
Facile synthesis of Ag-doped ZnCdS nanocrystals and transformation into Ag-doped ZnCdSSe nanocrystals with Se treatment†
Abstract
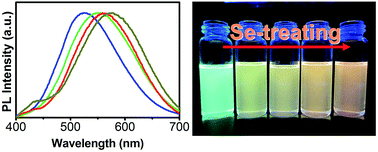
High-quality, pure, and color-tunable Ag:ZnCdS quantum dots (d-dots) are prepared by optimization of the experimental conditions including Ag-doping concentration and Zn/Cd precursor ratio. Highly emissive Ag:ZnCdS/ZnS core/shell d-dots with photoluminescence quantum yield (PL QY) as high as 58% are constructed in situ by the growth of a ZnS shell around the crude Ag:ZnCdS solution, which is the highest PL QY reported to date for Ag-based semiconductor d-dots. The emission color of a Ag:ZnCdS d-dot can be tuned toward a larger red region by simple Se treatment at high temperature (220–260 °C). With Se treatment, Ag:ZnCdS alloyed d-dots are transformed into Ag:ZnCdSSe alloyed d-dots, and the corresponding optical changes of d-dots in this process are investigated systematically. This strategy provides a versatile approach for the preparation of other multinary semiconductor nanocrystals.
 Please wait while we load your content...
Please wait while we load your content...

