
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mass-analyzed-threshold-ionization (MATI) spectroscopy of 1,2,3-substituted halogenated benzenes via different intermediate vibrational states in the S1 state
Sascha
Krüger
,
Frank
Witte
,
Jan
Helfrich
and
Jürgen
Grotemeyer
*
Institut für Physikalische Chemie, Christian-Albrechts-Universität Kiel, Max-Eyth Str. 1, 24118 Kiel, Germany. E-mail: grote@phc.uni-kiel.de
First published on 25th November 2014
Abstract
For the first time, two color resonant mass analyzed threshold ionization (MATI) spectroscopy has been applied in order to investigate the ionic properties of 1,3-dichloro-2-fluoro-benzene (1,3,2-DCFB) and 1,3-difluoro-2-chloro-benzene (1,3,2-DFCB) radical cations in their electronic ground state. The ionic ground state of the different samples has been investigated via different S1 intermediate states and compared to 1,2,3-trichlorobenzene measured in previous work. Additionally quantum chemical calculations at DFT (density functional theory) and TDDFT (time-dependent density functional theory) level of theory have been performed to support experimental findings. From the MATI spectra the adiabatic ionization energies of 1,3-dichloro-2-fluorobenzene and 1,3-fluoro-2-chlorobenzene could be determined to be 75.242 ± 6 cm−1 and 75.627 ± 6 cm−1, respectively. Several vibrational modes of both compounds have been assigned by comparison of the experimental and theoretical results.
1. Introduction
Halogenated aromatic molecules became a topic of great interest due to their widespread presence in industrial processes and hence presence in the environment as potentially toxic and cancerogenic pollutants. Moreover halogenated benzenes are important model substances to validate theoretical concepts, such as vibronic coupling and others. As one of the spectroscopically best-investigated molecules of all, benzene can serve as an outstanding reference system to study the influence of tailored perturbations, such as careful choice of substitution pattern, to the (ro)vibronic structure. As an immediate effect a shift in transition energies, ionization energy, molecular geometry and vibrational frequencies can be observed. The characteristics of these effects strongly depend on the number, type and localization of the substituents. As the magnitude of these effects are rather small (from a few wavenumbers up to a hundred wavenumbers), high resolution techniques, such as mass analyzed threshold ionization (MATI) or zero kinetic energy (ZEKE) spectroscopy are ideally suited to investigate such phenomena.1–4 Chloro- and fluoro-benzenes have been subject to various studies investigating the vibronic properties of the first electronic excited state (S1) as well as cationic ground state (D0).1,5–13 In this paper we will focus particularly on out-of-plane b1-symmetric modes. Modes of this symmetry are connected to interesting phenomena observed in a whole variety of halobenzenes. For example, difluorobenzenes exhibit large frequency lowering of b1 (b3u under D2h in case of p-DFB) symmetric modes in going from S0 to S1 or from D0 to S1,14 respectively. The intensity gain of this symmetry forbidden mode was interpreted in terms of the pseudo-Jahn–Teller effect (PJTE). The PJTE is based on the idea that a geometrical distortion blurs the difference in symmetry between two or more electronic states of virtually equal energy. In 1,2,4,5-tetrafluorobenzene (D2h) it was found that the b2g-symmetric mode 11 shows an unusual strong activity in the form of a long progression (v2n11). Recent results show that the planarity of this molecule is distorted along the eigenvector of the 11 mode during excitation to 1B2uS1 state. It is an interesting question to elucidate if these phenomena can be extended to further congeners and identified as a general feature. In particular in the case of the molecular geometry change it is crucial to determine the involved vibrational modes and participating electronic states. For this purpose, the experimental data are compared to theoretical calculations, which enable the possibility of assigning the observed vibrational modes. In this paper we present mass-selected two color two photon Resonance Enhanced Multi Photon Ionization (2C2P-REMPI) spectra of the S1 ← S0 transition and MATI spectra via different S1-intermediate states of 1,3-dichloro-2-fluorobenzene and 1,3-dichloro-2-fluorobenzene. Some assignments of 1,2,3-trichlorobenzene reported previously1 were reconsidered and compared to the newly gained knowledge.2. Experimental setup
The experimental setup consists of a homemade time-of-flight (TOF) mass spectrometer as described in detail previously.1,15,16 Briefly, the spectrometer consists of a standard second order corrected reflectron time-of-flight mass spectrometer equipped with single stage ion source. The laser system used for excitation and ionization consists of two dye lasers (Laser Analytical Systems LDL 205, Lambda Physics FL 3002). Each dye laser is pumped by a dedicated Nd:YAG laser (Lumonics HY-1200, Continuum Surelite II). A Quantum Composers 9600+ delay pulse generator fed by an external clock operated at 10 Hz controls flash lamp and Q-switch delays. The output of each dye laser is frequency-doubled by a BBO-I crystal yielding tunable ranges from 245 to 285 nm. Wavelength calibration of both dye lasers is performed by recording an opto-galvanic spectrum with a neon hollow cathode lamp yielding accuracy better than 2 cm−1. Initially a supersonic molecular beam of sample molecules and seed gas (argon) with a backing pressure of approximately 2 bar is expanded via a pulsed jet valve (General Valves Series 9) into the ion source. To obtain a sufficient vapor pressure the sample is heated to approximately 80 °C. Excitation or ionization is accomplished by multi photon absorption under field-free conditions. In the MATI modus, promptly generated ions are discriminated against Rydberg neutrals through the application of a weak electrical field of 1–2 V cm−1 by a subsequent delay (∼100 ns) to multi-photon excitation. Finally, a high voltage pulse (890 V cm−1) switched by a Behlke HS56-01 fast thyristor ionizes the Rydberg neutrals and accelerates them into the mass spectrometer. In REMPI modus, the generated ions are accelerated directly into the TOF without the retarding field. The ion signal is detected by a conventional dual micro-channel-plate detector and transferred to a LeCroy 534M digital oscilloscope. A computer, linked to the oscilloscope by a GPIB connection performs the data acquisition and processing. The 123-DCFB was purchased from Aldrich, 126-DFCB from ABCR and were used without further purification.3. Quantum-chemical calculations
Quantum chemical calculations were performed in order to assign the observed vibrational bands and to support the experimental findings. The software package Turbomole17–20 was used for all quantum chemical calculations. Geometry optimizations and subsequent frequency analyses for molecules in the electronic ground state (S0) and the cationic ground state (D0) were conducted at the density functional theory (DFT) level of theory with both the gradient corrected functional BP86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21a and the hybrid functional B3LYP.21b The triple basis set TZVPP21c has been applied to all DFT calculations. Calculations for the S1 were done analogously using the time dependent density functional theory (TDDFT). For comparison reasons, geometry calculations and frequency analyses were also treated at the coupled cluster (CC2) level of theory for the S0- and S1-state using the Ahlrichs basis set cc-pVTZ.21d The nomenclature used for assignment of vibrational bands is according to Varsanyi and Szoke,22 which is derived from Wilson's notation of the Benzene modes.23
21a and the hybrid functional B3LYP.21b The triple basis set TZVPP21c has been applied to all DFT calculations. Calculations for the S1 were done analogously using the time dependent density functional theory (TDDFT). For comparison reasons, geometry calculations and frequency analyses were also treated at the coupled cluster (CC2) level of theory for the S0- and S1-state using the Ahlrichs basis set cc-pVTZ.21d The nomenclature used for assignment of vibrational bands is according to Varsanyi and Szoke,22 which is derived from Wilson's notation of the Benzene modes.23
We refrained from the use of scaling factors to fit the calculated frequencies.
4. Experimental results
4.1. 1,3-Dichloro-2-fluorobenzene (1,3,2-DCFB)
460 ± 2 cm−1.
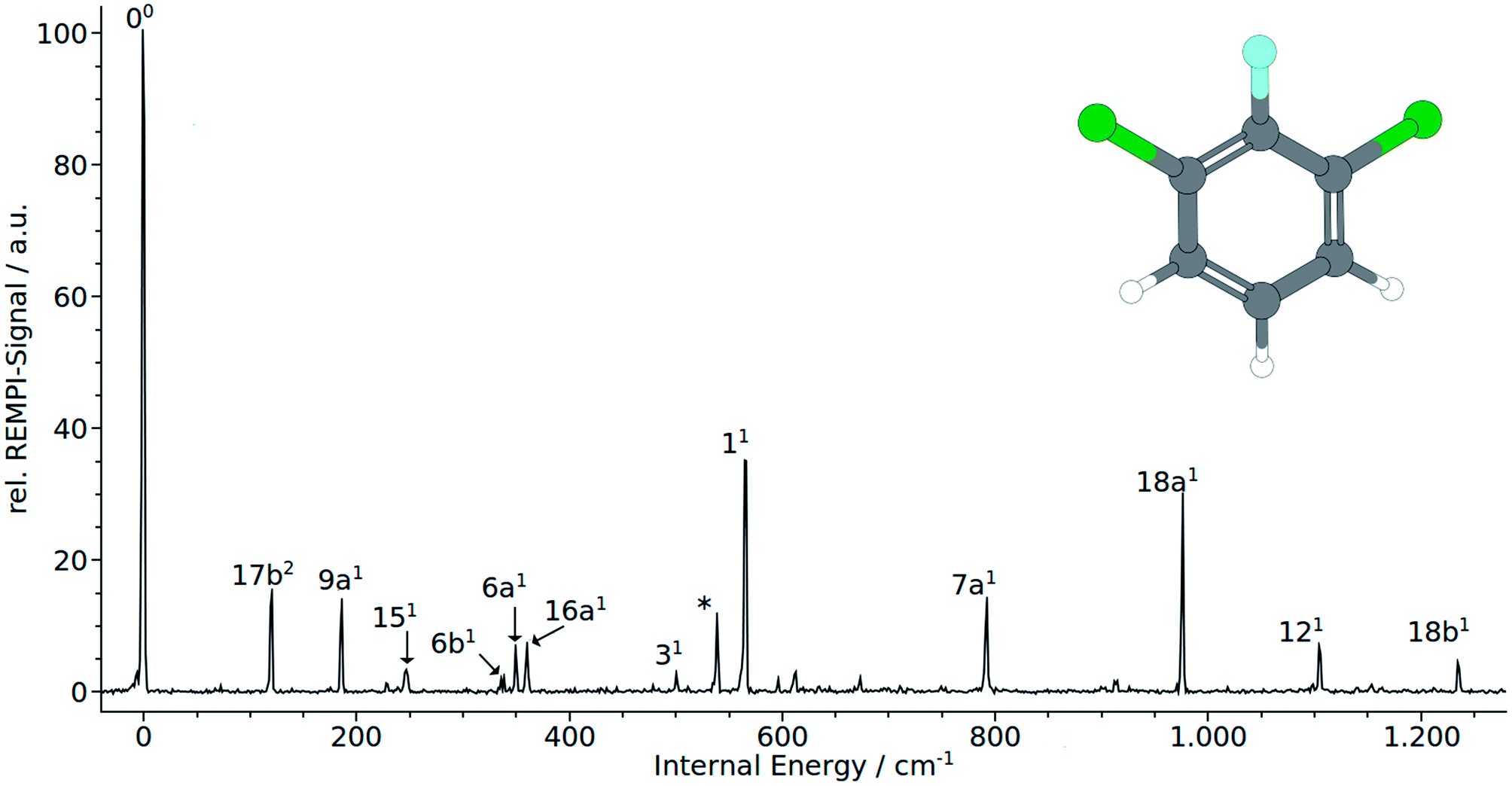 | ||
| Fig. 1 (1 + 1′) REMPI spectrum of 1,3-dichloro-2-fluoro-benzene (1,3,2-DCFB). | ||
Normal coordinate analysis for 1,3,2-DCFB yields following distribution of fundamental modes under C2v symmetry: Γvib = 11 × a1, 10 × b2, 3 × a2, 6 × b1. Clearly, the spectrum is dominated by the total symmetric modes 11 (565 cm−1) and 18a (977 cm−1). Moreover the a1-symmetric modes 9a1, 6a1, 7a1, 121 (186 cm−1, 349 cm−1, 792 cm−1, 1104 cm−1) could be identified with lower intensity. With 16a1 (360 cm−1) also an a2-symmetric mode could be assigned. As expected, no modes of b1-symmetry could be assigned to the spectrum in accordance with selection rules. Particular attention should be paid to the low frequency band at 120 cm−1. A band with such a low frequency can with certainty be assigned to an out-of-plain mode. Based on correlation with MATI spectra we assigned the first even overtone of the b1-symmetric mode 17b to this band. The frequency of 52 cm−1 calculated with the coupled cluster method is in reasonable agreement with the experimental value for the half of the overtone for 17b2. The bands at 247 cm−1 (151), 337 cm−1 (6b1), 500 cm−1 (31) and 1236 cm−1 (18b1) were assigned to b2-symmetric modes. The latter two experimental values are in excellent agreement with both calculated values (TDDFT, CC2) for the first excited singlet state of 1,2,3-DCFB. Comparing the calculated values for the 151 and 6b1 mode a major deviation becomes apparent, just like in the case of 17b. All performed calculations show good agreement with the experiment for the modes 9a1, 6a1, 11, 7a1, 18a1, 121, 16a1, whereas the best results were obtained with the CC2 level of theory (see Table 1).
| S0(C2v) | S1 | (C2v) | (C2v) | (CS) | D0 | (C2v) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wilson | Sym. | exp. | B3LYP | BP86 | CC2 | exp. | B3LYP | BP86 | CC2 | exp. | B3LYP | BP86 |
| a IR-band.24 b From combination band. c From overtone. | ||||||||||||
| 9a | a1 | 256a | 192 | 186 | 189 | 186 | 241 | 232 | 183 | 190 | 195 | 189 |
| 6a | a1 | 376a | 379 | 369 | 380 | 349 | 355 | 361 | 343 | 361 | 381 | 372 |
| 1 | a1 | 597a | 601 | 584 | 596 | 565 | 538 | 522 | 558 | 578 | 603 | 586 |
| 7a | a1 | 803a | 839 | 813 | 829 | 792 | 831 | 815 | 791 | 798 | 820 | 797 |
| 18a | a1 | 1058a | 1092 | 1061 | 1080 | 977 | 1013 | 998 | 991 | 1047 | 1027 | |
| 12 | a1 | 1112a | 1116 | 1083 | 1124 | 1104 | 1077 | 1046 | 1079 | 1143 | 1104 | |
| 13 | a1 | 1205a | 1277 | 1240 | 1277 | 1314 | 1320 | 1242 | 1339 | 1295 | ||
| 19a | a1 | 1462a | 1490 | 1444 | 1485 | 1213 | 1229 | 1383 | 1459 | 1412 | ||
| 8a | a1 | 1572a | 1613 | 1564 | 1608 | 1509 | 1476 | 1512 | 1618 | 1567 | ||
| 20a | a1 | 3084a | 3189 | 3119 | 3220 | 3159 | 3080 | 3210 | 3197 | 3126 | ||
| 2 | a1 | 3212 | 3142 | 3243 | 3220 | 3147 | 3246 | 3218 | 3147 | |||
| 10a | a2 | 215a,b | 208 | 201 | 207 | 120 | 123 | 71 | 174 | 180 | 173 | |
| 16a | a2 | 530a,b | 553 | 534 | 518 | 360 | 458 | 495 | 313 | 512 | 498 | 476 |
| 17a | a2 | 894a | 921 | 876 | 890 | 1045 | 1060 | 616 | 936 | 897 | ||
| 17b | b1 | 115a,b | 107 | 103 | 107 | 60c | 31 | 38 | 52 | 95 | 88 | 85 |
| 10b | b1 | 260a,b | 282 | 271 | 278 | 270c | 263 | 259 | 178 | 264 | 274 | 265 |
| 16b | b1 | 513a | 533 | 512 | 542 | 502 | 454 | 364 | 452 | 455 | 441 | |
| 4 | b1 | 705a | 732 | 699 | 680 | 766 | 755 | 460 | 732 | 743 | 715 | |
| 11 | b1 | 770a | 794 | 758 | 771 | 847 | 827 | 601 | 678 | 826 | 792 | |
| 5 | b1 | 965a | 983 | 937 | 949 | 1004 | 966 | 782 | 1009 | 966 | ||
| 15 | b2 | 279a | 258 | 248 | 252 | 247 | 62 | 61 | 227 | 242 | 233 | 253 |
| 6b | b2 | 401a | 404 | 393 | 401 | 337 | 202 | 209 | 319 | 331 | 300 | 331 |
| 3 | b2 | 539a | 545 | 528 | 538 | 500 | 500 | 498 | 519 | 471 | 513 | 504 |
| 7b | b2 | 829a | 798 | 779 | 810 | 698 | 683 | 759 | 827 | 812 | 796 | |
| 9b | b2 | 1154a | 1180 | 1145 | 1170 | 1177 | 1144 | 1105 | 1083 | 1104 | ||
| 18b | b2 | 1205a,b | 1233 | 1193 | 1223 | 1236 | 1250 | 1185 | 1170 | 1330 | 1192 | |
| 14 | b2 | 1255a | 1313 | 1322 | 1421 | 1413 | 1360 | 1497 | 1226 | 1296 | ||
| 19b | b2 | 1446a | 1481 | 1432 | 1465 | 1446 | 1407 | 1389 | 1514 | 1466 | ||
| 8b | b2 | 1583a | 1615 | 1566 | 1608 | 1574 | 1545 | 1683 | 1371 | 1384 | ||
| 20b | b2 | 3084a | 3207 | 3137 | 3235 | 3188 | 3177 | 3141 | 3215 | 3144 | ||
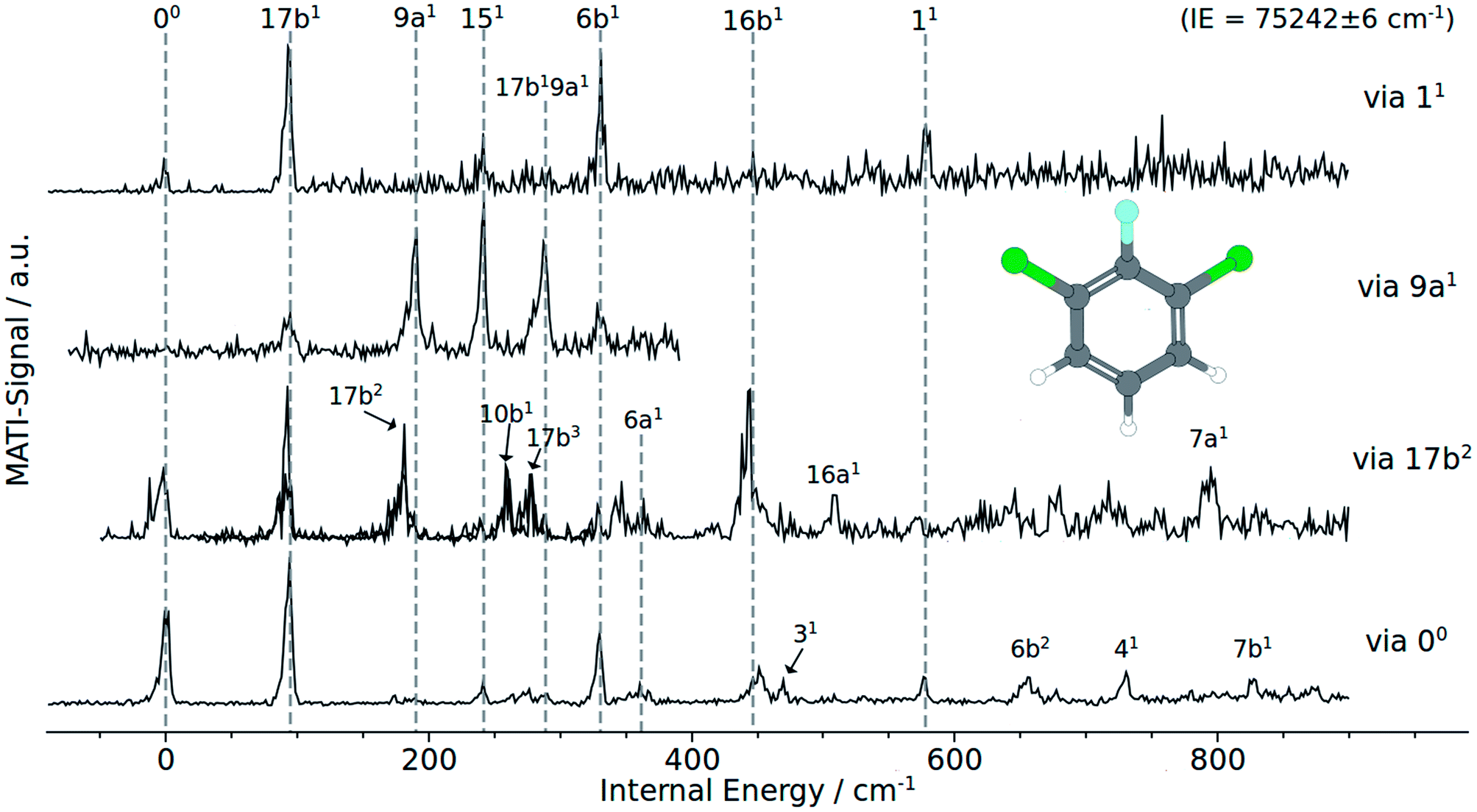 | ||
| Fig. 2 MATI spectra of 1,3,2-DCFB of different vibrational intermediate states of the first excited electronic state: (a) via 11, (b) via 9a1, (c) via 17b2, (d) via the electronic origin (00). | ||
Besides the 00 transition, the MATI spectrum obtained via the electronic origin exhibits several more additional resonances. The most prominent peak in the spectrum is the band assigned to the 17b mode, which is constituting a violation of the Δv = 0 propensity rule. Additionally, the mode 17b1 at 95 cm−1 appears as a combination band (17b19a1) at 288 cm−1. Moreover the spectrum is conspicuously rich in b1-symmetric modes. In addition to 17b1 we assigned the 10b1 (264 cm−1), 16b1 (452 cm−1), 111 (678 cm−1) and 41 (732 cm−1). Another out-of-plane vibration (10a1) with a2-symmetry and low intensity has been assigned to the band at 174 cm−1. The bands at 361 cm−1, 578 cm−1 have been assigned to the total symmetric modes 6a1 and 11, respectively. b2 symmetric modes were identified at 242 cm−1 (151), 331 cm−1 (6b1), 471 cm−1 (31) and 827 cm−1 (7b1). The band at 657 cm−1 fits the overtone 6b1.
The MATI spectrum obtained via the S117b2 shows a short, three-membered regular progression with transitions corresponding to the excitation of one, two and three quanta. In contradiction to the Δv = 0 propensity rule, the band labeled 17b1 is the most prominent peak in the spectrum. It is notable that the 17b mode also appears in the combination bands 17b19a1 (288 cm−1) and 16b16b1 (423 cm−1). Also apparent is the richness in b1-symmetric modes, first and foremost the intense 16b1 (448 cm−1), but also the 10b1 (264 cm−1), 111 (680 cm−1), 41 (728 cm−1). The MATI spectrum obtained via the S19a1 mode also shows a breakdown of the Δv = 0 propensity rule. Not the vertical transition into the D09a1 state is the dominating one, but the band at 242 cm−1 (151). In addition the 17b mode appears again, also as a combination in 17b19a1. A further band at 331 cm−1 could be identified with 6b1. The measured values are in good accordance with the calculated values (195 and 189 cm−1 for the 9a1, 233 and 253 cm−1 for the151 and also 300 and 331 cm−1 for the 6b1).
The MATI spectrum via S111 continues the series of MATI spectra characterized by a breakdown of the Δv = 0 propensity rule in favor for the vibronic transition into the D017b1 state. Further active modes that could be assigned in the recorded range up to 750 cm−1 are the 151 (241 cm−1), 6b1 (331 cm−1) and 11 (578 cm−1). The experimentally determined frequency for the 11 mode is in good accordance with the calculated values of 578 cm−1 or 603 cm−1 respectively. It should be noticed that, owing to the heavy substituents, the displacement pattern of the ‘ring-breathing-mode’ shows a striking deviation from the original benzene pattern. It resembles clearly the pattern of mode 6a found for benzene. The assignment of the band at 578 cm−1 to the mode 6a is excluded since it was already doubtlessly assigned to the band at 361 cm−1.
4.2. 1,3-Difluoro-2-chloro-benzene (1,3,2-DFCB)
449 ± 2 cm−1.
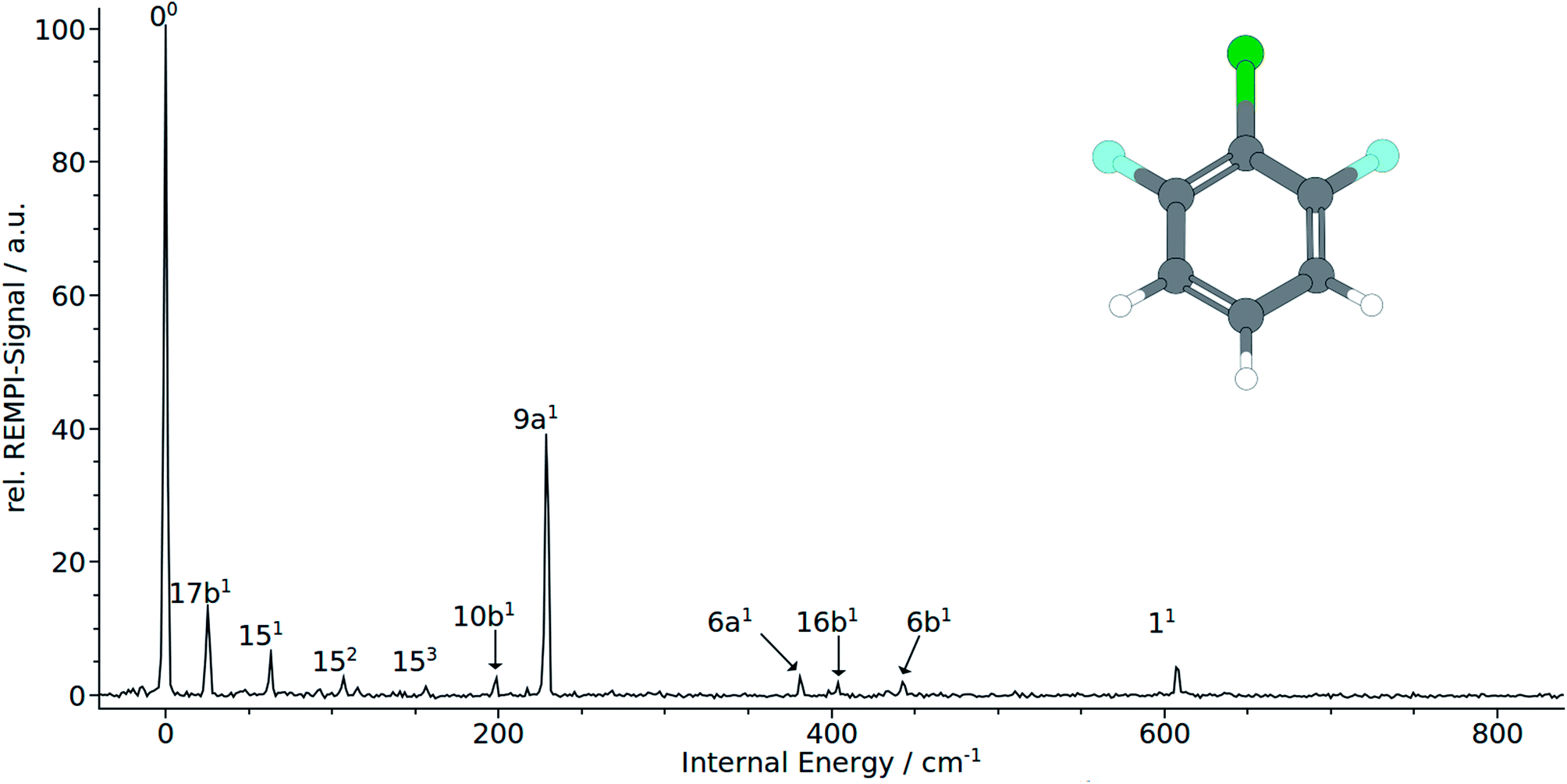 | ||
| Fig. 3 (1 + 1′) REMPI spectrum of 1,3-difluoro-2-chloro-benzene (1,3,2-DFCB). | ||
The electronic origin and the total symmetric mode 9a1 at 228 cm−1 dominates clearly the measured REMPI spectrum. It should be noted that the remaining bands at 381 cm−1 and 607 cm−1 that were both assigned to the total symmetric modes 61 and 11, respectively. Quite unusual for total symmetric modes we found poor consistency between calculated and observed bands: calculation substantially underestimate the 6a1 and 11 with 188 cm−1 to 202 cm−1 and 488 cm−1 to 517 cm−1, respectively. The 9a1 has been overestimated with 364 cm−1 to 374 cm−1. Nevertheless the correlation with MATI spectra backups the assignment of the total symmetric modes. Moreover, the calculations suggest the mode 7a1, that cannot be seen in the spectrum, to appear in the spectrum between 710 cm−1 and 750 cm−1. As expected, no modes of a2 symmetry could be assigned to the spectrum in accordance with selection rules. In contrast to 1,2,3-TCB and 1,3,2-DCFB, transitions to b1-symmetric modes are allowed in 1,3,2-DFCB. The bands at 25, 98 and 404 cm−1 could be identified with the b1-symmetric modes 17b1, 10b1 and 16b1. In this case, the calculated frequencies are in good agreement with the observed ones. The 17b1 has been predicted to appear between 31 cm−1 and 51 cm−1, the 10b1 between 176 cm−1 and 215 cm−1 and the 16b1 between 351 cm−1 and 428 cm−1. With 151 and 6b1 two a2-symmetric modes could be assigned. Noteworthy is the appearance of the 15 mode as a short, three-membered progression of overtones (151, 152, 153). The measured frequencies of 443 cm−1 for 6b1 and 63 cm−1 for 151 are well reproduced by the B3LYP calculation with 439 and 64 cm−1. The BP86 calculation predicts the 151 correctly with 67 cm−1, while it slightly overestimates the 6b1 with 463 cm−1. The CC2 calculation gives good results in predicting the 6b1 with 439 cm−1, while it overestimates the 151 with 154 cm−1.
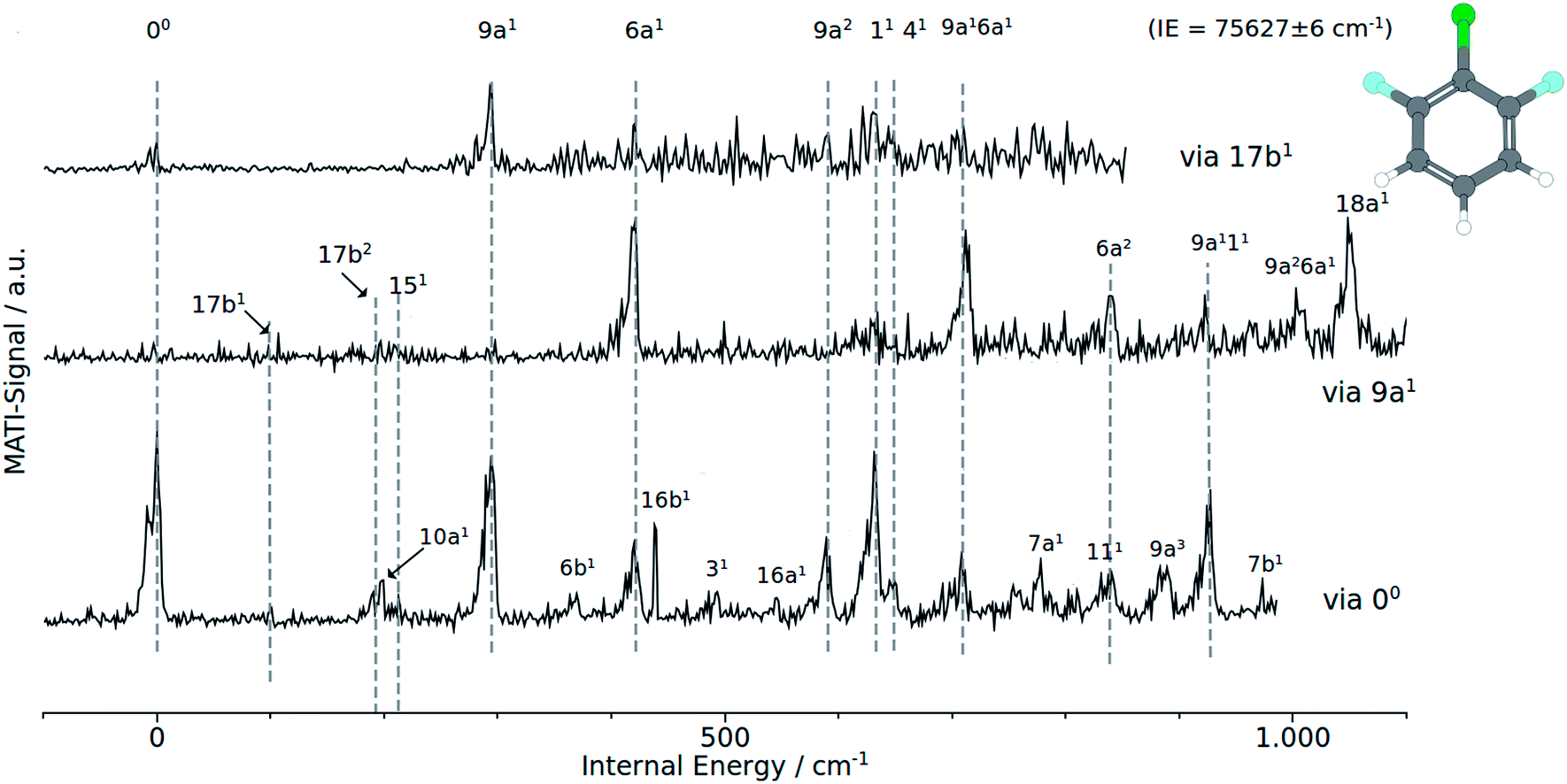 | ||
| Fig. 4 MATI spectra of 1,2,6 DCFB of different vibrational intermediate states of the first excited electronic state: (a) via 9a1, (b) via 17b2, (c) via the electronic origin (00). | ||
In accordance with the Δv = 0 propensity rule, the MATI spectrum obtained via the electronic origin is dominated by the 00-band. Furthermore the spectrum is characterized by strong activity of the two total symmetric modes 9a1 and 6a1. Within the recorded range of 1000 cm−1, the spectrum exhibits a progression in 9a composed of the first three overtones 9a1 (293 cm−1), 9a2 (588 cm−1) and 9a3 (882 cm−1). The 6a appears as ground vibration 6a1 (418 cm−1), first overtone 6a2 (839 cm−1) as well as combination vibration 9a16a1 (706 cm−1).
Additionally we assigned the band at 925 cm−1 to a combination band of modes 9a111. The relative intensive band at 630 cm−1 has been identified with the mode 11. The shoulder at 649 cm−1 in the peak labeled with 11 has been assigned to the mode 41. Exhibiting a similar weak intensity as the 41, another b1-symmetrical mode (7a1) could be identified at 778 cm−1. In the view of previous work,1 the assignment of the bands at 95 cm−1 and 196 cm−1 to the modes 17b1 and 17b2 respectively seems appropriate. Assignments that are more tentative are the 16b1 and 111 to the bands at 439 cm−1 and 839 cm−1. The a2-symmetrical modes 10a1 and 16a1 are assigned to the weak bands at 201 cm−1 and 546 cm−1. With exception of 41 all calculated frequencies are in good accordance with the experimentally determined ones.
Provided the validity of the Δv = 0 propensity rule, the MATI spectrum obtained via the S117b1 mode ought to be characterized by a towering 17b1 band. Contrary to expectations, the band 17b1 could not be observed in the spectrum shown in Fig. 4. Instead, the spectrum shows the 9a1 at 291 cm−1 as the most intensive resonance absorption. As can be seen from the correlation between the MATI spectra in Fig. 4, the first overtone 9a2 appears at 586 cm−1 as well. The signals at 419 cm−1 and 629 cm−1 are identified as the 6a1 and 11 total symmetric modes. Obviously, the signals for the 11-vibration is broadened. Based on the MATI spectrum of the 00 transition, we assigned this signal both to the 11 and 41 vibrations, the latter of which gives rise to the shoulder. The MATI spectrum obtained via the S19a1 shows three prominent peaks: the 6a1, the 9a16a1 and the 18a1. Without folding the spectrum with the laser power, no absolute statement can be made in regards to the peak intensities. The three peaks are of nearly equal intensity (approx. 5% difference), so it is hard to tell which one is the highest in intensity. Nevertheless it can be stated that in contrary to the predictions of the propensity rule, transitions to 6a1 and 9a16a1 are at least as same as intensive while the vertical transition into the D09a1 state is scarcely visible in the spectrum. Nevertheless the mode 9a appears in a series of overtones 9a16a1 (710 cm−1), 9a111 (924 cm−1) and 9a26a1 (1003 cm−1). Remarkably, the spectrum exhibits a strong activity in 6a modes: the bands assigned to the modes 6a1 and 9a16a1 are among the three most intense bands in the spectrum. The 6a1 vibration shows progression activity (6a1 and 6a2 in the recorded range). In addition, the 6a1 mode contributes to overtone bands as already discussed above. The weak features at 97, 193 and 213 cm−1 have been assigned on the basis of our previous studies7,8 to the out-of-plane modes 17b1, 17b2 and in-plane mode 151.
5. Discussion
5.1. 1,3,2-DCFB
Compared to electronic and cationic ground states the 17b exhibits a drastically decreased frequency in the first electronic excited state (see Table 1). Such a frequency lowering suggests a geometrical distortion along the eigenvector of 17b in going from S0 to S1 or from D0 to S1, respectively. As considered earlier by Tsuchiya et al.14 for difluorobenzene it is highly suggested that this phenomenon is the result of strong vibronic coupling between the S1 and nearby states. But the most important indication for a such a distortion shows up in the MATI spectra via the electronic origin and, first and foremost, via the 17b2. The latter exhibits a three-membered progression of the mode 17b. Not just that this progression shows a shift of the Franck–Condon maximum between S1 and D0, the fact that the transition from D017b1 ← S117b2 is favored could be interpreted as a replanarisation of the molecular geometry in going from S1 to D0 along that mode.Also during ionization via the vibrationless level of the first excited state, the transition D017b1 ← S00 is clearly favored over D000 ← S100 (see MATI via 00Fig. 2). A major role of the 17b mode during ionization is aggravated by the fact the 17b also appears as combination bands (17b19a1 in each of the recorded MATI spectra, 17b16b1 in the MATI via 17b2). The strong activity of the mode 16b1 in the MATI via 17b2 and the general, unusual appearance of multiple other out-of-plane modes throughout the MATI spectra give rise to the assumption that the geometry in the S1 is a product of a distortion along several modes. The CC2 computed geometry optimization seem to support the statements derived from the experimental data: the geometry shown in Fig. 5 is obviously not just the product of a distortion along a single (out-of-plane) normal mode (Table 2).
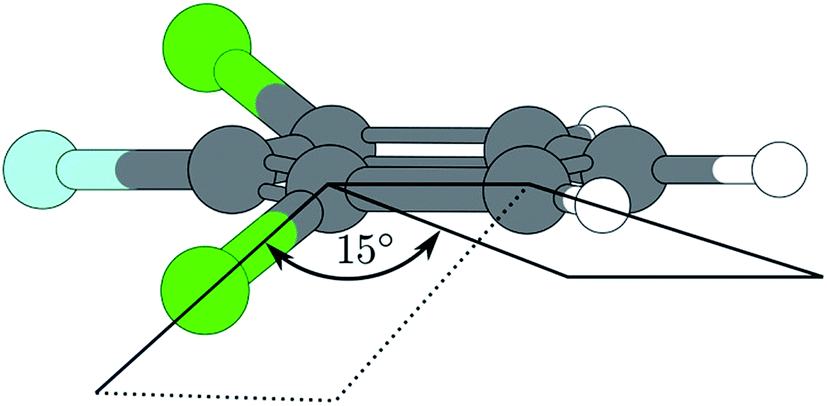 | ||
| Fig. 5 Excited state geometry of 1,3,2-DCFB obtained by CC2-optimization. | ||
| S0(C2v) | S1 | (C2v) | (C2v) | (CS) | D0 | (C2v) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wilson | Sym. | exp. | B3LYP | BP86 | CC2 | exp. | B3LYP | BP86 | CC2 | exp. | B3LYP | BP86 |
| 9a | a1 | 301 | 290 | 295 | 228 | 374 | 364 | 367 | 293 | 304 | 294 | |
| 6a | a1 | 437 | 425 | 436 | 381 | 198 | 202 | 188 | 418 | 435 | 422 | |
| 1 | a1 | 617 | 602 | 618 | 607 | 517 | 500 | 488 | 630 | 636 | 620 | |
| 7a | a1 | 783 | 760 | 776 | 748 | 729 | 713 | 778 | 784 | 761 | ||
| 19a | a1 | 1072 | 1142 | 1124 | 1506 | 1462 | 1360 | 1360 | 1318 | |||
| 18a | a1 | 1121 | 1042 | 1065 | 1113 | 1083 | 1059 | 1045 | 1021 | |||
| 12 | a1 | 1314 | 1275 | 1317 | 1005 | 984 | 949 | 1150 | 1114 | |||
| 13 | a1 | 1494 | 1448 | 1491 | 1294 | 1252 | 1268 | 1413 | 1372 | |||
| 8a | a1 | 1637 | 1587 | 1635 | 1577 | 1542 | 1507 | 1665 | 1613 | |||
| 20a | a1 | 3191 | 3121 | 3224 | 3201 | 3133 | 3212 | 3199 | 3128 | |||
| 2 | a1 | 3212 | 3143 | 3248 | 3223 | 3155 | 3266 | 3217 | 3148 | |||
| 10a | a2 | 249 | 239 | 247 | 194 | 189 | 183 | 201 | 218 | 209 | ||
| 16a | a2 | 597 | 573 | 588 | 762 | 766 | 519 | 546 | 565 | 541 | ||
| 17a | a2 | 893 | 850 | 873 | 1043 | 1107 | 788 | 920 | 880 | |||
| 17b | b1 | 124 | 119 | 125 | 25 | 31 | 41 | 51 | 97 | 98 | 94 | |
| 10b | b1 | 275 | 264 | 273 | 198 | 215 | 214 | 176 | 280 | 289 | 277 | |
| 16b | b1 | 530 | 510 | 521 | 404 | 428 | 407 | 351 | 439 | 434 | 415 | |
| 4 | b1 | 717 | 684 | 675 | 805 | 779 | 649 | 649 | 729 | 700 | ||
| 11 | b1 | 792 | 754 | 772 | 640 | 632 | 548 | 839 | 837 | 802 | ||
| 5 | b1 | 968 | 922 | 944 | 979 | 946 | 781 | 1000 | 954 | |||
| 15 | b2 | 212 | 204 | 208 | 63 | 64 | 67 | 154 | 213 | 221 | 212 | |
| 6b | b2 | 509 | 494 | 501 | 443 | 451 | 463 | 439 | 367 | 386 | 369 | |
| 3 | b2 | 555 | 534 | 543 | 511 | 501 | 492 | 489 | 539 | 522 | ||
| 7b | b2 | 1020 | 990 | 1014 | 979 | 958 | 948 | 982 | 1018 | 986 | ||
| 9b | b2 | 1179 | 1088 | 1167 | 1137 | 1128 | 1133 | 1110 | 1087 | |||
| 18b | b2 | 1266 | 1226 | 1262 | 1208 | 1171 | 1194 | 1303 | 1259 | |||
| 14 | b2 | 1322 | 1331 | 1424 | 1380 | 1393 | 1456 | 1354 | 1316 | |||
| 19b | b2 | 1503 | 1456 | 1495 | 1441 | 1382 | 1381 | 1548 | 1500 | |||
| 8b | b2 | 1626 | 1577 | 1623 | 1477 | 1451 | 1680 | 1381 | 1370 | |||
| 20b | b2 | 3206 | 3137 | 3242 | 3207 | 3139 | 3215 | 3214 | 3145 | |||
The quantum chemical calculations predict a S1(π* ← π) transition into the first excited state for 1,3,2-DCFB. However, due to σ*-orbitals localized on the Halogen–Carbon bond, it is possible for modes of appropriate symmetry to induce coupling to (πσ*)-states. In accordance with the assumption of a vibronic coupling effect we prevalently observed a substantial decrease in frequency comparing the S0 state and the S1 state of the neutral for formal forbidden modes with strong influence of halogen atoms on their displacement pattern. For the modes 15, 6b, 3 (b2 symmetry) and 16a (a2 symmetry) we observed such a decrease.
The decrease in frequency for the modes 15, 6b, 3 is explainable in accordance with the Herzberg–Teller (HT) effect. However, the frequency lowering of the symmetry allowed mode 17b2 could not be described in terms of the HT effect in a sufficient way. It seems that the distortion along 17b blurs the symmetry differences between the (πσ*)- and (ππ*)-states which would be an indication for a pseudo Jahn–Teller effect.
5.2. 1,3,2-DFCB
In 1,3,2-DFCB the vibration 15 appears in the REMPI spectrum as a progression-forming and largely frequency lowered mode. Remarkably, the progression reveals a negative anharmonicity. The occurrence of the 15 as a b2 symmetric mode is explained in terms of the Herzberg–Teller effect. Besides the mode 15, also the modes 6a and 9a exhibit a largely lowered frequency in the first excited state S1. Both modes are likely to contribute in pseudo Jahn–Teller distortion in going from the electronic ground to excited state or from excited state to ionic ground state, respectively. This is seen from the different MATI spectra as well as the different calculated structures given in Tables 3–8. The MATI spectrum via the electronic origin exhibits a harmonic, three-membered progression of mode 9a (9a1, 9a2, 9a3), whereby the band 9a1 is almost as intense as the 0–0 transition. Such a shift of the Franck–Condon maximum is a clear indication for a distortion along the eigenvector of this mode during ionization.|
|
|||
|---|---|---|---|
| B3LYP/TZVPP | C 2v | C 2 | C 2v |
| Bond length [Å] | S0 | S1 | D0 |
| C1–C2 | 1.391 | 1.364 | 1.438 |
| C2–C3 | 1.391 | 1.445 | 1.438 |
| C3–C4 | 1.389 | 1.403 | 1.373 |
| C4–C5 | 1.389 | 1.370 | 1.407 |
| C5–C6 | 1.389 | 1.370 | 1.407 |
| C6–C1 | 1.389 | 1.380 | 1.373 |
| C1–Cl7 | 1.739 | 2.358 | 1.698 |
| C2–F8 | 1.334 | 1.325 | 1.290 |
| C3–Cl9 | 1.739 | 1.700 | 1.698 |
| C4–H10 | 1.080 | 1.080 | 1.081 |
| C5–H11 | 1.081 | 1.082 | 1.082 |
| C6–H12 | 1.080 | 1.085 | 1.081 |
|
|||
| Bond angles [°] | |||
| C1–C2–C3 | 120.125 | 123.523 | 122.001 |
| C2–C3–C4 | 119.972 | 119.825 | 118.077 |
| C3–C4–C5 | 119.648 | 117.525 | 119.488 |
| C4–C5–C6 | 120.635 | 120.436 | 122.869 |
| C5–C6–C1 | 119.648 | 123.643 | 119.488 |
| C6–C1–C2 | 119.972 | 115.049 | 118.077 |
| Cl7–C1–C2 | 119.336 | 131.346 | 118.378 |
| C1–C2–F8 | 119.938 | 121.023 | 118.996 |
| C4–C3–Cl9 | 120.692 | 120.051 | 123.541 |
|
|||
| Dihedral angle [°] | |||
| Cl7–C1–C6–H12 | 0 | −0.018 | 0 |
| Cl7–C1–C2–F8 | 0 | 0.016 | 0 |
| Cl9–C3–C4–H10 | 0 | −0.005 | 0 |
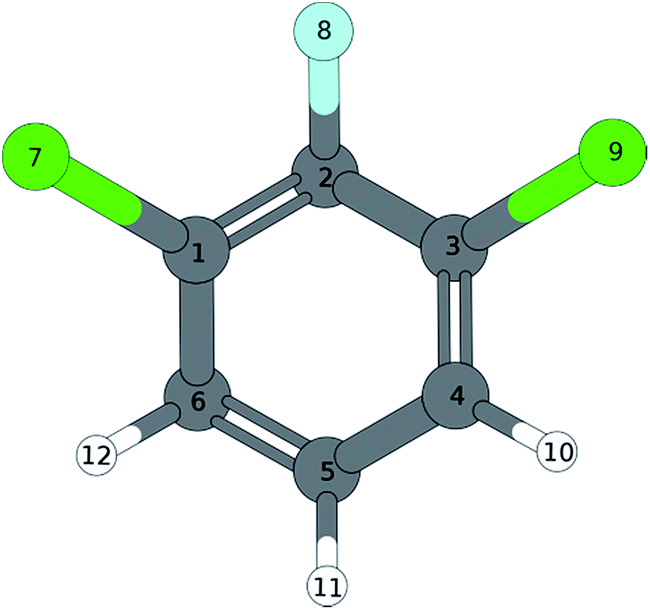
|
|
|||
|---|---|---|---|
| BP86/TZVPP | C 2v | C 2 | C 2v |
| Bond length [Å] | S0 | S1 | D0 |
| C1–C2 | 1.400 | 1.377 | 1.444 |
| C2–C3 | 1.400 | 1.445 | 1.444 |
| C3–C4 | 1.396 | 1.411 | 1.382 |
| C4–C5 | 1.395 | 1.379 | 1.411 |
| C5–C6 | 1.395 | 1.434 | 1.411 |
| C6–C1 | 1.396 | 1.395 | 1.382 |
| C1–Cl7 | 1.740 | 2.325 | 1.701 |
| C2–F8 | 1.341 | 1.335 | 1.300 |
| C3–Cl9 | 1.740 | 1.708 | 1.701 |
| C4–H10 | 1.088 | 1.088 | 1.089 |
| C5–H11 | 1.089 | 1.090 | 1.090 |
| C6–H12 | 1.088 | 1.094 | 1.089 |
|
|||
| Bond angles [°] | |||
| C1–C2–C3 | 120.007 | 123.861 | 121.791 |
| C2–C3–C4 | 120.019 | 120.100 | 118.195 |
| C3–C4–C5 | 119.613 | 117.257 | 119.447 |
| C4–C5–C6 | 120.728 | 120.361 | 122.865 |
| C5–C6–C1 | 119.613 | 124.461 | 119.477 |
| C6–C1–C2 | 120.019 | 113.959 | 118.195 |
| Cl7–C1–C2 | 119.198 | 130.690 | 118.269 |
| C1–C2–F8 | 119.938 | 121.023 | 118.996 |
| C4–C3–Cl9 | 120.783 | 119.621 | 123.535 |
|
|||
| Dihedral angle [°] | |||
| Cl7–C1–C6–H12 | 0 | −0.030 | 0 |
| Cl7–C1–C2–F8 | 0 | 0.038 | 0 |
| Cl9–C3–C4–H10 | 0 | −0.006 | 0 |
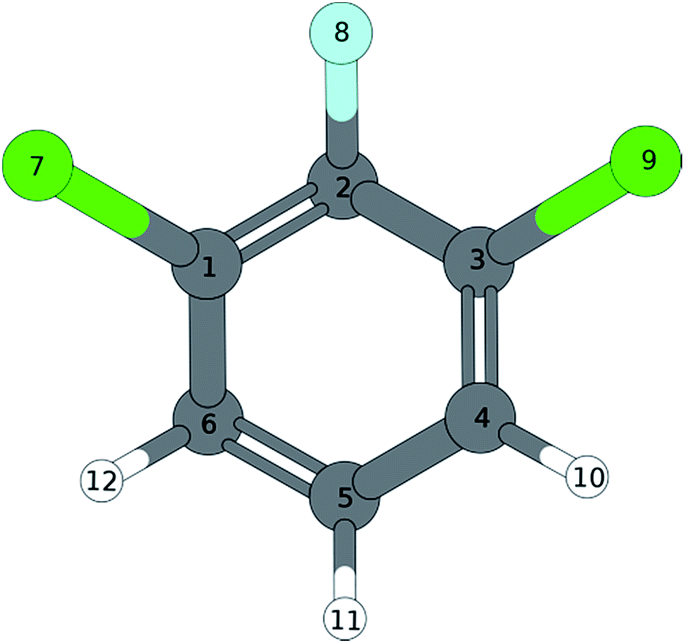
|
|
||
|---|---|---|
| BP86/TZVPP | C 2v | C 2 |
| Bond length [Å] | S0 | S1 |
| C1–C2 | 1.395 | 1.432 |
| C2–C3 | 1.395 | 1.432 |
| C3–C4 | 1.393 | 1.426 |
| C4–C5 | 1.393 | 1.421 |
| C5–C6 | 1.393 | 1.421 |
| C6–C1 | 1.393 | 1.426 |
| C1–Cl7 | 1.725 | 1.719 |
| C2–F8 | 1.333 | 1.326 |
| C3–Cl9 | 1.725 | 1.719 |
| C4–H10 | 1.080 | 1.079 |
| C5–H11 | 1.081 | 1.082 |
| C6–H12 | 1.080 | 1.079 |
|
||
| Bond angles [°] | ||
| C1–C2–C3 | 119.992 | 121.970 |
| C2–C3–C4 | 120.078 | 118.706 |
| C3–C4–C5 | 119.618 | 118.910 |
| C4–C5–C6 | 120.617 | 122.532 |
| C5–C6–C1 | 119.618 | 118.910 |
| C6–C1–C2 | 120.078 | 118.706 |
| Cl7–C1–C2 | 119.105 | 118.817 |
| C1–C2–F8 | 120.004 | 119.015 |
| C4–C3–Cl9 | 110.817 | 120.760 |
|
||
| Dihedral angle [°] | ||
| Cl7–C1–C6–H12 | 0 | −17.842 |
| Cl7–C1–C2–F8 | 0 | 17.222 |
| Cl9–C3–C4–H10 | 0 | −17.842 |
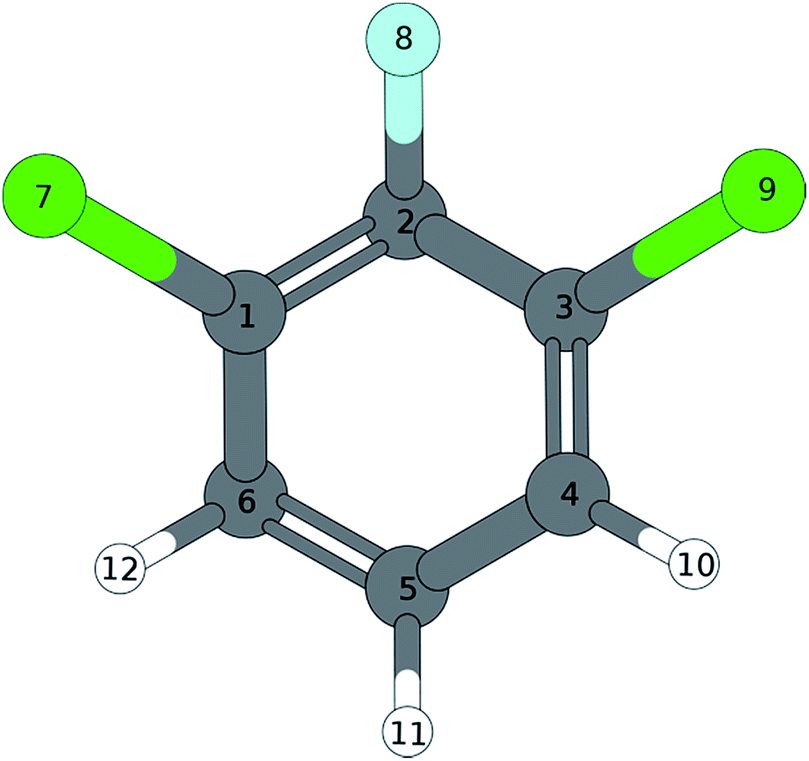
|
|
|||
|---|---|---|---|
| B3LYP/TZVPP | C 2v | C 2v | C 2v |
| Bond length [Å] | S0 | S1 | D0 |
| C1–C2 | 1.393 | 1.365 | 1.440 |
| C2–C3 | 1.393 | 1.365 | 1.440 |
| C3–C4 | 1.384 | 1.444 | 1.365 |
| C4–C5 | 1.389 | 1.384 | 1.409 |
| C5–C6 | 1.389 | 1.384 | 1.409 |
| C6–C1 | 1.384 | 1.444 | 1.365 |
| C1–F7 | 1.340 | 1.326 | 1.309 |
| C2–Cl8 | 1.729 | 2.361 | 1.665 |
| C3–F9 | 1.340 | 1.326 | 1.309 |
| C4–H10 | 1.081 | 1.081 | 1.081 |
| C5–H11 | 1.081 | 1.080 | 1.082 |
| C6–H12 | 1.081 | 1.081 | 1.081 |
|
|||
| Bond angles [°] | |||
| C1–C2–C3 | 117.528 | 111.693 | 118.393 |
| C2–C3–C4 | 121.919 | 126.328 | 120.838 |
| C3–C4–C5 | 118.997 | 118.962 | 118.528 |
| C4–C5–C6 | 120.640 | 117.726 | 122.876 |
| C5–C6–C1 | 118.997 | 118.962 | 118.528 |
| C6–C1–C2 | 117.528 | 126.328 | 120.838 |
| F7–C1–C2 | 118.843 | 120.164 | 117.338 |
| C1–C2–Cl8 | 121.236 | 124.155 | 120.803 |
| C4–C3–F9 | 119.237 | 113.508 | 121.823 |
|
|||
| Dihedral angle [°] | |||
| F7–C1–C6–H12 | 0 | 0.027 | 0 |
| F7–C1–C2–Cl8 | 0 | −0.204 | 0 |
| F9–C3–C4–H10 | 0 | −0.032 | 0 |
|
|
|||
|---|---|---|---|
| BP86/TZVPP | C 2v | C 2v | C 2v |
| Bond length [Å] | S0 | S1 | D0 |
| C1–C2 | 1.401 | 1.378 | 1.446 |
| C2–C3 | 1.401 | 1.378 | 1.446 |
| C3–C4 | 1.391 | 1.446 | 1.374 |
| C4–C5 | 1.396 | 1.391 | 1.414 |
| C5–C6 | 1.396 | 1.391 | 1.414 |
| C6–C1 | 1.391 | 1.446 | 1.374 |
| C1–F7 | 1.348 | 1.337 | 1.318 |
| C2–Cl8 | 1.730 | 2.332 | 1.671 |
| C3–F9 | 1.348 | 1.337 | 1.318 |
| C4–H10 | 1.089 | 1.089 | 1.089 |
| C5–H11 | 1.089 | 1.087 | 1.090 |
| C6–H12 | 1.089 | 1.089 | 1.089 |
|
|||
| Bond angles [°] | |||
| C1–C2–C3 | 117.514 | 110.326 | 118.508 |
| C2–C3–C4 | 121.885 | 127.177 | 120.727 |
| C3–C4–C5 | 118.999 | 118.870 | 118.543 |
| C4–C5–C6 | 120.718 | 117.579 | 122.956 |
| C5–C6–C1 | 118.999 | 118.870 | 118.543 |
| C6–C1–C2 | 117.514 | 127.177 | 120.727 |
| F7–C1–C2 | 118.763 | 119.276 | 117.392 |
| C1–C2–Cl8 | 121.242 | 124.836 | 120.740 |
| C4–C3–F9 | 119.349 | 113.548 | 121.870 |
|
|||
| Dihedral angle [°] | |||
| F7–C1–C6–H12 | 0 | 0.041 | 0 |
| F7–C1–C2–Cl8 | 0 | −0.191 | 0 |
| F9–C3–C4–H10 | 0 | −0.032 | 0 |
|
|
||
|---|---|---|
| B3LYP/TZVPP | C 2v | C 2v |
| Bond length [Å] | S0 | S1 |
| C1–C2 | 1.396 | 1.416 |
| C2–C3 | 1.396 | 1.416 |
| C3–C4 | 1.387 | 1.431 |
| C4–C5 | 1.394 | 1.407 |
| C5–C6 | 1.394 | 1.407 |
| C6–C1 | 1.387 | 1.431 |
| C1–F7 | 1.339 | 1.337 |
| C2–Cl8 | 1.716 | 1.825 |
| C3–F9 | 1.339 | 1.337 |
| C4–H10 | 1.080 | 1.825 |
| C5–H11 | 1.081 | 1.337 |
| C6–H12 | 1.080 | 1.083 |
|
||
| Bond angles [°] | ||
| C1–C2–C3 | 117.753 | 110.391 |
| C2–C3–C4 | 121.786 | 125.833 |
| C3–C4–C5 | 118.972 | 120.624 |
| C4–C5–C6 | 120.732 | 116.095 |
| C5–C6–C1 | 118.972 | 120.624 |
| C6–C1–C2 | 121.786 | 125.833 |
| F7–C1–C2 | 118.680 | 118.508 |
| C1–C2–Cl8 | 121.124 | 118.556 |
| C4–C3–F9 | 119.534 | 115.651 |
|
||
| Dihedral angle [°] | ||
| F7–C1–C6–H12 | 0 | 0.912 |
| F7–C1–C2–Cl8 | 0 | 46.945 |
| F9–C3–C4–H10 | 0 | −0.912 |
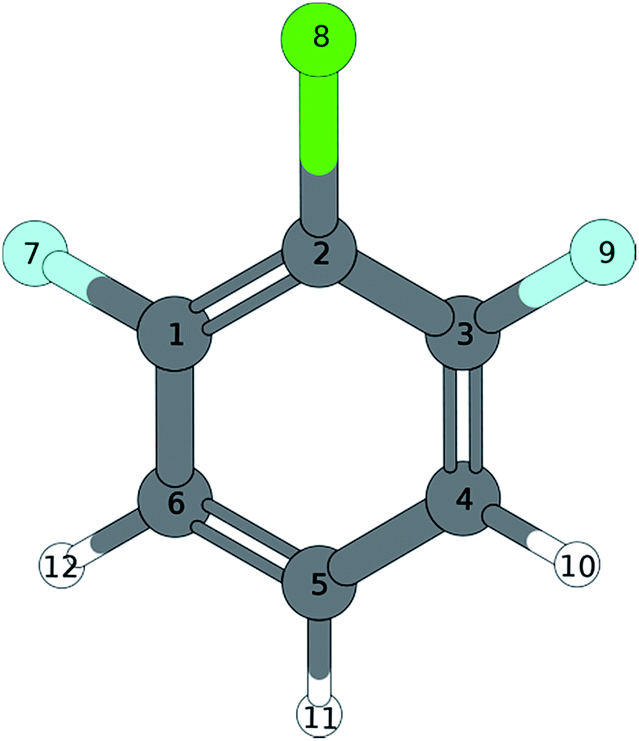
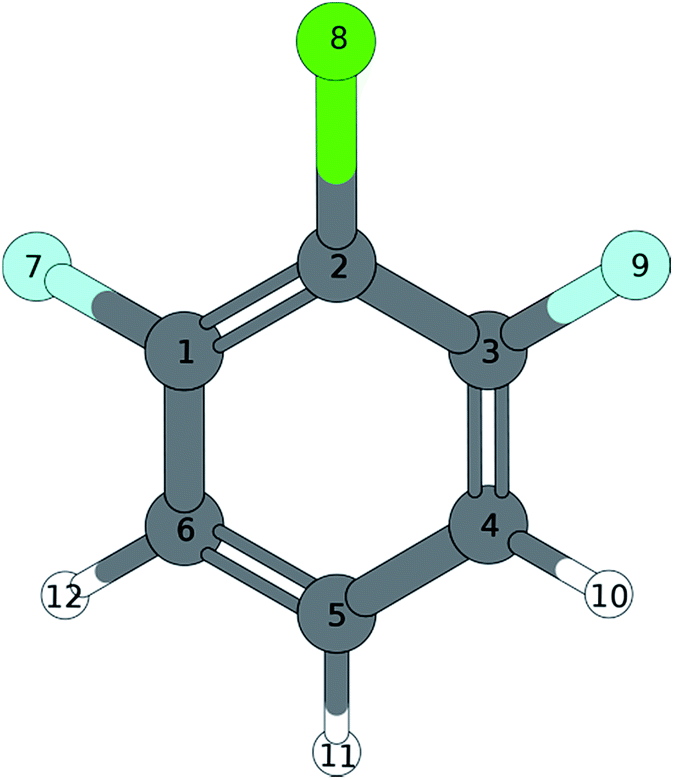
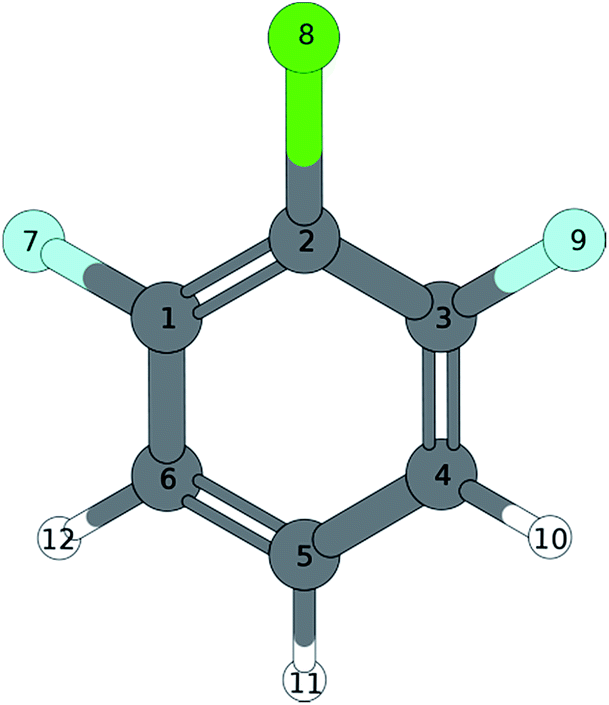
A similar situation is found in the MATI spectrum via 9a1. Here, in contradiction to the Δv = 0 propensity rule, the transition into the S16a1 state is the most favorable while the vertical transition into the 9a1 is scarcely observable. Nevertheless the mode 9a is strongly present in overtones (9a16a1, 9a111, 9a26a1). The geometry found by the TDDFT calculations (Fig. 6a; Tables 6 and 7) support the experimental findings in predicting a C2v-symmetric structure distorted along the 6a. The CC2 calculation suggests a 17b-like distorted structure (Fig. 6b; Table 8).
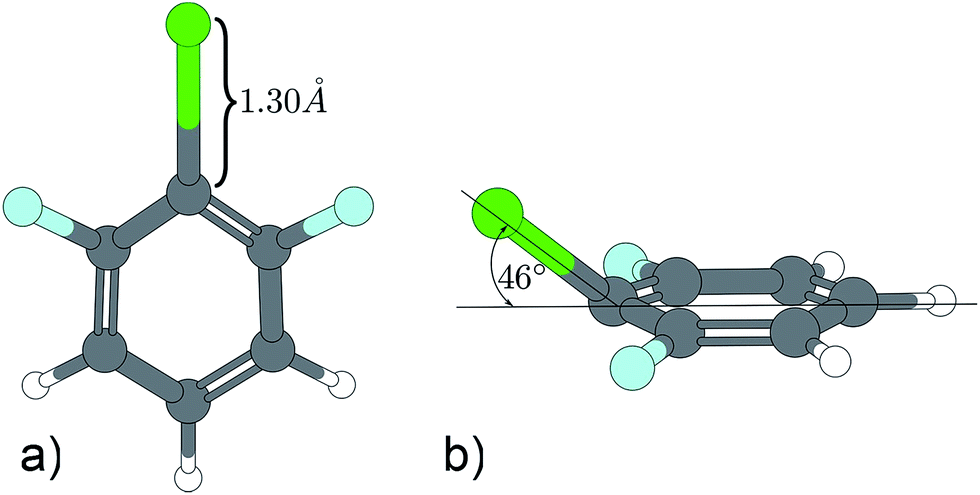 | ||
| Fig. 6 Excited state geometry of 1,3,2-DFCB obtained by different quantum mechanical methods. (a) TDDFT, (b) CC2. | ||
The CC2 calculation suggests a 17b-like distorted structure (Fig. 6b). Considering the vanishing low activity of 17b in the MATI spectra, the suggestion seems incorrect. Nevertheless the substantial decrease in frequency for 6a1 and 9a1 comparing the D0 state and the S1 state is accurately reproduced by all quantum chemical methods employed. Comparing experimental and calculated results for 1,3,2-DFCB, the unusual, striking deviation (up to 49%) for a1-symmetrical S1-frequencies was particularly noticeable. A poor reproduction of the molecular equilibrium structure in the S1 (wrong minimum on the PES) could be one possible explanation.
A contrary indication is the fact that we find very similar frequencies for widely differing structures predicted by TDDFT (planar structure) and CC2 method (out-of-plane-structure). The planar structure resembles the experimental observations well, but since all frequency analyses were performed in harmonic approximation, a strong anharmonicity for this particular vibrations could be another explanation for this findings.
6. Conclusion
Comparing the results from two compounds presented in this paper with results obtained from different isomeric trichloro-benzenes,1 the increasing number of fluorine atoms lead to a progressive decrease in some of the observed frequencies in going from S0 to S1 and from D0 to S1, respectively. This is especially true for modes with a strong fluorine participation in their vibrational pattern like 17b (cf.Table 1). We interpreted this as a strong indication for an out-of-plane distortion during excitation or a replanarisation during ionization along the eigenvector of those modes. This phenomenon could be attributed to an above, bounding S(πσ*) state which is stabilized by an increasing number of fluorine atoms.12 It can be concluded that the fluorine atoms contribute a significant share in form of σ* ← π character to the transition.For 1,3,2-DCFB the quantum chemical calculations gave excitation energies (EE) of 5.06 eV (B3LYP/TZVPP), 4.71 eV (BP86/TZVPP) and 4.44 eV (CC2/cc-pVTZ). The CC2 value reproduces the experimental value of 4.52 eV with a deviation of 0.08 eV best.
The experimental value for the ionization energy (IE) of 9.32 eV is underestimated by both DFT methods with 9.00 eV (B3LYP/TZVPP) and 8.97 eV (BP86/TZVPP). (The coupled cluster method CC2 is not suited for the ionic species!). In this case the frequency analysis performed by the CC2 method showed to be most appropriate to reproduce the experimental frequencies.
For 1,3,2-DFCB the quantum chemical calculations gave excitation energies (EE) of 5.23 eV (B3LYP/TZVPP), 4.90 eV (BP86/TZVPP) and 3.88 eV (CC2/cc-pVTZ). The TDDFT value reproduces the experimental value of 4.64 eV best, whereas BP86 underestimates and B3LYP overestimates the experimental value. Both DFT methods with 9.03 eV (B3LYP/TZVPP) and 9.02 eV (BP86/TZVPP) underestimate the experimental value for the ionization energy (IE) of 9.38 eV. Contrary to the 1,3,2 DCFB the frequency analysis performed by the DFT methods results in a better reproduction of the experimental frequencies for the 1,3,2 DFCB.
Furthermore the S1 ← S0 electronic excitation energies (EE) and D0 ← S0 adiabatic ionization energies (IE) could be determined very exactly:
EE(1,3,2-DFCB) = 36460 ± 2 cm−1, IE(1,3,2-DFCB) = 75.242 ± 6 cm−1;
EE(1,3,2-DCFB) = 37449 ± 2 cm−1, IE(1,3,2-DCFB) = 75.627 ± 6 cm−1.
References
- F. Witte, R. Mikko, F. Gunzer and J. Grotemeyer, Mass analyzed threshold ionization (mati) spectroscopy of trichlorobenzenes via different intermediate vibrational states in the s1 state, Int. J. Mass Spectrom., 2011, 306, 129–137, DOI:10.1016/j.ijms.2010.10.002.
- L. Yuan, C. Li, J. Lin, S. Yang and W. Tzeng, Mass analyzed threshold ionization spectroscopy of o-fluorophenol and o-methoxyphenol cations and influence of the nature and relative location of substituents, Chem. Phys., 2006, 323, 429–438, DOI:10.1016/j.chemphys.2005.10.004.
- K. Müller-Dethlefs, M. Sander and E. W. Schlag, Two-colour photoionization resonance spectroscopy of NO: complete separation of rotational levels of NO+ at the ionization threshold, Chem. Phys. Lett., 1984, 112, 291–294, DOI:10.1016/0009-2614(84)85743-7.
- L. Zhu and P. Johnson, Mass analyzed threshold ionization spectroscopy, J. Chem. Phys., 1991, 94(8), 5769–5771, DOI:10.1063/1.460460.
- K. Walter, U. Boesl and E. W. Schlag, Molecular ion spectroscopy: resonance-enhanced multiphoton dissociation spectra of the fluorobenzene cation, Chem. Phys. Lett., 1989, 162(4, 5), 261–268, DOI:10.1016/0009-2614(89)87041-1.
- G. Reiser, D. Rieger, T. G. Wright, K. Müller-Dethlefs and E. W. Schlag, Zero-kinetic-energy (ZEKE) photoelectron spectroscopy of p-difluorobenzene via different intermediate vibrational levels in the s1 state, J. Phys. Chem., 1993, 97, 4335–4343, DOI:10.1021/j100119a015.
- A. Gaber, M. Riese and J. Grotemeyer, Mass analyzed threshold ionization spectroscopy of o-, m-, and p-dichlorobenzenes. Influence of the chlorine position on vibrational spectra and ionization energy, J. Phys. Chem. A, 2008, 112, 425–434, DOI:10.1021/jp074802t.
- A. Gaber, M. Riese and J. Grotemeyer, Detailed analysis of the cation ground state of three dichlorobenzenes by mass analyzed threshold ionization spectroscopy, Phys. Chem. Chem. Phys., 2008, 10, 1168–1176, 10.1039/B715496H.
- C. Weickhardt, R. Zimmermann, K. W. Schramm, U. Boesl and E. W. Schlag, Laser mass spectrometry of the di-, tri- and tetrachlorobenzenes: isomer-selective ionization and detection, Rapid Commun. Mass Spectrom., 1994, 8, 381–384, DOI:10.1002/rcm.1290080508.
- C. H. Kwon and M. S. Kim, One-photon mass-analyzed threshold ionization spectroscopy of 1,3,5-trifluorobenzene: the Jahn–Teller effect and vibrational analysis for the molecular cation in the ground electronic state, J. Chem. Phys., 2004, 121(6), 2622–2629, DOI:10.1063/1.1765655.
- S. Sato, K. Ikeda and K. Kimura, Zeke photo electron spectroscopy and ab initio force-field calculation of 1,2,4,5-tetrafluorobenzene, J. Electron. Spectrosc. Relat. Phenom., 1998, 88–91, 137–142, DOI:10.1016/S0368-2048(97)00256-9.
- M. Z. Zgierski, T. Fujiwara and E. C. Lim, Photophysics of aromatic molecules with low-lying pi sigma * states: fluorinated benzenes, J. Chem. Phys., 2005, 122(14), 144312, DOI:10.1063/1.1873752.
- C. H. Kwon and M. S. Kim, Vacuum ultraviolet mass-analyzed threshold ionization spectroscopy of hexafluorobenzene: the Jahn–Teller effect and vibrational analysis, J. Chem. Phys., 2004, 120(24), 11578, DOI:10.1063/1.1753258.
- Y. Tsuchiya, K. Takazawa, M. Fujii and M. Ito, Electronic spectra of o-, m-, p-difluorobenzene cations: striking similarity in vibronic coupling between the neutral molecule and its cation, J. Phys. Chem., 1992, 96(1), 99–104, DOI:10.1021/j100180a022.
- F. Gunzer and J. Grotemeyer, New features in the mass analysed threshold ionization (MATI) spectra of alkyl benzenes, Phys. Chem. Chem. Phys., 2002, 24, 5966–5972, 10.1039/b208283g.
- F. Gunzer and J. Grotemeyer, Enhancing of the signal-to-noise ratio in MATI spectra, Int. J. Mass Spectrom., 2003, 228, 921–931, DOI:10.1016/S1387-3806(03)00195-7.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Electronic structure calculations on workstation computers: the program system turbomole, Chem. Phys. Lett., 1989, 162, 165–169, DOI:10.1016/0009-2614(89)85118-8.
- M. v. Arnim and R. Ahlrichs, Geometry optimization in generalized natural internal coordinates, J. Chem. Phys., 1999, 111, 9183–9190, DOI:10.1063/1.479510.
- M. Häser and R. Ahlrichs, Improvements on the direct SCF method, J. Comput. Chem., 1989, 10, 104–111, DOI:10.1002/jcc.540100111.
- O. Treutler and R. Ahlrichs, Efficient molecular numerical integration schemes, J. Chem. Phys., 1995, 102, 346–354, DOI:10.1063/1.469408.
- (a) A. D. Becke, Density functional calculations of molecular bond energies, J. Chem. Phys., 1986, 84, 4524–4529, DOI:10.1063/1.450025; (b) A. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098, DOI:10.1103/PhysRevA.38.3098; (c) F. Weigend, M. Häuser, H. Patzelt and R. Ahlrichs, RI-MP2: optimized auxiliary basis sets and demonstration of efficiency, Chem. Phys. Lett., 1998, 294, 143–152, DOI:10.1016/S0009-2614(98)00862-8; (d) F. Weigend, A. Köhn and C. Hättig, Efficient use of the correlation consistent basis sets in resolution of the identity MP2 calculations, J. Chem. Phys., 2002, 116, 3175–3183, DOI:10.1063/1.1445115.
- G. Varsanyi and S. Szoke, Vibrational Spectra of Benzene Derivatives, Academic Press, New York, London, 1969 Search PubMed.
- E. B. Wilson, The normal modes and frequencies of vibration of the regular plane hexagon model of the benzene molecule, J. Phys. Rev., 1934, 41, 706–714, DOI:10.1103/PhysRev.45.706.
- J. Green, D. Harrison and W. Kynaston, Vibrational spectra of benzene derivatives-XI 1,3,5- and 1,2,3-trisubstituted compounds, Spectrochim. Acta, Part A, 1971, 27(6), 793–806, DOI:10.1016/0584-8539(71)80158-7.
- M. Mohraz, J. Maier and E. Heilbronner, He(Iα) and He(IIα) photoelectron spectra of fluorinated chloro- and bromo-benzenes, J. Electron Spectrosc. Relat. Phenom., 1980, 19, 429–446, DOI:10.1016/0368-2048(80)80063-6.
| This journal is © The Royal Society of Chemistry 2015 |