DOI:
10.1039/C4RA12714E
(Paper)
RSC Adv., 2015,
5, 2239-2248
Two- and three-dimensional lanthanide-based coordination polymers assembled by the synergistic effect of various lanthanide radii and flexibility of a new binicotinate-containing ligand: in situ synthesis, structures, and properties†
Received
19th October 2014
, Accepted 28th November 2014
First published on 28th November 2014
Abstract
Ten lanthanide coordination polymers have been obtained by the hydrothermal reactions between 3,3′-dimethoxy-2,2′-bipyridine-6,6′-dicarboxylic acid (H2mbpdc) and rare earth ions, in which H2mbpdc undergoes an in situ ligand transformation reaction, giving 3,3′-hydroxy-2,2′-bipyridine-6,6′-dicarboxylic acid (hbpdcH4). The structures are governed by the synergistic effect of lanthanide contraction with diverse coordination modes and conformations of the ligand. They have three structural types from 3D to 2D polymers. Type I for large ions with the general formula [Ln(hbpdcH)(H2O)]n (Ln = LaIII (1), CeIII(2), PrIII (3)), possesses a 6-connected pcu network with unprecedented 2D entangled layers of warp-and-woof threads interwoven by left- and right-handed helical chains; type II for intermediate ions (Ln = EuIII (4), GdIII (5)) and type III for small ions (Ln = TbIII (6), DyIII (7), HoIII (8), ErIII (9), LuIII (10)), with general formula [Ln(hbpdcH)(H2O)2]n. The type II and III comprised 2D Ln-hbpdcH grids. A magnetic study of 6–8 indicated that the coupling interaction between Ln3+ ions is weak. In addition, the photophysical properties of Eu and Tb polymers at room temperature were investigated.
Introduction
Since Weissman first discovered the photoluminescence of Eu(III) complexes in the 1940s,1 extensive research on luminescent lanthanide complexes has been carried out.2–4 In particular, multidimensional lanthanide-based coordination polymers (Ln–CPs) have recently gained much attention owing to their intriguing and diverse architectures5 and potential applications in the area of catalysis, magnetism, gas storage, selective adsorption and separation, and luminescent sensors.6 However, as high and variable coordination numbers and special characteristics lanthanide ions make it difficult to control the preparation of lanthanide complexes, the assembly of high dimensional lanthanide-based coordination polymers is currently a formidable project in contrast to the fruitful production of metal–organic frameworks (MOFs) with d-transition metal ions.7 In general, chemists have used the coordination requirements of the metal ions, the shape and functionalities of the organic ligands, metal-to-ligand ratio, and possible counterion influence to design and synthesis of new MOFs. This strategy of crystal engineering can also be transposed successfully in lanthanide chemistry to generate novel lanthanide-based CPs. However, the assembly rule must concern the unusual coordination characteristics controlled by the radii of lanthanide ions.8,9 As a consequences, lanthanide contraction is an effective synthetic route to fabricate diverse lanthanide complexes.10 Recently, Liu et al. reported seven lanthanide-based CPs formed by 2,6-pydinedicarboxylic acid and oxalic acid, which display the progressive structural variation from high to low dimension with a decrease in the Ln3+ radii.11 This interesting phenomenon was also been observed in some previous reports, and many reports are concerned with the diversity of Ln-based framework structures controlled by lanthanide contraction effect to date.12,13
Apart from the lanthanide contraction effect, properties of the organic ligands such as coordination mode, geometry, relative orientation of the donor groups and flexibility also play a key role in constructing Ln-based frameworks with novel structural features.14 Generally, the utilization of low-density rigid organic building blocks to construct Ln–CPs often lead to the presence of solvent molecules coordinated to the Ln3+ ions that results in luminescence quenching and low thermal stability. On the other hand, through the judicious choice of multidentate linkers and effective synthetic strategy, lanthanide frameworks exhibiting intense and long-lived fluorescent emissions could be achieved. So far, numerous ligands used for such a purpose have been based on polycarboxylate ligands with aromatic rings. Especially, those containing both N- and O-donors can exhibit great adaptability while coordinated to metal centers.11–13 In this sense, pyridine–polycarboxylic acids,15 pyrazine–polycarboxylic acids,16 imidazole–polycarboxylic acids,17 binicotinic acids and their derivatives18 have been widely used as multidentate organic linkers to construct high-dimensional lanthanide CPs with fascinating architectures and potential properties. Indeed, the salient virtues inherent to the choice of binicotinic acid as multidentate ligands, including coordination polymer formation and structural reproducibility, have been highly lauded by Lombardi and others.19 Meanwhile, rich topologies, including coordination modes, packing fashions and dimensionalities of coordination solids may be resulted from the multidentate ligand.
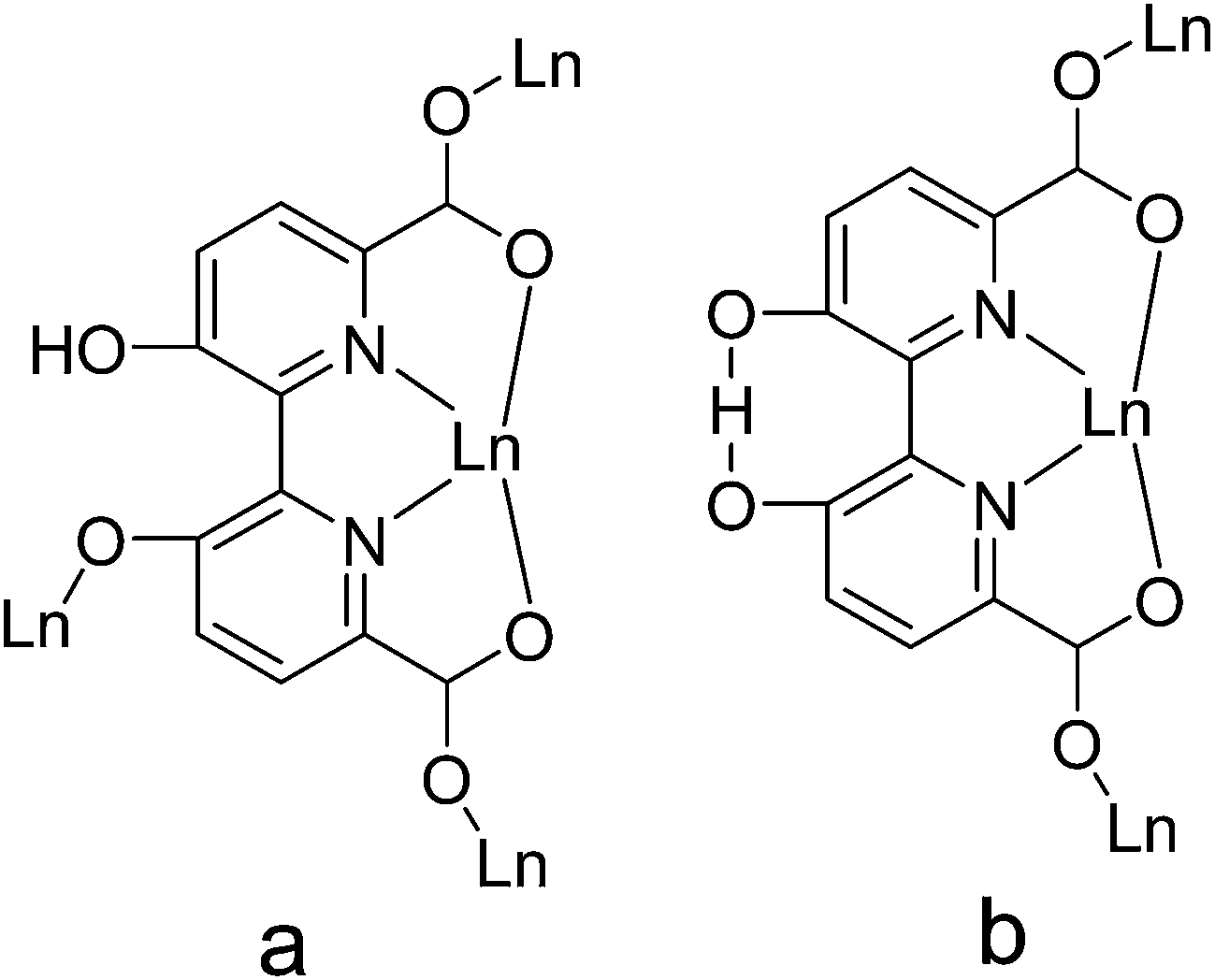
In this context, to fabricate novel Ln–CPs, the ligand 3,3′-dimethoxy-2,2′-bipyridine-6,6′-dicarboxylic acid (H2mbpdc) was selected on the basis of the following reasons: (i) it is multidentate organic ligand of up to eight donor atoms-N2O6, which can coordinate to the metal ions in a variety of coordination modes (see Scheme 1) leading to high-dimensional structures; (ii) the two pyridine units in H2mbpdc are linked together by a single C–C bond and they can rotate along the C–C bond to each other suggesting that the H2mbpdc ligand has flexibility to some extent and may offer the skew coordination orientation of the carboxyl groups providing the potential for the formation of helical structures; (iii) the H2mbpdc ligand could be transformed in situ into 3,3′-dihydroxy-2,2′-bipyridine-6,6′-dicarboxylic acid (hbpdcH4) during the hydrothermal process, and this may be used as a deliberate strategy to produce unusual MOF architectures; (iv) compared with other binicotinic derivatives, the substituents at 3,3′-positions were found to strongly effect the geometry and the energy of the bipyridine complexes,20 thus resulting in novel structures; (v) its hydroxyl group and carboxylate group may be partially or completely deprotonated resulting in rich coordination modes and formation of diverse architectures. Previous studies show that H2mbpdc is a good multidentate organic ligand for the construction of coordination polymers with Cu2+.21 However, its lanthanide complexes have never been reported to date. Following our systematic study on polycarboxylate containing lanthanide complexes, we decided to check the ability of lanthanide ions to afford high-dimensional complexes. Herein, the syntheses, structures and luminescent and magnetic properties of ten novel Ln-hbpdc CPs are reported. Single-crystal X-ray diffraction studies reveals that the ten CPs exhibit three different structure types: [Ln(hbpdcH)(H2O)]n (Ln = LaIII (1), CeIII (2), PrIII (3)) for type I, and [Ln(hbpdch)(H2O)2]n (Ln = EuIII (4), GdIII (5)) for type II, and [Ln(hbpdch)(H2O)2]n (TbIII (6), DyIII (7), HoIII (8), ErIII (9), LuIII (10)) for type III. Remarkably, the hbpdcH4 ligand in the ten Ln–CPs was in situ synthesized from H2mbpdc through a demethylation reaction.
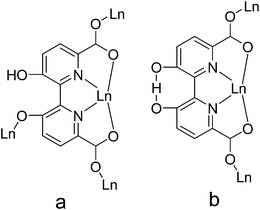 |
| | Scheme 1 The coordination modes of the hbpdcH4 ligand in complexes 1–10. | |
Results and discussion
Synthetic and spectral spects
Recent researches have demonstrated that the in situ ligand transformation reaction may occur during hydrothermal reaction. This may be ascribed to the features of hydrothermal synthesis, a special environment with high press and temperature, which encouraged to obtain novel CPs that are inaccessible or not easily achieved by the conventional methods.22 In this regard, the crystals of 1–10 were successfully synthesized by reacting H2mbpdc with the corresponding lanthanide nitrates at 180 °C for 3 days under hydrothermal conditions. It is noted that a new 3,3′-hydroxy-2,2′-bipyridine-6,6′-dicarboxylic acid (hbpdcH4) ligand was generated by an in situ ligand transformation reaction of demethylation of the H2mbpdc ligand during the hydrothermal reactions (see Scheme S1 in the ESI†). However, the suitable crystals were not obtained when we tried to synthesize the complexes by the use of traditional methods such as slow evaporating and diffusing at room temperature. Furthermore, some variations of the starting materials that may have influence on the assembly process including reactant molar ratio, metal salts and pH, have been investigated in details. No other kinds of crystals were obtained, however, suggesting the stability of the final structures in such temperature hydrothermal conditions.
Complexes 1–10 were stable towards oxygen and moisture, and were almost insoluble in common organic solvents such as CHCl3, MeOH, DMSO and DMF. For example, in the polymer 6, this result is further confirmed by the PXRD analyses (Fig. S1 in the ESI†). The elemental analyses of 1–10 were consistent with their chemical formula. The IR data of 1–10 are reported in the ESI.† It can be seen that the IR spectra show features attributable to the carboxylate stretching vibrations of the complexes. The absence of signals in the range 1760–1680 cm−1 indicates complete deprotonation of the dicarboxylate ligand. The characteristic bands of carboxylate groups are shown in the range 1533–1578 cm−1 for an asymmetric stretching and 1317–1483 cm−1 for a symmetric stretching. The broad bands at ca. 3320 cm−1 are corresponding to the vibration of the water molecules in the complexes.
Structure description
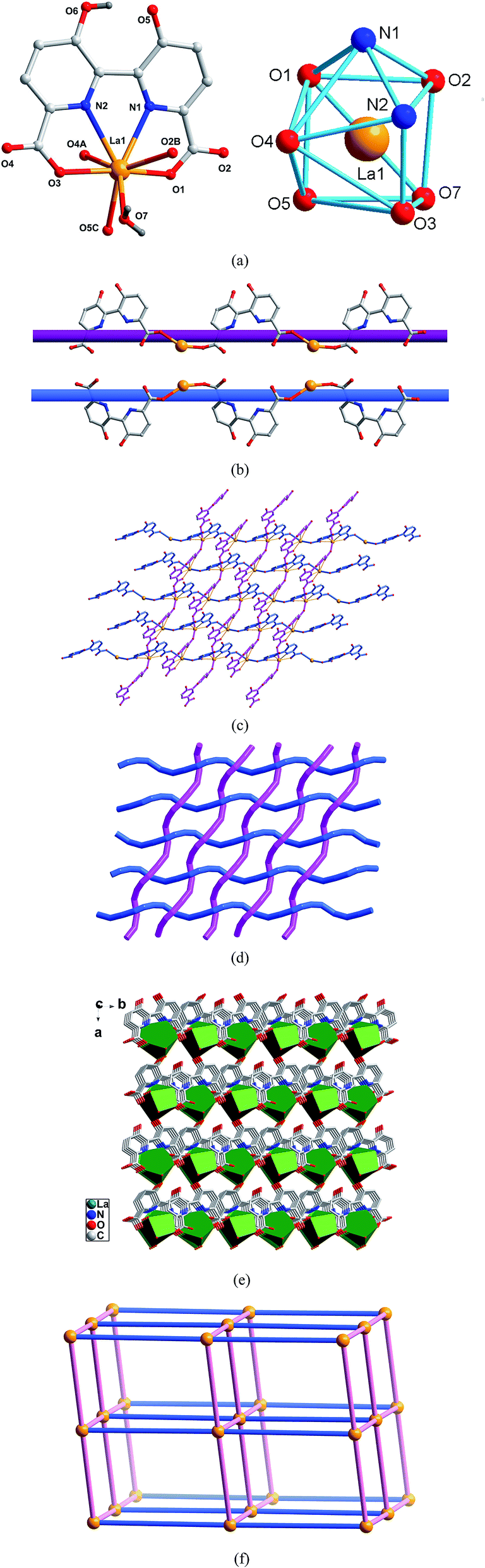
Crystal structure of type I [Ln(hbpdcH)(H2O)]n (Ln = LaIII (1), CeIII (2), PrIII (3)). X-Ray single crystal diffraction reveals that polymers 1–10 are isomorphous and possesses 3D network based on the linkage of unprecedented 2D entangled layers of warp and woof threads interwoven by left- and right-handed helical chains, hence only the structure of complex 1 will be representatively discussed in detail. As shown in Fig. 1a, the asymmetric unit of 1 consists of one unique La3+ ion, one hbptcH3− anion, one coordinated water molecule. Each La3+ ion is eight-coordinated, and the coordination geometry around La3+ ion may be described as a trigonally distorted dodecahedron. The La–O and La–N bond distances are in the range of 2.451(2)–2.638(3) Å and 2.692(2)–2.709(2) Å, respectively. The hbpdcH3− ligand acts as a μ2-bridge mode to connect La3+ ion into two kinds of infinite 1D helical chains, right- and left-handed, as shown in Fig. 1b. The 1D chains run in two nearly perpendicular directions and interweave in a ‘one-over/one-under’ fashion. Further, the interwoven chains are cross-linked via the chelation of the hbpdcH3− ligands with La ions, generating a 2D entangled woof and warp threaded layer with an uncommon topology, similar to the previous reports,23a as shown in Fig. 1c and d. This kind of cloth like structure is very rare in coordination polymers and exhibits unusual structural features being that the interwoven helical chains in complex 1 have different chirality: one is left-handed while the other is right-handed, though they have the same components and linking mode. It must be pointed that the equivalent coexistence of left- and right-handed helices in 1 might due to that during the formation of helical chains, there exist an equilibrium between helices with different chirality, and no factor can provide a sufficient energy difference to move the equilibrium between left-handed and right-handed helices to one side only. Schematic representation of the 2D interwoven network is shown in Fig. 1d. Furthermore, the interwoven network connects neighbouring ones with coordination bonds between hydroxyl O5 atoms and La ions to form a 3D architecture (Fig. 1e). In a topological point of view, each La1–hbpdcH mononuclear motif can be regarded as six-connected nodes to connect six adjacent ones, and thus the 3D network can be simplified as a 6-connected pcu net,23b as depicted in Fig. 1f.
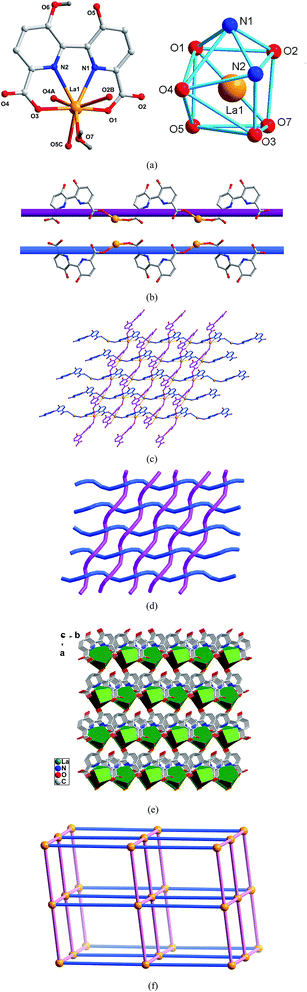 |
| | Fig. 1 (a) Asymmetric unit (left) and the square-antiprismatic coordination environments of the metal ions (right) in 1 (symmetry codes: (A) x, −y, z − 1/2; (B) x, −y + 1, z + 1/2; (C) x + 1/2, −y + 1/2, z + 1/2). (b) The right- and left-handed helical chains of 1. (c) The 2D entangled woof and warp threaded layer of 1. (d) Schematic representation of the 2D interwoven network. (e) The 3D network of 1. (f) Schematic representation of the pcu net for 1. | |
Polymers 2 and 3 are of the same structure as 1. The selected bond lengths and some nonbonding separations are listed in Table 1, which indicates that all the Ln–O and Ln–N bond lengths, together with the Ln⋯Ln separations decrease from complex 1 to complex 3, consistent with the radius contraction from La to Pr.
Table 1 Selected bond lengths for polymers 1–3a
| Bond |
Distance (Å) |
| 1·La |
2·Ce |
3·Pr |
| Symmetry transformations use to generate equivalent atoms (consistent with the symmetry transformations in Fig. 1a). (A): x + 1/2, −y + 1/2, z + 1/2; (B): x, −y, z + 1/2; (C): x, −y + 1, z − 1/2. |
| Ln–O(3A) |
2.451(2) |
2.416(4) |
2.406(2) |
| Ln–O(4) |
2.459(2) |
2.436(3) |
2.422(2) |
| Ln–O(1) |
2.468(2) |
2.441(3) |
2.427(2) |
| Ln–O(2B) |
2.477(2) |
2.450(3) |
2.434(2) |
| Ln–O(5C) |
2.535(2) |
2.509(3) |
2.488(2) |
| Ln–O(7) |
2.638(3) |
2.628(4) |
2.607(3) |
| Ln–N(1) |
2.692(2) |
2.666(3) |
2.647(3) |
| Ln–N(2) |
2.709(2) |
2.680(3) |
2.667(3) |
| Doubly bridged Ln1⋯Ln1B |
6.3144(9) |
6.2998(4) |
6.3005(9) |
| Doubly bridged Ln1⋯Ln1C |
6.4572(9) |
6.4210(4) |
6.3990(9) |
| Doubly bridged Ln1⋯Ln1A |
8.2290(2) |
8.1844(7) |
8.1725(2) |
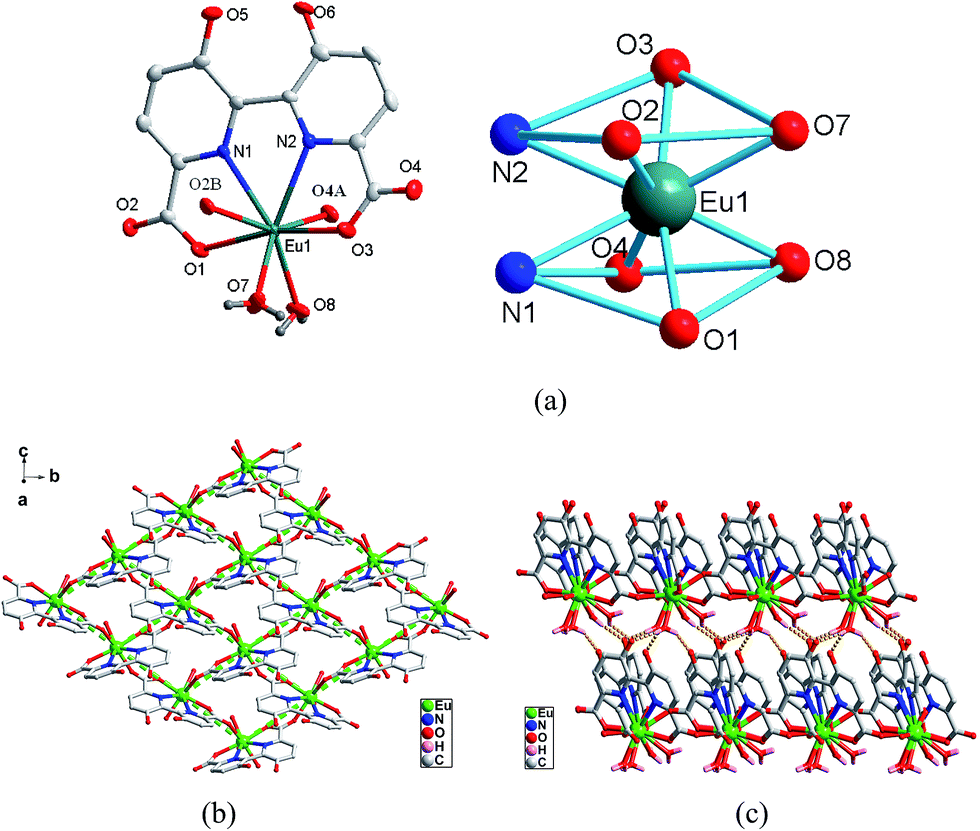
Crystal structure of type II [Ln(hbpdcH)(H2O)2]n (Ln = EuIII (4), GdIII (5)). The structure of type II, represented by complex 4 can be described as a 3D supramolecular architecture based on the linkage of 2D grid layers. As shown in Fig. 2a, the asymmetric unit of 4 consists of one unique Eu3+ ion, one hbptcH3− anion, two coordinated water molecules. Each Eu3+ ion is eight-coordinated, in a square antiprism. One hbpdcH3− ligand chelates to Eu1 through two carboxylate oxygen atoms and two adjacent nitrogen atoms (O1/N1, O3/N2), occupying four coordination sites of Eu1. The remaining four coordination sites of Eu3+ ions are taken by the oxygen atoms of two water molecules (O7, O8) and another two hbptcH3− ligands, each through a single carboxylate oxygen atom (O2, O4). The Eu–O bond distances are in the range of 2.340(3)–2.427(3) Å. In the structure, the hbpdcH3− ligand in a tetradentate chelating mode links Eu3+ ion into a mononuclear Eu-hbpdcH unit, which is further connected by the carboxyl of hbptcH3− in a bi-monodentate μ2–η1:η1-bridging mode, resulting in an alternating pattern of shorter and longer Eu⋯Eu distances (Fig. 2b). Thus, there are two different Eu⋯Eu distances; the shorter one is 6.184 Å and the longer one is 6.505 Å. In addition, there are three different Eu⋯Eu⋯Eu angles; those lying among 69.93, 74.16 and 107.96°. In this way, the structure is extended parallel to the a axis forming a 2D coordination polymer, while considering each Eu centre as a node, a 2D 44 grid is formed (Fig. 2b). Within the 4-membered rings, two coordinated water molecules are situated the same side of the ring, forming hydrogen bonds (see ESI† for detail) between their hydrogen atoms and the oxygen atoms of neighbouring uncoordinated hydroxyl groups. As a result, a supramolecular framework is formed, as shown in Fig. 2c.
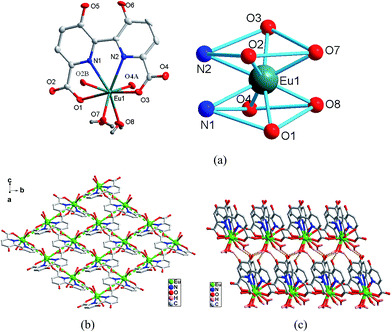 |
| | Fig. 2 (a) Asymmetric unit (left) and the square-antiprismatic coordination environments of the metal ions (right) in complex 4. (b) The 2D 44 grid of 4. (c) The 3D supramolecular framework of 4. | |
Polymer 5 is isomorphous with 4, and selected distances are listed in Table 2. As the ionic radii decreasing from Eu to Gd, all Ln–N and Ln–O bond distances decrease due to the lanthanide contraction, as do the Ln⋯Ln nonbonding separations spanned by the double μ2-carboxyl bridge.
Table 2 Selected bond lengths for polymers 4 and 5a
| Bond |
Distance (Å) |
| 4 Eu |
5 Gd |
| Symmetry transformations use to generate equivalent atoms (consistent with the symmetry transformations in Fig. 2a). (A): x, −y + 1, z − 1/2; (B): x, −y + 2, z + 1/2. |
| Ln–O(4A) |
2.340(3) |
2.330(3) |
| Ln–O(3) |
2.361(3) |
2.364(3) |
| Ln–O(1) |
2.393(3) |
2.398(3) |
| Ln–O(8) |
2.412(3) |
2.402(3) |
| Ln–O(2B) |
2.414(3) |
2.407(4) |
| Ln–O(7) |
2.427(3) |
2.420(3) |
| Ln–N(2) |
2.557(3) |
2.545(3) |
| Ln–N(1) |
2.603(3) |
2.592(3) |
| Doubly bridged Ln1⋯Ln1A |
6.184 |
6.174(4) |
| Doubly bridged Ln1⋯Ln1B |
6.505 |
6.499(4) |
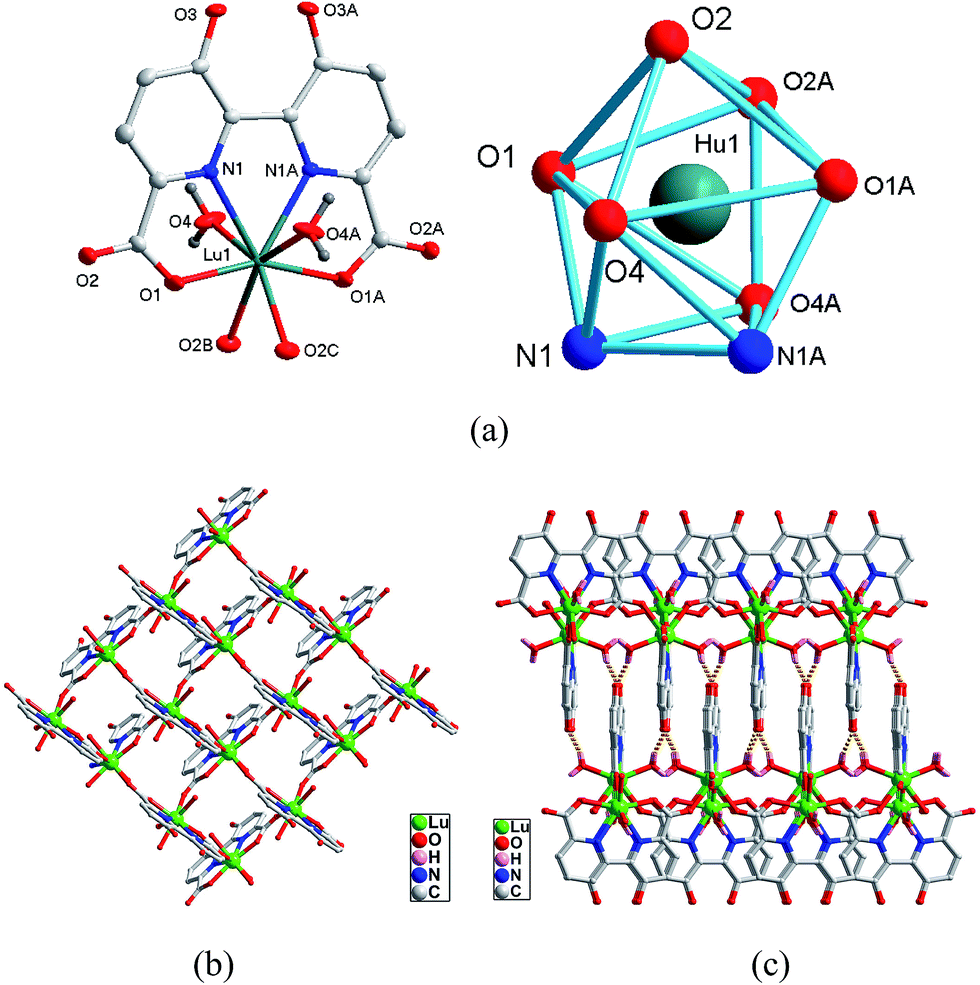
Crystal structure of type III [Ln(hbpdcH)0.5(H2O)]n (TbIII (6), DyIII (7), HoIII (8), ErIII (9), LuIII (10)). The crystal structures of complexes 6–10 are isomorphous, belonging to the orthorhombic system with space group Pbcn, only complex 10 was discussed in detail as a representative. As depicted in Fig. 3a, the asymmetric unit of complex 10 contains half unique Lu3+ ion, half hbpdcH3− anion, one coordinated water molecule. Each Lu3+ ion is eight-coordinated by two nitrogen atoms and two carboxylate oxygen atoms from one hbptcH3− ligand, two carboxylate oxygen atoms from the other two hbpdcH3− ligand and two coordinated water molecules. Its coordination geometry can be viewed as being a trigonally distorted dodecahedron. The Lu–O bond distances are in the range of 2.2327(19)–2.3931(18) Å. The average lengths of the Ln–O bonds in complexes 8 and 9 are 2.452(2) and 2.437(4) Å, respectively, in accordance with the decrease of the lanthanide metal radius.11–13
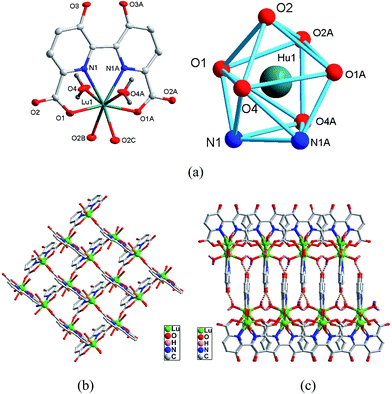 |
| | Fig. 3 Asymmetric unit (left) and the square-antiprismatic coordination environments of the metal ions (right) in complex 10. (b) The 2D 44 grid of 10. (c) The 3D supramolecular framework of 10. | |
As shown in Fig. 3b, the hbpdcH3− acts as a tetradentate chelating ligand to link links Lu3+ ion into a mononuclear Lu-hbptcH unit. Subsequently, the Lu-hbpdcH units are further interconnected by the carboxyl of the hbptcH3− ligands in a bi-monodentate μ2-η1:η1-bridging mode to generate the 2D 44 grid, similar to those observed in polymers 4 and 5. It is noted that all the Lu⋯Lu distances between the adjacent Lu-hbpdcH units are 6.0081(5) Å. Moreover, such 2D layers are connected by the extensive hydrogen bonds (see ESI† for detail) to generate a 3D supramolecular architecture, as shown in Fig. 3c.
Polymers 6, 7, 8, and 9 are isomorphous with 10. The subseries of complexes of type III structure also show the effect of lanthanide contraction: Ln–O bond lengths and Ln⋯Ln nonbonding distances decrease along with the decrease of ionic radii from Tb to Lu (Table 3).
Table 3 Selected bond lengths for polymers 6–10a
| Bond |
Distance (Å) |
| 6 Tb |
7 Dy |
8 Ho |
9 Er |
10 Lu |
| Symmetry transformations use to generate equivalent atoms (consistent with the symmetry transformations in Fig. 3a). (A): −x, y, −z + 3/2; (B): −x + 1/2, −y + 1/2, z + 1/2; (C): x − 1/2, −y + 1/2, −z + 1; (1#): −x, −y, −z + 1; (2#): x − 1/2, −y + 1/2, −z + 1. |
| Ln–O(1) |
2.286(2) |
2.272(3) |
2.271(2) |
2.255(2) |
2.2327(19) |
| Ln–O(1A) |
2.286(2) |
2.272(3) |
2.271(2) |
2.255(2) |
2.2327(19) |
| Ln–O(4) |
2.359(2) |
2.345(3) |
2.343(2) |
2.323(2) |
2.294(2) |
| Ln–O(4A) |
2.359(2) |
2.345(3) |
2.343(2) |
2.323(2) |
2.294(2) |
| Ln–O(2B) |
2.447(2) |
2.432(3) |
2.430(2) |
2.417(2) |
2.3931(18) |
| Ln–O(2C) |
2.447(2) |
2.432(3) |
2.430(2) |
2.417(2) |
2.3931(18) |
| Ln–N(1A) |
2.508(2) |
2.494(3) |
2.486(2) |
2.469(2) |
2.443(2) |
| Ln–N(1) |
2.508(2) |
2.494(3) |
2.486(2) |
2.469(2) |
2.443(2) |
| Doubly bridged Ln1⋯Ln1A |
6.0694(4) |
6.0483(4) |
6.0561(3) |
6.0343(3) |
6.0081(5) |
Comparison of the crystal structures
Although polymers 6–10 were synthesized from the same starting materials and experimental method, it is interesting to observe that they present a clear progressive change in three different types of crystal structures. Type I structure results from the large La, Ce and Pr ions with 3D networks constructed from the left-handed and right-handed helices bridged by the organic ligands, respectively; the intermediate ions, Eu and Gd, form a type II structure with the distinct 2D 44 grid, and the small ones, Tb, Dy, Ho, Er, and Lu, afford a type III structure which possesses the same 2D 44 grid as that observed in type II. Thus, with the increasing atomic number of lanthanides, the structures of ten coordination polymers changed from 3D to 2D, and they display three different side views of the 3D (1–3), 2D (4 and 5), and 2D (6–10), as depicted in Fig. S2 (in the ESI†). Most importantly, the result of comparation of the average Ln–O and Ln–N bond distances among the ten polymers (Tables 1–3) shows that the average bond lengths between the lanthanide and oxygen atoms are decreasing continuously from 2.638(3) to 2.233(19) Å, and the average bond distances between the lanthanide and nitrogen atoms are also decreasing continuously from 2.709(2) to 2.443(2) Å. In view of the above-mentions, it is the lanthanide contraction effect that results in forming the different structure, similar to the previous reports.11–13
Thermal behaviors and photoluminescent properties
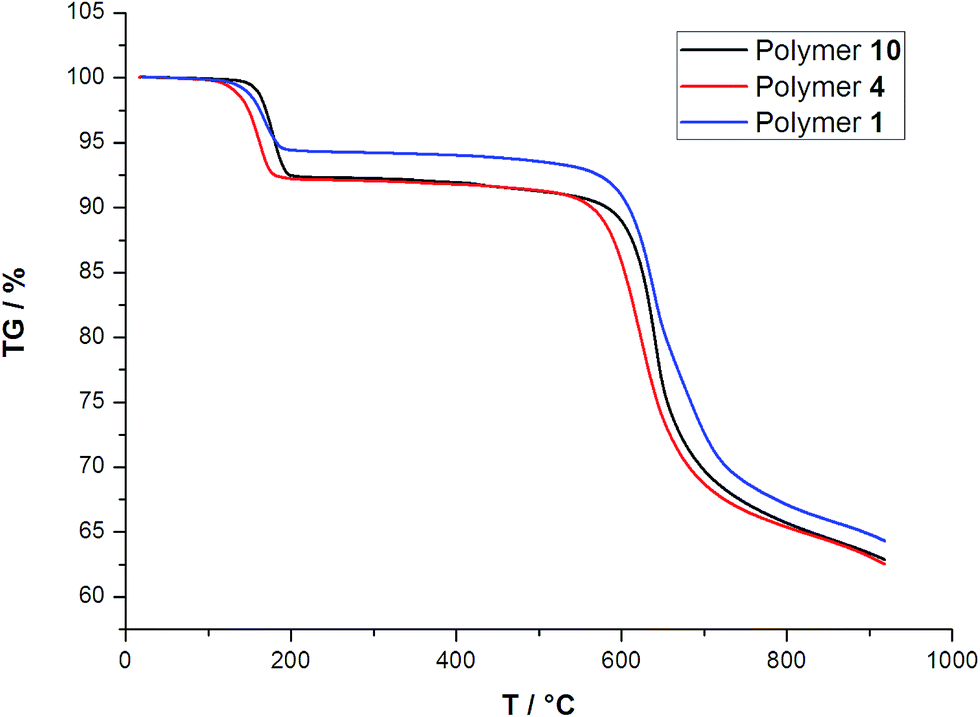
The thermal stability of the ten polymers was investigated through TGA experiments in the temperature range of 25–900 °C under a flow of nitrogen. Since the progressive types of structures of these polymers, only three polymers in each type are selected, La of type I, Eu for type II, and Lu of type III for description. As depicted in Fig. 4, the three types I, II, and III show similar thermal behaviors.
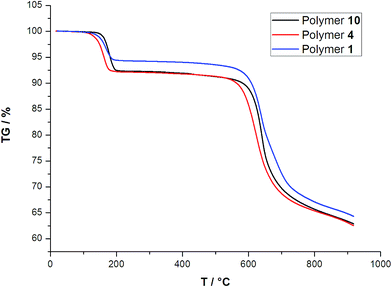 |
| | Fig. 4 Thermogravimetric analyses (TGA) curves of polymers 1, 4 and 10 under N2 atmosphere. | |
The TGA curves of type I, type II and type III display initial weight losses up to 137, 121 and 155 °C, respectively. The observed weight losses of 4.54% for type I, 8.05% for type II, and 7% for type III are in agreement with calculated value for the loss of coordinated water molecules (4.19% for type I, 8.35% for type II, and 7.43 for type III). The anhydrous phases are stable between 200 and 560 °C, and then the framework collapses. For example, in the polymer 10, this result is further confirmed by the PXRD analyses (Fig. S3 in the ESI†). It can be seen that, under 450 °C, the PXRD patterns of the different temperature are almost the same to that simulated from the single-crystal data, indicating the integrity of the framework of polymer 10 is robust to the exclusion of water.
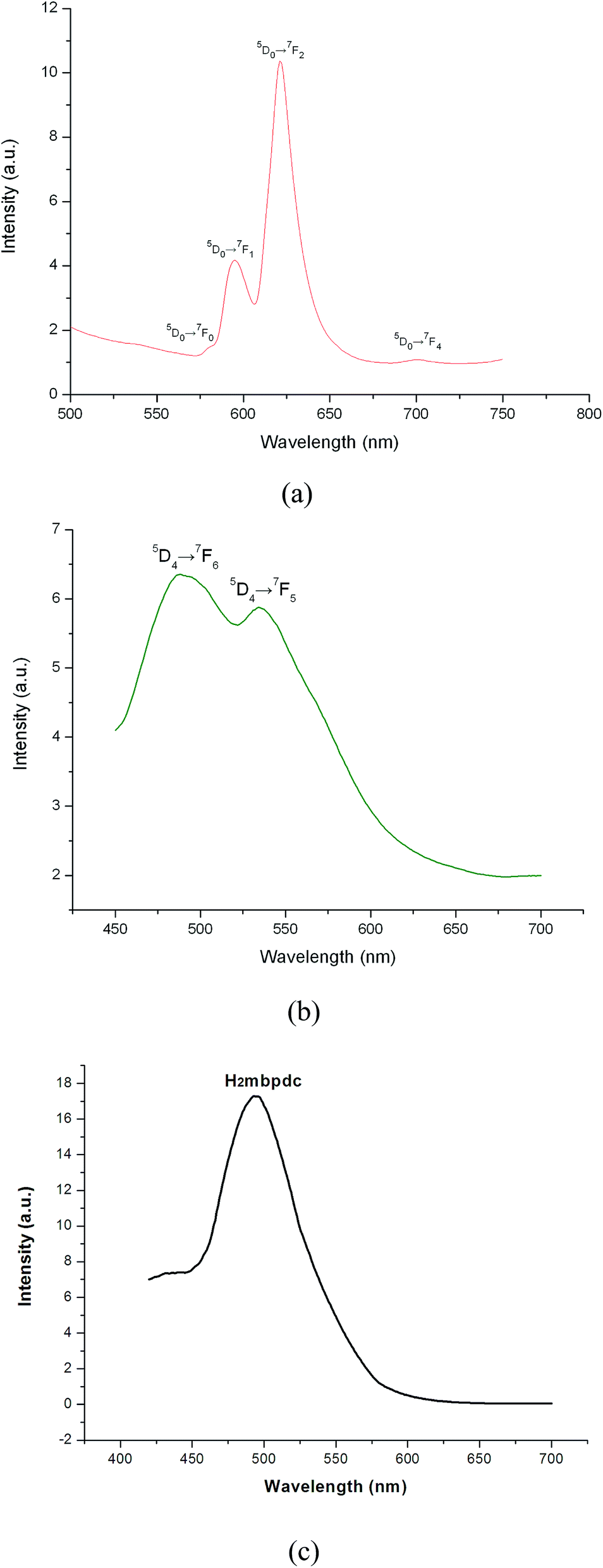
The luminescent spectra of 4, 6 and ligand in the solid were recorded at room temperature, as shown in Fig. 5. For the polymer 4, irradiated at 385 nm, the characteristic emissions of Eu(III) are obtained distinctly at the peaks of 580, 595, 621 and 701 nm, corresponding to 5D0 → 7F0, 5D0 → 7F1, 5D0 → 7F2, and 5D0 → 7F4 transitions. However, no broad emission band from the free ligand is observed, which indicates that the ligand transfers the absorbed energy effectively to the emitting level of the metal center. The symmetry forbidden transition of 5D0 → 7F0 (580 nm) shows that the Eu3+ ions in polymer 4 have the noncentrosymmetric coordination environment.24 As is well known, the 5D0 → 7F1 transition is magnetic dipole in nature and less sensitive to its environment, while 5D0 → 7F2 is electric dipole in origin and its intensity is strongly influenced by the crystal field.25,26 It is noted that the intensity of the 5D0 → 7F2 transition is ca. 2.8 times of the intensity of the 5D0 → 7F1 transition, which suggests that the Eu3+ ions occupy low symmetry coordination site with no inversion center, in consistent with the result from crystallographic analyses. In addition, the red emission of 5D0 → 7F2 transition is the most intense, which indicates that the H2mbpdc ligands are suitable for sensitization of red luminescence for Eu3+ ion under the experimental conditions, as observed in the literature.24 When H2mbpdc ligand is introduced to sensitize the Tb3+ ion, the emission spectrum of 6 exhibits the two characteristic emission bands of Tb3+ ion. They are assigned to the 5D4 → 7FJ (J = 5, 6), 5D4 → 7F6 (489 nm) and 5D4 → 7F5 (535 nm) transitions. However, there is a very intense broad background in the excitation spectrum of 6, which means there exists intramolecular π → π* transition of ligands. This luminescent phenomenon was observed in terbium complexes.27
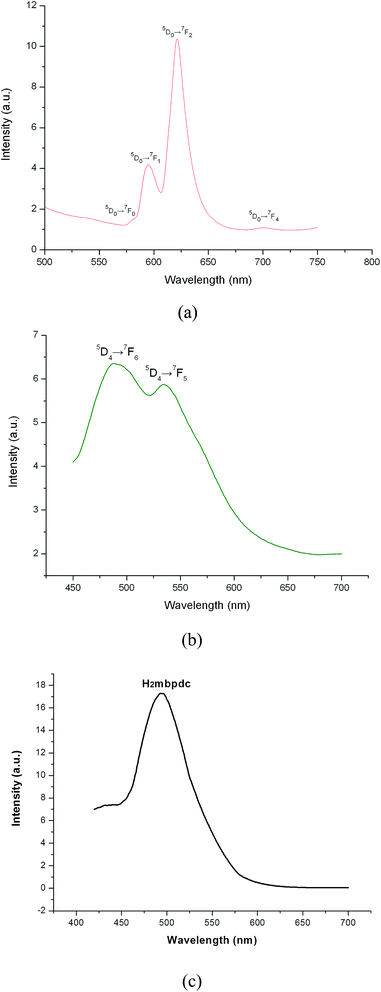 |
| | Fig. 5 Solid-state emission spectra of 4 (a), 6 (b) and ligand (c) at room temperature. | |
Magnetic properties
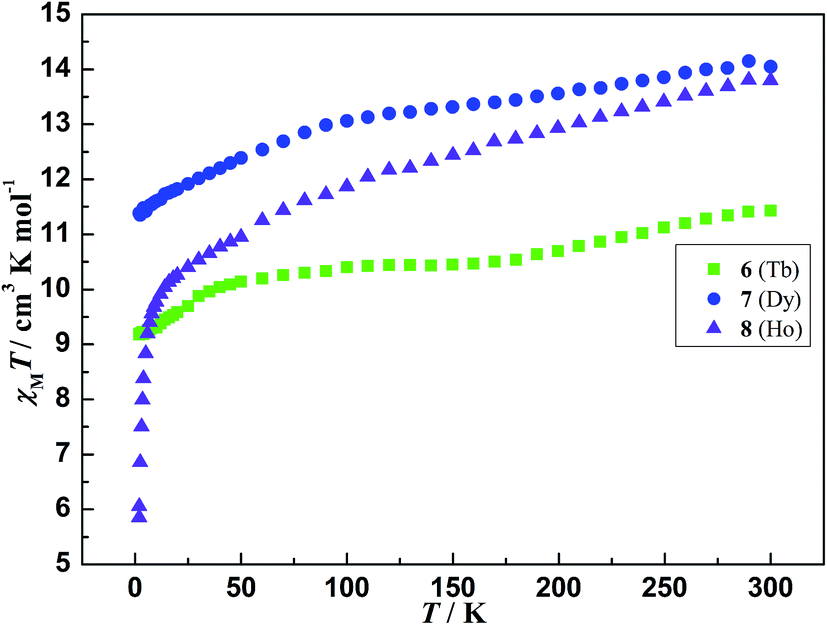
The temperature-dependent magnetic susceptibilities of 6–8 were measured with an applied magnetic field of 1000 Oe at the temperatures range of 2–300 K. The χMT value of 11.43, 14.05 and 13.80 cm3 K mol−1 at room temperature for 6–8 (Fig. 6), respectively, is slightly lower than the corresponding expected value of 11.82, 14.17 and 14.07 cm3 K mol−1 for one isolated Tb3+ (S = 3, L = 3, 7F6, g = 3/2),28 Dy3+ (S = 5/2, L = 5, 6H15/2, g = 4/3)29 and Ho3+ (S = 2, L = 6, 5I8, g = 5/4).30 As the temperature cooling, the χMT values of 6 and 7 gradually declines to 9.18 and 11.38 cm3 K mol−1 at 2 K, respectively. The value of 8 slowly decreases to 10.38 cm3 K mol−1 at 25 K. and then sharply drops to 5.87 cm3 K mol−1 at 2 K with further cooling. Although compounds 6, 7 and 8 are isomorphous, the different magnetic behavior at low temperature may originate from their different MJ sublevels.
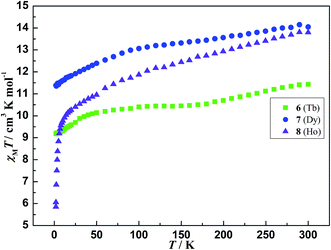 |
| | Fig. 6 The plots of χMT vs. T for compounds 6, 7 and 8. | |
As well known, the coupling between the lanthanide ions is relatively weak due to the efficient shielding of the unpaired electrons in their 4f orbitals. Significant coupling of lanthanide ions usually involves one or two atom bridges. For compounds 6–8, the carboxylic bridges amongst metal ions poorly mediate the magnetic interactions. The metal ions are quite far apart from each other with the shortest Ln⋯Ln distances of 6.0694(4) Å, 6.0483(4) Å, and 6.0561(3) Å, respectively, indicating that magnetic coupling between the lanthanide ions is likely to be rather weak. On the other hand, the inter-electronic repulsion and the spin-orbit coupling lead to the 4fn electron configurations of the lanthanide ions splitting into 2S + 1LJ states and further into MJ sublevels due to the crystal field perturbation. These sublevels are thermally populated at room temperature,31 and the progressive depopulations occur when the temperature decreases, causing the χMT value of lanthanide ions drop. As a result, the overall behaviors of χMT for 6–8 are mainly ascribed to thermal depopulation of the excited MJ sublevels split by the crystal field.
Conclusions
In summary, we have synthesized ten novel coordination polymers based on lanthanide ions and 3,3′-dimethoxy-2,2′-bipyridine-6,6′-dicarboxylic acid (H2mbpdc) through an in situ ligand transformation reaction. These polymers display three new structural types, from three to two-dimensional frameworks with respective nodes as the Ln3+ radii decrease. Polymers 1–3 exhibit highly ordered 3D frameworks with unprecedented 2D entangled layers of warp and woof threads interwoven by left- and right-handed helical chains (type I). While 4–10 show 2D 44 grids (type II and type III). Moreover, polymers 4 and 6 exhibit strong red and green photoluminescence at room temperature, respectively. The magnetic properties of compounds 6–8 were also studied by measuring their magnetic susceptibility over the temperature range of 2–300 K.
Experimental section
Materials and physical techniques
All chemicals were used as received without further purification. 3,3′-Dimethoxy-2,2′-bipyridine-6,6′-dicarboxylic acid (H2mbpdc) was prepared by the method described in the literature.21 The hydrothermal syntheses of 1–10 were carried out in 23 mL Teflon-lined autoclaves under autogenous. Element analyses for C, H, and N were performed on a Flash 1112 elemental analyzer. The infrared spectra were recorded on a Nicolet Avatar-360 spectrophotometer with pressed KBr pellets. Powder X-ray diffraction (PXRD) patterns were recorded using a Rigaku D/Max-IIIA diffractometer from 5 to 50° with a step size of 0.01052° and a scan speed of 0.4 s preset time. TGA (thermal gravimetric analyses) were carried out under nitrogen on a SDT Q600 with a heating rate of 2 °C min−1. Luminescent spectra were recorded with a Hitachi F4500 fluorescence spectrophotometer. The variable-temperature magnetic susceptibility measurements were carried out on polycrystalline samples on a Quantum Design MPMS-7 SQUID magnetometer in the temperature range of 2–300 K. Diamagnetic corrections were made with Pascal's constants for all of the constituent atoms.
Synthesis of the coordination Polymers
All ten polymers were prepared by the same synthetic routes, as follows: an aqueous mixture containing Ln(NO3)3·6H2O (Ln = La, Ce, Pr, Eu, Gd, Tb, Dy, Ho, Er, and Lu, 0.2 mmol), and H2mbpdc ligand (18.0 mg, 0.2 mmol) was allowed to place in a Parr Teflon lined stainless steel vessel (23 mL), and the vessel was sealed and heated to 180 °C for 3 days, then cooled to room temperature at a rate of 5 °C h−1. The suitable crystals of 1–10 were collected directly. For 1, yield: 0.036 g, 42% based on La. Anal. calcd (%) for C12H7LaN2O7: C, 33.51; H, 1.64; N, 6.51. Found: C, 33.69; H, 1.81; N, 6.67. For 2, yield: 0.037 g, 43% based on Ce. Anal. calcd (%) for C12H7CeN2O7: C, 33.42; H, 1.64; N, 6.49. Found: C, 33.58; H, 1.74; N, 6.76. For 3, yield: 0.048 g, 55% based on Pr. Anal. calcd (%) for C12H7PrN2O7: C, 33.46; H, 1.63; N, 6.48. Found: C, 33.56; H, 1.74; N, 6.68. For 4, yield: 0.044 g, 48% based on Eu. Anal. calcd (%) for C12H9EuN2O8: C, 31.25; H, 1.97; N, 6.07. Found: C, 31.45; H, 2.12; N, 6.18. For 5, yield: 0.056 g, 60% based on Gd. Anal. calcd (%) for C12H9GdN2O8: C, 30.90; H, 1.94; N, 6.01. Found: C, 31.25; H, 2.15; N, 6.19. For 6, yield: 0.056 g, 66% based on Tb. Anal. calcd (%) for C12H9TbN2O8: C, 30.79; H, 1.94; N, 5.98. Found: C, 31.05; H, 2.17; N, 6.15. For 7, yield: 0.064 g, 68% based on Dy. Anal. calcd (%) for C12H9DyN2O8: C, 30.55; H, 1.92; N, 5.94. Found: C, 30.85; H, 2.12; N, 6.17. For 8, yield: 0.064 g, 63% based on Ho. Anal. calcd (%) for C12H9HoN2O8: C, 30.40; H, 1.91; N, 5.91. Found: C, 30.65; H, 2.09; N, 6.11. For 9, yield: 0.066 g, 69% based on Er. Anal. calcd (%) for C12H9ErN2O8: C, 30.25; H, 1.90; N, 5.88. Found: C, 30.45; H, 2.04; N, 5.98. For 10, yield: 0.062 g, 64% based on Lu. Anal. calcd (%) for C12H9LuN2O8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C, 29.77; H, 1.87; N, 5.79. Found: C, 30.45; H, 2.04; N, 5.98.
:C, 29.77; H, 1.87; N, 5.79. Found: C, 30.45; H, 2.04; N, 5.98.
Crystallographic analyses
Crystallographic data of 1–10 were collected on a Bruker Smart Apex-II CCD area detector at room temperature (graphite-monochromated, Mo Kα-radiation, ω- and θ-scan technique, λ = 0.71073 Å). An empirical absorption correction was applied. The structures were solved by direct methods and refined on F2 using SHELXTL package.32,33 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were set in calculated positions and refined as riding atoms with a common isotropic thermal parameter. The crystallographic data for 1–10 are listed in Table 4.
Table 4 Crystallographic data and structure refinement summary for polymers 1–10
| |
1 |
2 |
3 |
4 |
5 |
| Formula |
C12H7LaN2O7 |
C12H7CeN2O7 |
C12H7PrN2O7 |
C12H9EuN2O8 |
C12H9GdN2O8 |
| M |
430.11 |
431.32 |
432.11 |
461.17 |
466.46 |
| Cryst syst |
Monocline |
Monocline |
Monocline |
Monocline |
Monocline |
| Space group |
Cc |
Cc |
Cc |
Cc |
Cc |
| a (Å) |
17.724(3) |
17.604(16) |
17.570(3) |
17.909(3) |
17.858(3) |
| b (Å) |
10.6818(15) |
10.624(9) |
10.6061(15) |
10.2645(15) |
10.2355(15) |
| c (Å) |
7.0003(10) |
6.996(6) |
6.9846(10) |
7.4561(11) |
7.4678(11) |
| α (°) |
90 |
90 |
90 |
90 |
90 |
| β (°) |
111.839(10) |
111.663(8) |
111.5980(10) |
96.9570(10) |
96.909(2) |
| γ (°) |
90 |
90 |
90 |
90 |
90 |
| V (Å3) |
1230.2(3) |
1216.1(19) |
1210.2(3) |
1360.5(3) |
1355.1(3) |
| Z |
4 |
4 |
4 |
4 |
4 |
| Dcalc (g cm−3) |
2.322 |
2.356 |
2.372 |
2.251 |
2.286 |
| μ (mm−1) |
3.514 |
3.785 |
4.069 |
4.658 |
4.942 |
| F(000) |
824 |
828 |
832 |
888 |
892 |
| Data/params |
4627/2254 |
4455/2229 |
4500/2208 |
4096/2296 |
4382/2425 |
| GOF on F2 |
1.022 |
1.080 |
1.086 |
1.024 |
1.128 |
| R1(I > 2σ) |
0.0143 |
0.0149 |
0.0148 |
0.0171 |
0.0181 |
| wR2(I > 2σ) |
0.0386 |
0.0397 |
0.0379 |
0.0419 |
0.0469 |
| Dimensionality |
3D network |
2D grid |
| |
6 |
7 |
8 |
9 |
10 |
| Formula |
C12H9TbN2O8 |
C12H9DyN2O8 |
C12H9HoN2O8 |
C12H9ErN2O8 |
C12H9LuN2O8 |
| M |
468.13 |
471.71 |
474.14 |
476.47 |
484.18 |
| Cryst syst |
Orthorhombic |
Orthorhombic |
Orthorhombic |
Orthorhombic |
Orthorhombic |
| Space group |
Pbcn |
Pbcn |
Pbcn |
Pbcn |
Pbcn |
| a (Å) |
8.4886(11) |
8.439(2) |
8.446(4) |
8.4022(15) |
8.3414(10) |
| b (Å) |
20.972(3) |
20.933(5) |
21.009(10) |
20.939(4) |
20.948(3) |
| c (Å) |
8.0409(10) |
8.016(2) |
8.016(4) |
7.9883(14) |
7.9372(10) |
| V (Å3) |
1431.4(3) |
1416.0(6) |
1422.4(12) |
1405.4(4) |
1386.9(3) |
| Z |
4 |
4 |
4 |
4 |
4 |
| Dcalc (g cm−3) |
2.172 |
2.213 |
2.214 |
2.252 |
2.319 |
| μ (mm−1) |
4.986 |
5.323 |
5.608 |
6.018 |
7.165 |
| F(000) |
896 |
900 |
904 |
908 |
920 |
| Data/params |
9879/1335 |
9575/1314 |
8996/1323 |
9727/1304 |
5910/2925 |
| GOF on F2 |
1.036 |
1.061 |
1.086 |
1.046 |
1.079 |
| R1(I > 2σ) |
0.0168 |
0.0179 |
0.0148 |
0.0147 |
0.0150 |
| wR2(I > 2σ) |
0.0397 |
0.0395 |
0.0379 |
0.0319 |
0.0368 |
| Dimensionality |
2D grid |
Acknowledgements
We are grateful to the Natural Science Foundations of China (Grant no. 21372112 and 21272109) and the science and technology innovation team support programs of Henan Province University (no. 2012IRTSTHN019) for financial support.
References
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS PubMed.
-
(a) D. Parker, Coord. Chem. Rev., 2000, 205, 109–130 CrossRef CAS;
(b) N. Sabbatini, M. Guardigli and J. M. Lehn, Coord. Chem. Rev., 1993, 123, 201–228 CrossRef CAS;
(c) J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357–2368 CrossRef CAS PubMed.
-
(a) H. C. Aspinall, Chem. Rev., 2002, 102, 1807–1850 CrossRef CAS PubMed;
(b) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187–2209 CrossRef CAS PubMed;
(c) R. J. Hill, D. L. Long, N. R. Champness, P. Hubberstey and M. Schroder, Acc. Chem. Res., 2005, 38, 335–348 CrossRef CAS PubMed;
(d) J. C. G. Bunzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
-
(a) L. Pan, K. M. Adams, H. E. Hernandez, X. Wang, C. Zheng, Y. Hattori and K. Kaneko, J. Am. Chem. Soc., 2003, 125, 3062–3067 CrossRef CAS PubMed;
(b) J. B. Yu, L. Zhou, H. J. Zhang, Y. X. Zheng, H. R. Li, R. P. Deng, Z. P. Peng and Z. F. Li, Inorg. Chem., 2005, 44, 1611–1618 CrossRef CAS PubMed;
(c) J. Yang, Q. Yue, G. D. Li, J. J. Cao, G. H. Li and J. S. Chen, Inorg. Chem., 2006, 45, 2857–2865 CrossRef CAS PubMed;
(d) L. Z. Zhang, W. Gu, B. Li, X. Liu and D. Z. Liao, Inorg. Chem., 2007, 46, 622–624 CrossRef CAS PubMed;
(e) X. Q. Zhao, P. Cui, B. Zhao, W. Shi and P. Cheng, Dalton Trans., 2011, 40, 805–819 RSC;
(f) P. F. Shi, B. Zhao, G. Xiong, Y. L. Hou and P. Cheng, Chem. Commun., 2012, 48, 8231–8233 RSC.
-
(a) Z. Zheng, Chem. Commun., 2001, 2521–2529 RSC;
(b) M. R. Bürgstein, M. T. Gamer and P. W. Roesky, J. Am. Chem. Soc., 2004, 126, 5213–5218 CrossRef PubMed;
(c) R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS PubMed;
(d) L. Candillas-Delgado, O. Fabelo, C. Ruiz-Perez, F. S. Delgado, M. Julve, M. Hernandez-Molina, M. Milagros Laz and P. Lorenzo-luis, Cryst. Growth Des., 2006, 6, 87–93 CrossRef;
(e) E. Deiters, B. Song, A. S. Chauvin, C. D. B. Vandevyver, F. Gumy and J. C. G. Bunzli, Chem.–Eur. J., 2009, 15, 885–900 CrossRef CAS PubMed;
(f) P. F. Shi, Y. Z. Zheng, X. Q. Zhao, G. Xiong, B. Zhao, F. F. Wan and P. Cheng, Chem.–Eur. J., 2012, 18, 15086–15091 CrossRef CAS PubMed;
(g) P. F. Shi, G. Xiong, B. Zhao, Z. Y. Zhang and P. Cheng, Chem. Commun., 2013, 49, 2338–2340 RSC.
-
(a) J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357–2368 CrossRef CAS PubMed;
(b) N. Marques, A. Sella and J. Takats, Chem. Rev., 2002, 102, 2137–2160 CrossRef CAS PubMed;
(c) J. C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC;
(d) A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS PubMed;
(e) H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed;
(f) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC;
(g) J. C. G. Bunzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
-
(a) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed;
(b) J. C. G. Bünzli and C. Piguet, Chem. Rev., 2002, 102, 1897–1928 CrossRef PubMed;
(c) F. A. A. Paz and J. Klinowski, Chem. Commun., 2003, 1484–1485 Search PubMed;
(d) X. Guo, G. Zhu, Q. Fang, M. Xue, G. Tian, J. Sun, X. Li and S. Qiu, Inorg. Chem., 2005, 44, 3850–3855 CrossRef CAS PubMed;
(e) J. Xia, B. Zhao, H. S. Wang, W. Shi, Y. Ma, H. B. Song, P. Cheng, D. Z. Liao and S. P. Yan, Inorg. Chem., 2007, 46, 3450–3458 CrossRef CAS PubMed;
(f) L. Huang, L. J. Han, W. J. Feng, L. Zheng, Z. B. Zhang, Y. Xu, Q. Chen, D. R. Zhu and S. Y. Niu, Cryst. Growth Des., 2010, 10, 2548–2552 CrossRef CAS.
-
(a) L. Pan, X. Huang, J. Li, Y. Wu and N. Zheng, Angew. Chem., Int. Ed., 2000, 39, 527–530 CrossRef CAS;
(b) A. Dimos, D. Tsaousis, A. Michaelides, S. Skoulika, S. Golhen, L. Ouahab, C. Didierjean and A. Aubry, Chem. Mater., 2002, 14, 2616–2622 CrossRef CAS;
(c) Z. He, E. Gao, Z. Wang, C. Yan and M. Kurmoo, Inorg. Chem., 2005, 44, 862–874 CrossRef CAS;
(d) F. N. Shi, L. Cunha-Silva, R. A. Sá Ferreira, L. Mafra, T. Trindade, L. D. Carlos, F. A. Almeida Paz and J. Rocha, J. Am. Chem. Soc., 2008, 130, 150–167 CrossRef CAS PubMed.
-
(a) W. Shi, X. Chen, Y. Zhao, B. Zhao, P. Cheng, A. Yu, H. Song, H. Wang, D. Liao, S. Yan and Z. Jiang, Chem.–Eur. J., 2005, 11, 5031–5039 CrossRef CAS PubMed;
(b) J. W. Cheng, S. T. Zheng and G. Y. Yang, Dalton Trans., 2007, 4059–4066 RSC.
-
(a) F. A. Cotton, G. Wilkinson, C. A. Murrillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley, New York, 6th edn, 1999, p. 1108 Search PubMed;
(b) S. Mizukami, H. Houjou, M. Kanesato and K. Hiratani, Chem.–Eur. J., 2003, 9, 1521–1528 CrossRef CAS PubMed;
(c) B. Liu, W. P. Wu, L. Hou and Y. Y. Wang, Chem. Commun., 2014, 50, 8731–8734 RSC;
(d) L. N. Jia, L. Hou, L. Wei, X. J. Jing, B. Liu, Y. Y. Wang and Q. Z. Shi, Cryst. Growth Des., 2013, 13, 1570–1576 CrossRef CAS.
- C. M. Liu, M. Xiong, D. Q. Zhang, M. Du and D. B. Zhu, Dalton Trans., 2009, 5666–5672 RSC.
-
(a) M. S. Liu, Q. Y. Yu, Y. P. Cai, C. Y. Su, X. M. Lin, X. X. Zhou and J. W. Cai, Cryst. Growth Des., 2008, 8, 4083–4091 CrossRef CAS;
(b) Y. W. Wang, Y. L. Zhang, W. Dou, A. J. Zhang, W. W. Qin and W. S. Liu, Dalton Trans., 2010, 39, 9013–9021 RSC;
(c) W. G. Lu, L. Jiang and T. B. Lu, Cryst. Growth Des., 2010, 10, 4310–4318 CrossRef CAS;
(d) J. Lin, P. Chai, K. Diefenbach, M. Shatruk and T. E. Albrecht-Schmitt, Chem. Mater., 2014, 26, 2187–2194 CrossRef CAS;
(e) J. Lin, K. Diefenbach, N. Kikugawa, R. E. Baumbach and T. E. Albrecht-Schmitt, Inorg. Chem., 2014, 53, 8555–8564 CrossRef CAS PubMed.
-
(a) A. Dimos, D. Tsaousis, A. Michaelides, S. Skoulika, S. Golhen, L. Ouahab, C. Didierjean and A. Aubry, Chem. Mater., 2002, 14, 2616–2622 CrossRef CAS;
(b) S. Mizukami, H. Houjou, M. Kanesato and K. Hiratani, Chem.–Eur. J., 2003, 9, 1521–1528 CrossRef CAS PubMed;
(c) Z. He, E. Gao, Z. Wang, C. Yan and M. Kurmoo, Inorg. Chem., 2005, 44, 862–874 CrossRef CAS;
(d) J. W. Cheng, T. S. Zheng and G. Y. Yang, Dalton Trans., 2007, 4059–4066 RSC;
(e) Y. H. Wan, X. J. Zheng, F. Q. Wang, X. Y. Zhou, K. Z. Wang and L. P. Jin, CrystEngComm, 2009, 11, 278–283 RSC.
-
(a) A. Huignard, T. Gacoin and J. P. Boilot, Chem. Mater., 2000, 12, 1090–1094 CrossRef CAS;
(b) W. L. Fan, Y. X. Bu, X. Y. Song, S. X. Sun and X. Zhao, Cryst. Growth Des., 2007, 7, 2361–2366 CrossRef CAS;
(c) C. Marchal, Y. Filinchuk, X. Y. Chen, D. Imbert and M. Mazzanti, Chem.–Eur. J., 2009, 15, 5273–5288 CrossRef CAS PubMed;
(d) L. W. Qian, J. Zhu, Z. Chen, Y. C. Gui, Q. Gong, Y. P. Yuan, J. T. Zai and X. F. Qiang, Chem.–Eur. J., 2009, 15, 1233–1240 CrossRef CAS PubMed;
(e) Z. J. Zhang, S. Y. Zhang, Y. Li, Z. Niu, W. Shi and P. Cheng, CrystEngComm, 2010, 12, 1809–1815 RSC.
-
(a) J. Jia, X. Lin, A. J. Blake, N. R. Champness, P. Hubberstey, L. Shao, G. Walker, C. Wilson and M. Schröder, Inorg. Chem., 2006, 45, 8838–8840 CrossRef CAS PubMed;
(b) B. L. Chen, L. B. Wang, Y. Q. Xiao, F. R. Fronczek, M. Xue, Y. J. Cui and G. D. Qian, Angew. Chem., Int. Ed., 2009, 48, 500–503 CrossRef CAS PubMed;
(c) S. L. Xiang, J. Huang, L. Li, J. Y. Zhang, L. Jiang, X. J. Kuang and C. Y. Su, Inorg. Chem., 2011, 50, 1743–1748 CrossRef CAS PubMed.
-
(a) F. Q. Wang, X. J. Zheng, Y. H. Wan, C. Y. Sun, Z. M. Wang, K. Z. Wang and L. P. Jin, Inorg. Chem., 2007, 46, 2956–2958 CrossRef CAS PubMed;
(b) D. F. Weng, X. J. Zheng, X. B. Chen, L. Li and L. P. Jin, Eur. J. Inorg. Chem., 2007, 3410–3415 CrossRef CAS;
(c) P. C. R. Soares-Santos, L. Cunha-Silva, F. A. Almeida Paz, R. A. S. Ferreira, J. Rocha, L. D. Carlos and H. I. S. Nogueira, Inorg. Chem., 2010, 49, 3428–3440 CrossRef CAS PubMed.
-
(a) X. Li, B. L. Wu, C. Y. Niu, Y. Y. Niu and H. Y. Zhang, Cryst. Growth Des., 2009, 9, 3423–3421 CrossRef CAS;
(b) W. Y. Wang, Z. L. Yang, C. J. Wang, H. J. Lu, S. Q. Zang and G. Li, CrystEngComm, 2011, 13, 4895–4902 RSC;
(c) S. L. Cai, S. R. Zheng, Z. Z. Wen, J. Fan and W. G. Zhang, Cryst. Growth Des., 2012, 12, 2355–2361 CrossRef CAS.
-
(a) N. R. Kelly, S. Goetz, S. R. Batten and P. E. Kruger, CrystEngComm, 2008, 10, 68–78 RSC;
(b) N. R. Kelly, S. Goetz, S. R. Batten and P. E. Kruger, CrystEngComm, 2008, 10, 1018–1026 RSC;
(c) B. M. Ji, D. S. Deng, X. He, B. Liu, S. B. Miao, N. Ma, W. Z. Wang, L. G. Ji, P. Liu and X. F. Li, Inorg. Chem., 2012, 51, 2170–2177 CrossRef CAS PubMed.
-
(a) J. R. Lombardi and B. Davis, Chem. Rev., 2002, 102, 2431–2460 CrossRef CAS PubMed;
(b) J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS PubMed.
-
(a) T. J. J. Kinnunen, M. Haukka and T. A. Pakkanen, J. Organomet. Chem., 2002, 654, 8–15 CrossRef CAS;
(b) U. Dawid, F. P. Pruchnik and R. Starosta, Dalton Trans., 2009, 3348–3353 RSC.
- B. M. Ji, D. S. Deng, H. H. Lan, C. X. Du, S. L. Pan and B. Liu, Cryst. Growth Des., 2010, 10, 2851–2853 CAS.
-
(a) D. S. Deng, L. L. Liu, B. M. Ji, G. J. Yin and C. X. Du, Cryst. Growth Des., 2012, 12, 5338–5342 CrossRef CAS;
(b) H. B. Zhu and S. H. Gou, Coord. Chem. Rev., 2011, 255, 318–338 CrossRef CAS PubMed;
(c) H. Zhao, Z. R. Qu, H. Y. Ye and R. G. Xiong, Chem. Soc. Rev., 2008, 37, 84–100 RSC;
(d) X. D. Chen, H. F. Wu and M. Du, Chem. Commun., 2008, 1296–1298 RSC;
(e) X. M. Chen and M. L. Tong, Acc. Chem. Res., 2007, 40, 162–170 CrossRef CAS PubMed.
-
(a) Y. H. Feng, Y. Guo, Y. O. Yang, Z. Q. Liu, D. Z. Liao, P. Cheng, S. P. Yan and Z. H. Jiang, Chem. Commun., 2007, 3643–3645 RSC;
(b) X. F. Wang, Y. B. Zhang, X. N. Cheng and X. M. Chen, CrystEngComm, 2008, 10, 753–758 RSC.
-
(a) B. Zhao, X. Y. Chen, P. Cheng, D. Z. Liao, S. P. Yan and Z. H. Jiang, J. Am. Chem. Soc., 2004, 126, 15394–15395 CrossRef CAS PubMed;
(b) X. J. Gu and D. F. Xue, CrystEngComm, 2007, 9, 471–477 RSC.
- G. Vicentini, L. B. Zinner, J. Zukerman-Schpector and K. Zinner, Coord. Chem. Rev., 2000, 196, 353–382 CrossRef CAS.
-
(a) A. F. Kirby, D. Foster and F. S. Richardson, Chem. Phys. Lett., 1983, 95, 507–512 CrossRef CAS;
(b) J. C. G. Bünzli and G. R. Choppin, in Lanthanide Probes in life, Chemical and Earth Science. Theory and Practice, Elsevier Scientific Publishers, Amsterdam, The Netherlands, 1989, ch. 7 (ref:1.2 IC) Search PubMed.
-
(a) S. Raphael, M. L. P. Reddy, A. H. Cowley and M. Findlater, Eur. J. Inorg. Chem., 2008, 4387–4394 CrossRef CAS;
(b) X. Yang, R. A. Jones and W. K. Wong, Chem. Commun., 2008, 3266–3268 RSC.
- C. A. Black, J. S. Costa, W. T. Fu, C. Massera, O. Roubeau, S. J. Teat, G. Aromi, P. Gamez and J. Reedijk, Inorg. Chem., 2009, 48, 1062–1068 CrossRef CAS PubMed.
-
(a) Q. Y. Liu, W. F. Wang, Y. L. Wang, Z. M. Shan, M. S. Wang and J. K. Tang, Inorg. Chem., 2012, 51, 2381–2392 CrossRef CAS PubMed;
(b) G. F. Xu, Q. L. Wang, P. Gamez, Y. Ma, R. Clerac, J. K. Tang, S. P. Yan, P. Cheng and D. Z. Liao, Chem. Commun., 2010, 46, 1506–1508 RSC;
(c) Y. Liu, Z. Chen, J. Ren, X. Q. Zhao, P. Cheng and B. Zhao, Inorg. Chem., 2012, 51, 7433–7435 CrossRef CAS PubMed.
- Z. Majeed, K. C. Mondal, G. E. Kostakis, Y. Lan, C. E. Anson and A. K. Powell, Chem. Commun., 2010, 46, 2551–2553 RSC.
- M. Fang, B. Zhao, Y. Zuo, J. Chen, W. Shi, J. Liang and P. Cheng, Dalton Trans., 2009, 7765–7770 RSC.
- SHELXTL, version 5.1, Bruker AXS, Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SHELXS-97 and SHELXL-97, Program for X-ray Crystal Structure Solution, University of Göettingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 975869–975877. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra12714e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.