 volume ratio of ethylene carbonate (EC)/dimethyl carbonate (DMC). Arbin battery testing system (BT-2000) was employed with a constant current density ranging from 0.9 to 0.7 mA cm−2 with the cut-off voltage between 3.5 to 5 V. The electrochemical behaviour of sample was evaluated by cyclic voltammetry (Potentiostat 263A) at a voltage sweep rate of 0.1 mV s−1.
Results and discussion
Fig. 1 shows SEM image of as-calcined powder samples AQ, NAQ and NAQ-P prepared by the different solvent systems (i.e. aqueous, non-aqueous and non-aqueous with polymer assisted). A well-shaped octahedral particle was observed in both samples of NAQ-P and AQ with the comparable size around 300 nm. Nevertheless, the chunky-like powder was found in sample NAQ with an average particle size about 4 μm. The measured particle size is widely ranged from 0.5 to 10 μm with a serious powder aggregation. | ||
| Fig. 1 SEM images of as-fabricated sample: (a) NAQ, (b) AQ, and (c) NAQ-P. | ||
The composition of as-fabricated samples was confirmed by XPS analysis and the calculated Mn/Ni stoichiometry is displayed in Table 1. The calculation is based on the equation: (n1/n2) = (I1/S1)/(I2/S2), where n is atomic concentration, I is integrated peak area and S is relative sensitivity factor (R.S.F.) of elements (S = 2.66 for Mn 2p3/2 and 4.04 for Ni 2p3/2).28 The correspondence pattern with the fitting curve is provided in ESI.† The result shows that the atomic ratio of Mn/Ni in three samples are close to 3, coincidences to our original design of LiNi0.5Mn1.5O4. It was also noticed an obvious chloride contamination in sample NAQ. Yet, none of any chloride contents can be detected in both samples AQ and NAQ-P as evidenced in Fig. 2.
| Sample | Integrated peak area in elemental XPS spectrum | Atomic ratio of Mn/Ni | Relative atomic% of Cl | ||
|---|---|---|---|---|---|
| Mn 2p3/2 | Ni 2p3/2 | Cl 2p3/2 | |||
| AQ | 8510.6 | 4312.7 | 0 | 2.98 | n/a |
| NAQ | 7286.3 | 3913.5 | 353.7 | 2.82 | 7.48 |
| NAQ-P | 8903.1 | 4425.4 | 0 | 3.02 | n/a |
 | ||
| Fig. 2 XPS spectra of Cl 2p in as fabricated powder samples. | ||
The following mechanism may help to explain the variation on Cl contents. During co-precipitations, the spontaneous metathesis occurred between oxalic acid and metal salt, releasing hydrochloric acid according to the reaction: 2LiCl + NiCl2 + MnCl2 + 3H2C2O4 → Li2C2O4 + NiC2O4 + MnC2O4 + 6HCl. In DMAc solvent system, however, the undesired side reaction was produced as follows: CH3CON(CH3)2 + H2O + HCl → CH3COOH + (CH3)2NH2+Cl−. The N,N-disubstituted amide would hydrolyze in the presence of hydrochloric acid, leaving the intermediate compound “(CH3)2NH2Cl”.29 This compound is hard to remove through high temperature calcination process. Furthermore, the fore reaction easily take places if any water retains.
It is noteworthy that after mediating polyamic acid, none of any residual chlorides can be detected in the sample NAQ-P. The transformation is attributed to the intermolecular capping between metal–DMAc complex and polyamic acid in the precursor stage. A study suggested that DMAc behaves as a neutral ligand and forms a complex between metal ions proposed by reaction: MeClx + 2xDMAc(excess) → [Me(DMAc)2x]Clx + DMAc(excess).30
Due to the anionic valences of polymeric chain, carboxylic moieties of polyamic acid reacts with metal–DMAc to satisfy its cationic valencies. This process accompanied with liberating of gaseous HCl.31 The whole chelating process is illustrated in Fig. 3.
 | ||
| Fig. 3 Schematic illustration of polyamic acid capping with metal–DMAc complex in a co-precipitation route. | ||
Therefore, none of any residual chlorides was trapped in the resulting product (sample NAQ-P) after polymer pre-meditations. In addition to the remove of contamination, significant reduction of particle size was also obtained via introducing polymeric surfactants. Previous results strongly support the founding that polymers can form a barrier which hinders the particle growth and inhibits particle aggregations.32,33 The process is based on the chemical adsorption between metal oxalates and polymer surface, creating a shield to against van der Waals interactions between particles.34,35
Fig. 4 displays the X-ray diffraction pattern of powder samples fabricated by different solvent systems. Both samples of NAQ-P and AQ were indexed as a perfect cubic spinel structure (JCPDS: 32-0581) without any undesired secondary phases. Referring to the spectra derived from XPS, this result implies that the stoichiometric component – LiNi0.5Mn1.5O4 is successfully achieved. Nevertheless, the noticeable secondary phases were found in sample NAQ, together with unexpected phase separations such as Mn2O3 and LiNiOx. It is assumed that the pre-mediating of chloride impurity obscures the interatomic diffusion of metal ions during sintering process, leading to the destruction of spinel framework.
 | ||
| Fig. 4 X-ray diffraction patterns of powder sample: (a) NAQ, (b) AQ, and (c) NAQ-P. | ||
The dependence between ordered/disordered distributions of Ni/Mn atoms on the octahedral sites has been reported with a significant effect on its electrochemical properties.36,37 Spinel LiNi0.5Mn1.5O4 has two different crystallographic phases (P4332 and Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) each corresponds to the specific X-ray diffraction patterns (JCPDS #80-2184 and #32-0581). The calculated peak ratio of I111/I311 is a useful indicator to distinguish these two structures in spinel samples. The (311) diffraction peak reflects the displacement of A/B atoms in AB2O4 formula of antispinel structure,38 which suggests the tendency to form Fdm phase. As listed in Table 2, I111/I311 ratio of sample AQ and NAQ-P is 2.27 and 1.72, respectively. This indicates that the crystallographic phase of samples AQ and NAQ-P separately belongs to P4332 and Fdm. Moreover, the intensity ratio of I311/I400 reflects the degree of tetragonal distortion from cubic spinel structure.39 The phase transition of spinel LiNi0.5Mn1.5O4 from P4332 to Fdm is triggered by the creation of multi-valences Mn.40,41 The expansion of lattice parameter in sample NAQ-P is therefore associated with the numerous presence of Mn3+, due to bigger ionic radius of Mn3+ (0.65 Å) than Mn4+(0.54 Å).
m) each corresponds to the specific X-ray diffraction patterns (JCPDS #80-2184 and #32-0581). The calculated peak ratio of I111/I311 is a useful indicator to distinguish these two structures in spinel samples. The (311) diffraction peak reflects the displacement of A/B atoms in AB2O4 formula of antispinel structure,38 which suggests the tendency to form Fdm phase. As listed in Table 2, I111/I311 ratio of sample AQ and NAQ-P is 2.27 and 1.72, respectively. This indicates that the crystallographic phase of samples AQ and NAQ-P separately belongs to P4332 and Fdm. Moreover, the intensity ratio of I311/I400 reflects the degree of tetragonal distortion from cubic spinel structure.39 The phase transition of spinel LiNi0.5Mn1.5O4 from P4332 to Fdm is triggered by the creation of multi-valences Mn.40,41 The expansion of lattice parameter in sample NAQ-P is therefore associated with the numerous presence of Mn3+, due to bigger ionic radius of Mn3+ (0.65 Å) than Mn4+(0.54 Å).
| Sample | Lattice parameter (Å) | Relative intensity (normalized by I111) | Intensity ratio | |||
|---|---|---|---|---|---|---|
| I111 | I311 | I400 | I111/I311 | I311/I400 | ||
| AQ | 8.139 | 100 | 43.9 | 48.0 | 2.27 | 0.91 |
| NAQ-P | 8.199 | 100 | 57.9 | 56.7 | 1.72 | 1.02 |
| JCPDS #32-0581 | 8.173 | 100 | 55.0 | 60.0 | 1.67 | 0.92 |
| JCPDS #80-2184 | 8.170 | 100 | 38.2 | 42.6 | 2.61 | 0.89 |
To further understand the crystallographic properties of spinel samples, FTIR spectroscopy was employed in this study. It is proved to be an effective technique to differentiate ordered and disordered structures in LiNi0.5Mn1.5O4.42,43 Spinel LiNi0.5Mn1.5O4 with ordered cation occupancy exhibits a series of fingerprint bands at 432, 476, 501, 557, 588, 623, and 647 cm−1 as displayed in Fig. 5. Apparent shoulder bands at 647 and 432 cm−1 represents a higher degree of Ni/Mn ordering, associating to the P4332 phase.44 Moreover, an increased ratio of I588/I623 is also distinguished as a higher ordered Ni/Mn occupancy in spinel lattice.45 It clearly reveals a higher cation order of P4332 phase in sample AQ with respect to Fdm phase in sample NAQ-P, in consist of the result of X-ray analysis. The obvious phase transformation is correlated with the increasing amount of Mn3+ in spinel structure,46 as evidenced in the following cyclic voltammetry analysis.
 | ||
| Fig. 5 FTIR spectra of samples AQ and NAQ-P. | ||
Fig. 6(a) displays room temperature electrochemical performances of assembled NAQ-P and AQ half-cells. Both samples demonstrated a competitive capacity retention during charge–discharge in 1 C. A similar initial capacity around 128–125 mA h g−1 was delivered and losses about 15% after 250 cycles. With increasing discharge rate (1, 10, 20 C), the superior high current stability was delivered in sample NAQ-P rather than sample AQ as shown in Fig. 6(b). Sample NAQ-P outperforms about 85% initial capacity during 10 C discharging. Even at 20 C, it still remained a half of the specific discharge capacity around 60 mA h g−1. Table 3 lists the performance of LiNi0.5Mn1.5O4 cathodes in comparison with related literatures at room temperature. Among various co-precipitation routes, the optimal rate capability is revealed in this study while discharging the spinel electrode at 5 C and 10 C.
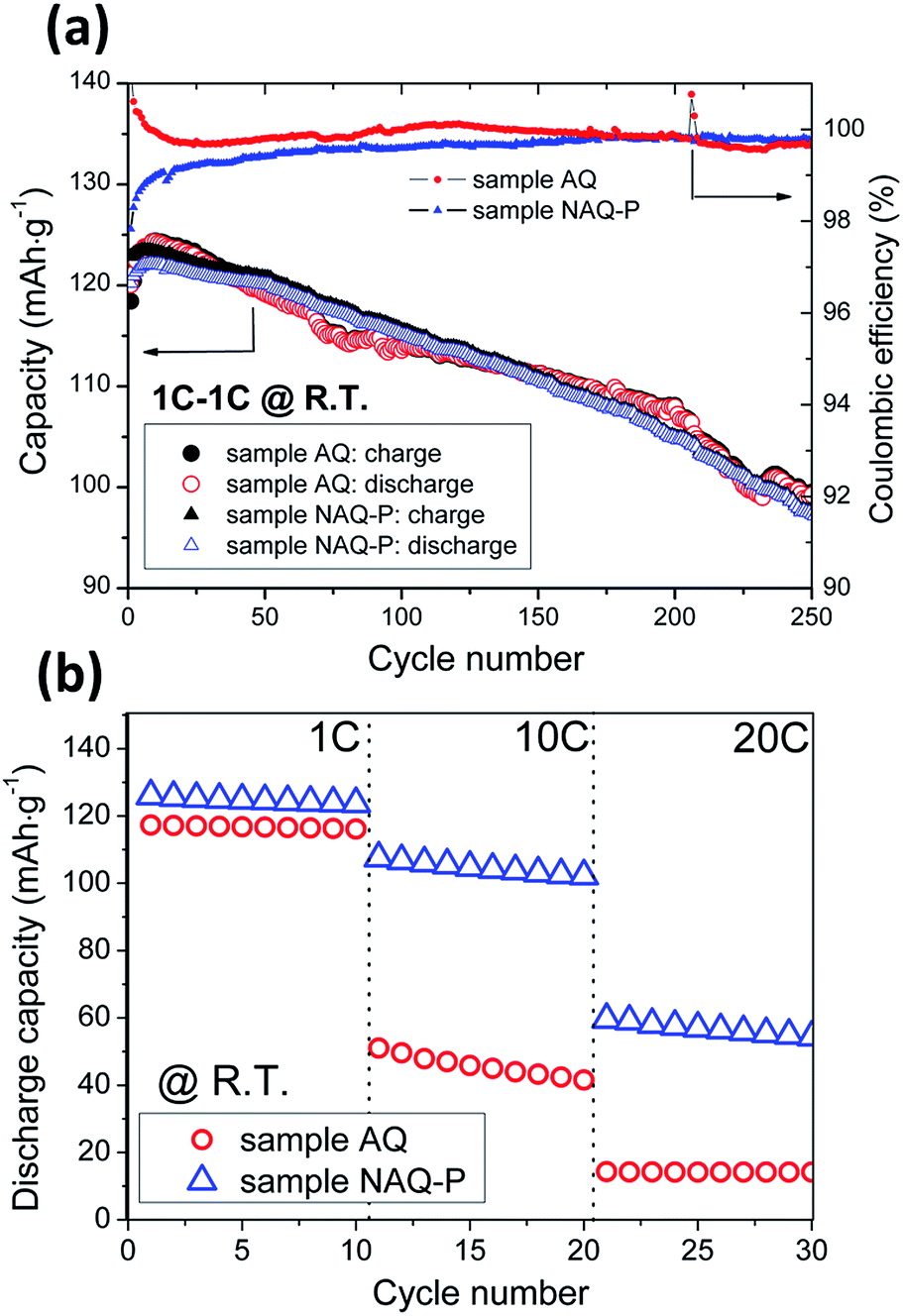 | ||
| Fig. 6 Electrochemical properties of samples AQ and NAQ-P in half-cells (a) cycling performance and (b) rate performances at room temperature. | ||
| Various co-precipitation route | Year published | Initial capacity in 1 C (mA h g−1) | C-rate capability (discharge rate: mA h g−1) |
|---|---|---|---|
| This work | 2014 | 125–120 | 5 C: 110–100 |
| 10 C: 65–60 | |||
| Ethanol33 | 2013 | 130–125 | 5 C: 90–100 |
| 10 C: 55–60 | |||
| Distilled water13 | 2010 | 132–130 | 5 C: 45–40 |
| 10 C: 25–20 | |||
| Distilled water14 | 2009 | 130–128 | 3 C: 125–110 |
| 5 C: 100–90 | |||
| Distilled water with ultrasonic-assisted16 | 2007 | 120–115 | 1 C: 120–115 |
| 2 C: 110–100 |
The cyclic voltammetry was applied for examining detail electrochemistry of assembled cells. The specialized reduction–oxidation peak of LiNi0.5Mn1.5O4 was around 4.7 V, corresponding to Ni2+/Ni4+ symmetric reaction. Additionally, an obvious redox peak at 4 V was recorded at sample NAQ-P as exhibited in Fig. 7. The small bump corresponds to Mn3+/Mn4+ symmetric reaction.47 Conclusively, the remarkable rate capability of sample NAQ-P is mainly attributed from two factors. An improved electric conductivity due to a higher Mn3+ contains and the site disorder of Ni/Mn in spinel structure.48 These two factors facilitate phase transformations during lithium intercalation which is inseparable in the spinel LiNi0.5Mn1.5O4 system.49,50
 | ||
| Fig. 7 Cyclic voltammetry of samples AQ and NAQ-P in scan rate of 0.1 mV s−1. | ||
Conclusions
The use of organic solvent in co-precipitation process is a newly adapted for synthesizing cathode compound materials. Through incorporation of specialized polymeric surfactant at the precursor stage, a well-crystallized fine powder with pure spinel phase was successfully obtained. Moreover, the residual halide impurities, such as chlorides, could be eliminated completely without additional filtrations. The adequate cycling stability is demonstrated and superior rate capability is derived as compared to the traditional aqueous system. Expand from this concept, a new class of co-precipitation method could be therefore developed. It clearly represents a feasible method for solvent control and an alternative use of polymeric surfactants. The accomplished compound product is potentially expected to be further used in functionalized lithium insertion material for future applications.Acknowledgements
The authors would like to thank the Material and Chemical Research Lab, Industrial Technology Research Institute, R.O.C. Taiwan for financially supporting this research under Contract no. 102A0127J4. The authors also thank the Fate and Transport of Environmental Contaminant Laboratory for the FTIR instrument support.References
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 CAS
.
- Z. Yang, J. Zhang, C. W. Michael, X. Lu, D. Choi, J. P. Lemmon and J. J. Liu, Chem. Rev., 2011, 111(5), 3577–3613 CrossRef CAS PubMed
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS
- R. Santhanam and B. Rambabu, J. Power Sources, 2010, 195, 5442–5451 CrossRef CAS PubMed
- M. Kunduraci, J. F. Al-Sharab and G. G. Amatucci, Chem. Mater., 2006, 18, 3585–3592 CrossRef CAS
- Z. Chen, H. Zhu, S. Ji, V. Linkov, J. Zhang and W. Zhu, J. Power Sources, 2009, 189, 507–510 CrossRef CAS PubMed
- Y. Lee, J. Mun, D. W. Kim, J. K. Lee and W. Choia, Electrochim. Acta, 2014, 115, 326–331 CrossRef CAS PubMed
- X. Zhang, F. Cheng, J. Yang and J. Chen, Nano Lett., 2013, 13(6), 2822–2825 CrossRef CAS PubMed
- X. Huang, Q. Zhang, J. Gan, H. Chang and Y. Yang, J. Electrochem. Soc., 2011, 158(2), A139–A145 CrossRef CAS PubMed
- J. H. Kim, S. T. Myung and Y. K. Sun, Electrochim. Acta, 2004, 49(2), 219–227 CrossRef CAS PubMed
- L. Zhang, X. Lv, Y. Wen, F. Wang and H. Su, J. Alloys Compd., 2009, 480, 802–805 CrossRef CAS PubMed
- S. H. Choi, Y. J. Hong and Y. C. Kang, Nanoscale, 2013, 5, 7867–7871 RSC
- D. Liu, J. Han and J. B. Goodenough, J. Power Sources, 2010, 195, 2918–2923 CrossRef CAS PubMed
- X. Fang, N. Ding, X. Y. Feng, Y. Lu and C. H. Chen, Electrochim. Acta, 2009, 54, 7471–7475 CrossRef CAS PubMed
- Y. H. Ding, H. M. Ren, Y. Y. Huang, F. H. Chang, X. He, J. Q. Fen and P. Zhang, Nanotechnology, 2013, 24, 375–401 Search PubMed
- T. F. Yi and X. G. Hu, J. Power Sources, 2007, 167, 185–191 CrossRef CAS PubMed
- Y. Fan, J. Wang, X. Ye and J. Zhang, J. Mater. Chem. Phys., 2007, 103, 19–23 CrossRef CAS PubMed
- Z. Li, N. A. Chernova, J. Feng, S. Upreti, F. Omenya and M. S. Whittingham, J. Electrochem. Soc., 2012, 159, A116–A120 CrossRef CAS PubMed
- M. Niederberger and G. Garnweitner, Chem.–Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed
- Y. W. Jun, J. S. Choi and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 3414–3439 CrossRef CAS PubMed
- M. Karmaoui, M. G. Willinger, L. Mafra, T. Herntricha and N. Pinna, Nanoscale, 2009, 1, 360–365 RSC
- X. R. Ye, D. Z. Jia, J. Q. Yu, X. Q. Xin and Z. Xue, Adv. Mater., 1999, 11, 941–942 CrossRef CAS
- A. Potthast, T. Rosenau, R. Buchner, T. Röder, G. Ebner, H. Bruglachner, H. Sixta and P. Kosma, Cellulose, 2002, 9(1), 41–53 CrossRef CAS
- G. Pistoia and B. Scrosati, Ric. Sci., 1967, 37, 1173–1175 CAS
- H. Yamada, M. Fukudome and N. Egawa, Method for Producing Polyimide Film, US6264866, 2001 Search PubMed
- H. Fang, L. Li and G. Li, J. Power Sources, 2007, 167, 223–227 CrossRef CAS PubMed
- N. M. Hagh and G. G. Amatucci, J. Power Sources, 2010, 195, 5005–5012 CrossRef CAS PubMed
- B. V. Cris, Handbooks of Monochromatic XPS Spectra: The Elements and Native Oxides, XPS International LLC: Mountain View, CA, USA, 2004, vol. 1 Search PubMed
- S. Zen and E. Kaji, Org. Synth., 1977, 57, 60 CrossRef CAS
- L. T. Taylor and R. K. Boggess, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 685 CrossRef
- C. Kundnani, A. K. Gupta, M. Arya, D. Shrivastava, P. Aseri, J. Keller and R. Bajpai, Polym. Eng. Sci., 2009, 49, 977–983 CAS
- Y. C. Jin and J. G. Duh, Mater. Lett., 2013, 93, 77–80 CrossRef CAS PubMed
- W. Liu, G. C. Farrington, F. Chaput and B. Dunn, J. Electrochem. Soc., 1996, 143(3), 879–884 CrossRef CAS PubMed
- J. Huang, N. Matsunaga, K. Shimanoe, N. Yamazoe and T. Kunitake, Chem. Mater., 2005, 17, 3513–3518 CrossRef CAS
- J. C. Arrebola, A. Caballero, M. Cruz, L. Hernán, J. Morales and E. R. Castellón, Adv. Funct. Mater., 2006, 16, 1904–1912 CrossRef CAS
- S. Ivanova, E. Zhecheva, R. Stoyanova, D. Nihtianova, S. Wegner, P. Tzvetkova and S. Simova, J. Phys. Chem. C, 2011, 115, 25170–25182 CAS
- J. Zheng, J. Xiao, X. Yu, L. Kovarik, M. Gu, F. Omenya, X. Chen, X. Q. Yang, J. Liu, G. L. Graff, M. S. Whittingham and J. G. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 13515–13521 RSC
- J. Feng, Z. Huang, C. Guo, N. A. Chernova, S. Upreti and M. S. Whittingham, ACS Appl. Mater. Interfaces, 2013, 5, 10227–10232 CAS
- Z. Bai, N. Fan, Z. Ju, C. Sun and Y. Qian, Mater. Lett., 2012, 76, 124–126 CrossRef CAS PubMed
- J. Xiao, X. Chen, P. V. Sushko, M. L. Sushko, L. Kovarik, J. Feng, Z. Deng, J. Zheng, G. L. Graff, Z. Nie, D. Choi, J. Liu, J. Zhang and M. S. Whittingham, Adv. Mater., 2012, 24, 2109–2116 CrossRef CAS PubMed
- H. Seyyedhosseinzadeh, F. Mahboubi and A. Azadmehr, Electrochim. Acta, 2013, 108, 867–875 CrossRef CAS PubMed
- K. Ariyoshi, Y. Iwakoshi, N. Nakayama and T. Ohzuku, J. Electrochem. Soc., 2004, 151, A296–A303 CrossRef CAS PubMed
- J. H. Kim, C. S. Yoon, S. T. Myung, J. Prakash and Y. K. Sun, Electrochem. Solid-State Lett., 2004, 7(7), A216–A220 CrossRef CAS PubMed
- T. F. Yi and Y. R. Zhu, Electrochim. Acta, 2008, 53, 3120–3126 CrossRef CAS PubMed
- X. Ma, B. Kang and G. Ceder, J. Electrochem. Soc., 2010, 157(8), A925–A931 CrossRef CAS PubMed
- Y. C. Jin, C. Y. Lin and J. G. Duh, Electrochim. Acta, 2012, 69, 45–50 CrossRef CAS PubMed
- D. Liu, W. Zhu, J. Trottier, C. Gagnon, F. Barray, A. Guerfi, A. Mauger, H. Groult, C. M. Julien, J. B. Goodenough and K. Zaghib, RSC Adv., 2014, 4, 154–167 RSC
- M. Kunduraci and G. G. Amatucci, J. Electrochem. Soc., 2006, 153(7), A1345–A1352 CrossRef CAS PubMed
- J. Xiao, X. Chen, P. V. Sushko, M. L. Sushko, L. Kovarik, J. Feng, Z. Deng, J. Zheng, G. L. Graff, Z. Nie, D. Choi, J. Liu, J. G. Zhang and M. S. Whittingham, Adv. Mater., 2012, 24(16), 2109–2116 CrossRef CAS PubMed
- J. H. Kim, S. T. Myung, C. S. Yoon, S. G. Kang and Y. K. Sun, Chem. Mater., 2004, 16, 906–914 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09334h |
| This journal is © The Royal Society of Chemistry 2015 |