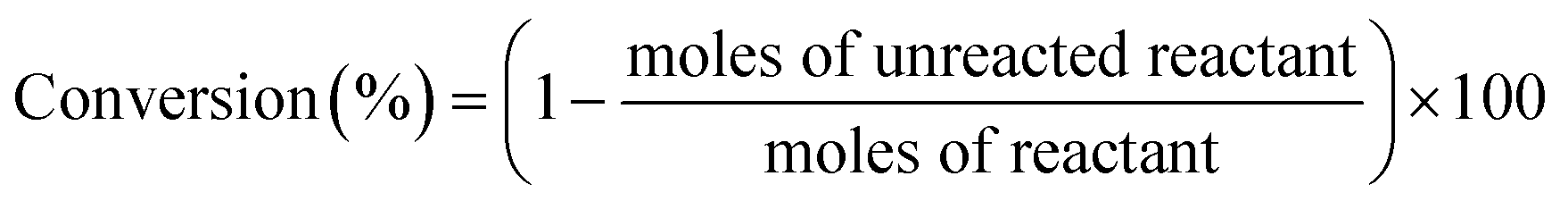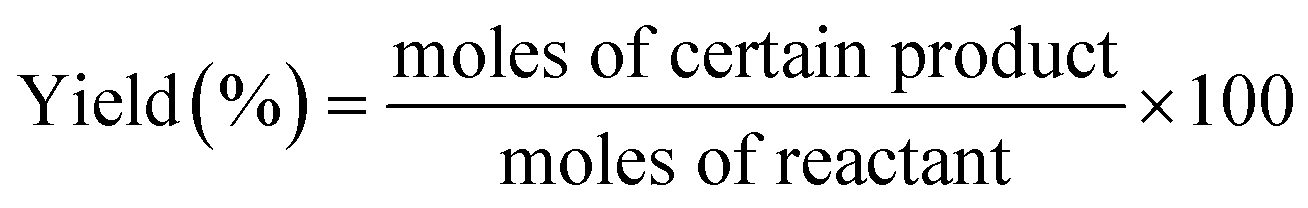DOI:
10.1039/C4RA10783G
(Paper)
RSC Adv., 2015,
5, 485-492
Preparation of 1,4-cyclohexanedimethanol by selective hydrogenation of a waste PET monomer bis(2-hydroxyethylene terephthalate)
Received
19th September 2014
, Accepted 24th November 2014
First published on 24th November 2014
Abstract
A new approach is developed for the preparation of 1,4-cyclohexanedimethanol (CHDM) by hydrogenation of bis(2-hydroxyethylene terephthalate) (BHET) obtained from waste poly(ethylene terephthalate) (PET). The influences of various reaction parameters including temperature, pressure and time, on the hydrogenation reaction were studied, and the 100% conversion of BHET and 78% yield of CHDM were achieved with Pd/C and Cu-based catalysts. X-ray diffraction, low temperature N2 adsorption–desorption and H2 temperature-programmed reduction were used to characterize the Cu-based catalysts, demonstrating that the Cu+/Cu0 species are the active sites. This study not only provides a new route for the production of CHDM, but also an approach for the efficient utilization of waste PET.
1. Introduction
1,4-Cyclohexanedimethanol (CHDM) is a highly valued and extensively used chemical in the polymer industry. It is used in paints and particularly as a modified material for fibers and polyesters1 such as poly(ethylene terephthalate-co-1,4-cylclohexylenedimethylene terephthalate) (PETG) with excellent performance such as thermal resistance, weather resistance and physical strength.2 Due to the wide application of the high performance polyesters, the demand of CHDM will be increasing significantly. Industrially, CHDM is prepared by a two-step process using two reactors. The first step is the highly exothermic conversion of dimethyl terephthalate (DMT) into dimethyl hexahydroterephthalate (DMHT) by the hydrogenation of the phenyl group using catalysts including Ni, Pd, Ru, Rh, added Pt, Ba, Ca and other additives in the temperature range of 433–453 K and an H2 pressure of 5–48 MPa.3–7 The second step is the conversion of DMHT into CHDM by the hydrogenation of the ester groups to hydroxyl groups using conventional Cu-based catalysts8,9 such as Cu–Cr or Cu–Zn catalysts,10 added Mn, Ba and other catalysts containing Ru11–13 and Fe-based catalysts bearing a PNP-pincer ligand14 in the temperature range of 473–573 K and the reaction pressure of 3–15 MPa.
There are some studies on the preparation of CHDM by other means. A new process for producing CHDM by the hydrogenation of terephthalic acid (TPA) into 1,4-cyclohexanedicarboxylic acid (CHDA) using Pd- or Ru-based catalysts, and further getting CHDM from CHDA15 was described. Recently, Thomas and co-workers reported that bimetallic nanoparticle catalysts,16 such as silica-supported Ru5Pt, Ru10Pt2, Ru6Pd6, Ru12Cu4 and a trimetallic cluster Ru5PtSn17 can promote the single-step conversion of DMT into CHDM under mild reaction conditions.18 However, both the conversion and selectivity are not so satisfactory.
However, the above raw materials are derived from petroleum industries and the hydrogenation processes are carried out in solvents such as ethyl acetate, tetrahydrofuran and methanol, which increase the difficulty in the separation of products and the cost of the process. Therefore, it is imperative to find a low-cost material and a solvent-free process for the synthesis of CHDM.
Herein, we developed a new solvent-free route to produce CHDM using the waste poly(ethylene terephthalate) (PET) derived monomer bis(2-hydroxyethylene terephthalate) (BHET) as the raw material. The low-cost material and the solvent-free process can reduce the cost and simplify the process significantly. BHET can be easily obtained from the glycolysis of waste PET, which was carried out conventionally in ethylene glycol solution under 393–463 K, 1–5 h and atmospheric pressure using various kinds of catalysts such as zinc acetate, cobalt acetate19–22 and ionic liquids.23–25 BHET has a potential for the conversion into chemicals of utility, which can be used as the raw material to synthesize the fatty amide derivatives,26 unsaturated polyester resin27 and hydrophobic textile dyestuffs.28 However, there are still few studies on the application of BHET. Therefore, transforming BHET easily obtained from waste PET into high value-added CHDM is of great significance, which contributes to the recycling of resources.
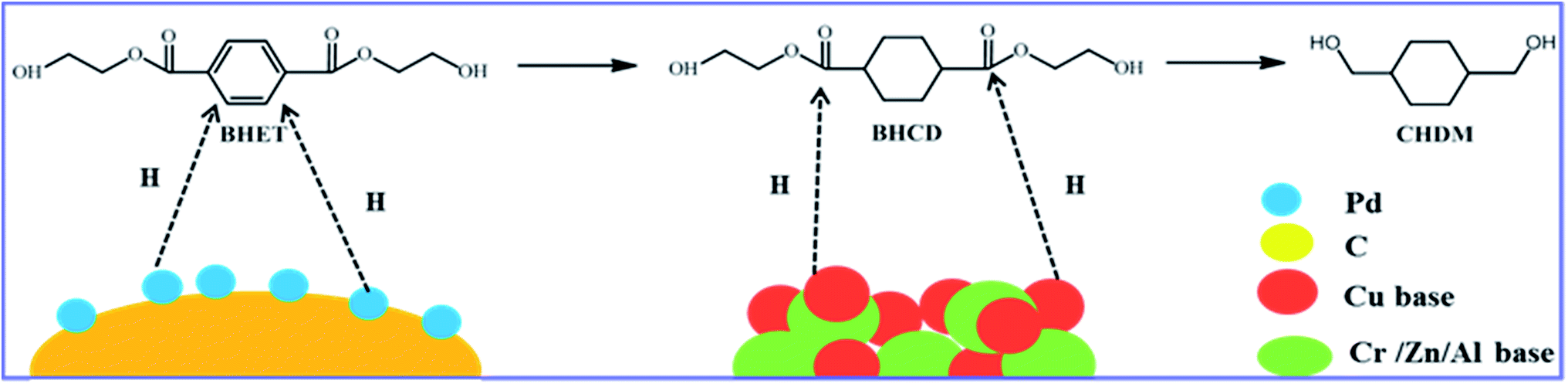
In this paper, it is found that BHET obtained from waste PET can be efficiently converted to CHDM as shown in Scheme 1, which has not been reported to date. It was carried out by a two-step process using the traditional catalyst Pd/C and Cu-based catalysts. In the first step, the phenyl group was selectively hydrogenated to cyclohexane catalyzed by Pd/C catalyst. In the second step, the ester groups were selectively hydrogenated to hydroxyl groups catalyzed by Cu-based catalysts. The aim of this paper is to provide a new method that can selectively hydrogenate BHET to CHDM. The influences of various reaction parameters, including reaction temperature, pressure and time, on the two hydrogenate reactions were investigated to get the optimal reaction conditions. Moreover, X-ray diffraction, low temperature N2 adsorption–desorption and H2 temperature-programmed reduction were used to characterize the Cu-based catalysts.
 |
| | Scheme 1 BHET obtained from waste PET is converted to CHDM. | |
2. Experimental
2.1. Materials
BHET (≥99%) was obtained by the degradation of the waste PET in our laboratory. Copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, ≥99%) and chromium(III) nitrate nonahydrate (Cr(NO3)3·9H2O, ≥99%) were obtained from Sinopharm Chemical Reagent Co., Ltd, China. Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, ≥99%), aluminum nitrate nonahydrate (Al(NO3)3·9H2O, ≥99%), and sodium carbonate (Na2CO3, ≥99%) were obtained from Xilong Chemical Co., Ltd, China. 10% Pd/C was obtained from J & K Scientific Ltd, China. The materials were used without any further treatment.
2.2. Preparation of Cu-based catalysts
2.2.1. Preparation of Cu–Cr catalyst. The Cu–Cr catalyst was prepared by a conventional co-precipitation method.29 In a typical synthesis, Cu(NO3)2·3H2O and Cr(NO3)3·9H2O with a desired molar ratio (Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr = 1:1) were dissolved in 50 mL deionized water at 333 K with stirring, the solution was precipitated by adding sodium carbonate until the pH of the solution reached 8. The precipitate was aged at 333 K for 3 h and then filtered and washed several times with deionized water. The obtained solid sample was dried at 393 K for 12 h in an oven and then calcined at 723 K for 4 h to provide the as-prepared oxide. The as-prepared oxide was reduced by 5% H2 in Ar at the programmed temperature which is rising from room temperature to 493 K staying 1 h by the rate of 4 K min−1, then to 623 K staying 2 h by the rate of 1 K min−1 to obtain the final Cu–Cr catalyst.
:Cr = 1:1) were dissolved in 50 mL deionized water at 333 K with stirring, the solution was precipitated by adding sodium carbonate until the pH of the solution reached 8. The precipitate was aged at 333 K for 3 h and then filtered and washed several times with deionized water. The obtained solid sample was dried at 393 K for 12 h in an oven and then calcined at 723 K for 4 h to provide the as-prepared oxide. The as-prepared oxide was reduced by 5% H2 in Ar at the programmed temperature which is rising from room temperature to 493 K staying 1 h by the rate of 4 K min−1, then to 623 K staying 2 h by the rate of 1 K min−1 to obtain the final Cu–Cr catalyst.
2.2.2. Preparation of Cu–Zn/Al2O3 catalyst. Firstly, an aqueous solution was prepared with Cu(NO3)2·3H2O, Zn(NO3)2·6H2O and Al(NO3)3·9H2O with molar ratio of Cu2+:Zn2+:Al3+ = 5:4:1,30 then the solution was precipitated by adding sodium carbonate until the pH of the solution reached 7 at 333 K with stirring. The precipitate was aged at 333 K for 3 h and then filtered and washed several times by deionized water. The obtained solid sample was dried at 393 K for 12 h in an oven and then calcined at 723 K for 4 h to provide the as-prepared oxide. The as-prepared oxide was reduced by H2 under 573 K before use.
2.3. Hydrogenation reaction of BHET to CHDM
All hydrogenation reactions were carried out in a 100 mL stainless steel high pressure reactor (MSC County Petroleum Research Instruments, Jiangsu, China), equipped with magnetic stirring. The hydrogenation reaction of BHET to CHDM was carried out by a two-step process as shown in Scheme 2. In the first step, the reactor was charged with 2.0 g of BHET as starting material and 0.1 g of 10% Pd/C catalyst.31 The reactor was then purged with H2 three times to remove air and then pressurized to the desired pressure of H2 and heated to the desired temperature under stirring of 400 rpm. After the reaction, the intermediate products which are mainly consisted of bis(2-hydroxyethyl) cyclohexane-1,4-dicarboxylate (BHCD) was obtained and then used as the raw material for the second step. In the second step, 2.0 g of the intermediate products and 0.1 g of Cu-based catalysts were placed in the reactor, the reactor was then purged with H2 three times to remove air and then pressurized to the desired pressure of H2 and heated to the desired temperature under stirring of 400 rpm, the target product CHDM was obtained.
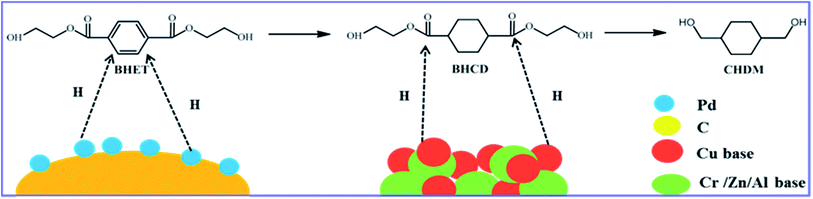 |
| | Scheme 2 The two-step process for the hydrogenation of BHET to CHDM without solvent. | |
2.4. Characterization and analysis
The structures and crystal phase compositions of the Cu–Cr and Cu–Zn/Al2O3 catalysts were determined by Powder X-ray diffraction (XRD) patterns with a Bruker D8-Advance X-ray powder diffractometer using Cu Kα radiation within a 2θ range of 5–85° in steps of 10°, operated at an accelerating voltage of 40 kV and an emission current of 200 mA.
Temperature-programmed reduction (TPR) of the Cu-based catalysts was carried out in a stream of 90% Ar and 10% H2 with a flowing rate of 50 mL min−1, and the samples were heated at 10 K min−1 from room temperature to 773 K.
The intermediate products of the first step were analyzed by HPLC (Agilent 1100) fitted with a refractive index detector and an ODS-BP column (0.46 mm × 250 mm). The mixture of acetonitrile and water with a ratio of 1:1 was used as mobile phase. The oven temperature for the determination of standards and samples was set at 313 K with a flow rate of 0.5 mL min−1. The final products from the second step were analyzed by gas chromatography on a GC-2014 instrument (Shimadzu Corporation) equipped with a HP-5 chromatographic column and a flame ionization detector (FID). The products were identified by GC (Agilent 6890N)-MS (Agilent 5975B) equipped with a HP-5ms UI chromatographic column. The conversion and yield of products were calculated by eqn (1) and (2) as follows:
| |
 | (1) |
| |
 | (2) |
3. Results and discussion
3.1. Hydrogenation of BHET to BHCD
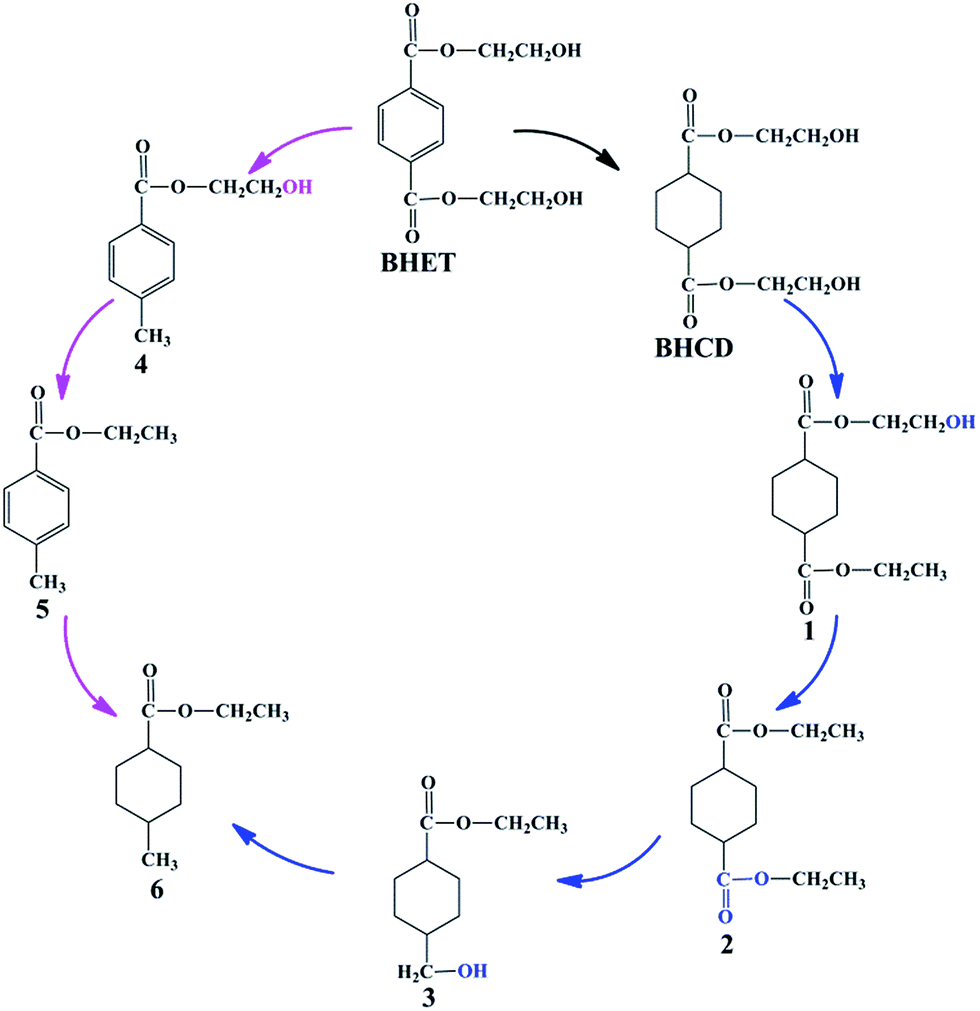
In the first step, BHET was converted to BHCD by the selective hydrogenation of phenyl group to cyclohexane group, which was performed on 10% Pd/C catalyst without solvent. In order to get the maximum direction of the main reaction, we investigated the influences of different reaction conditions on the reaction to get the optimal reaction conditions. Moreover, we report the possible reaction pathway for the hydrogenation of BHET to BHCD as shown in Scheme 3.
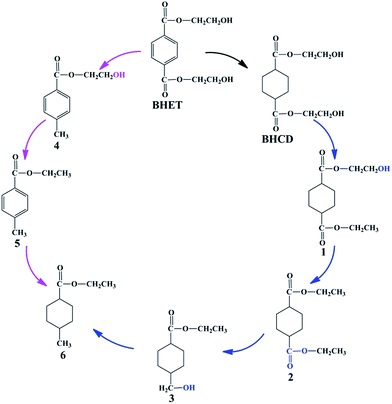 |
| | Scheme 3 The possible reaction pathway for the hydrogenation of BHET to BHCD. | |
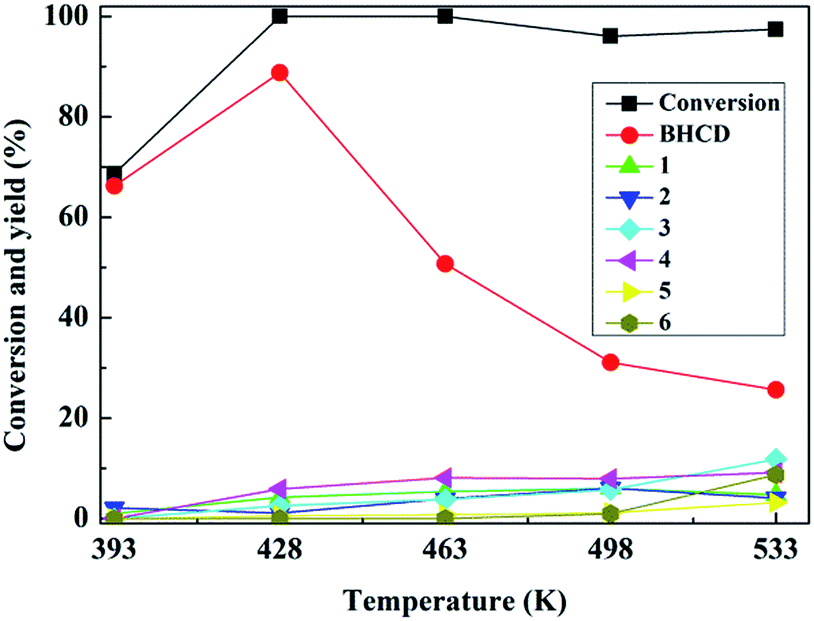
3.1.1. Influence of reaction temperature. The influence of reaction temperature on the conversion of BHET and yields of BHCD and main by-products is shown in Fig. 1. It displays that the reaction temperature is particularly important for the hydrogenation reaction. The conversion of BHET increased and reached almost 100% when the reaction temperature reached 428 K. The yield of BHCD initially increased and then decreased with the increase of the reaction temperature.
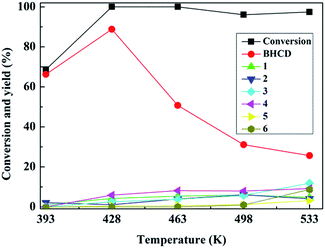 |
| | Fig. 1 Influence of reaction temperature on the conversion of BHET and yields of BHCD and main by-products. Reaction conditions: 2.0 g BHET, 0.1 g 10% Pd/C, 3.5 MPa, and 5 h. | |
Because the further increase of the temperature, the reaction shifted to side pathways including the hydrogenation of the ester and hydroxyl groups of BHET which is confirmed by the appearance of the by-products 4 and 5, and the excessive hydrogenation of the ester and hydroxyl groups of BHCD which is confirmed by the appearance of the by-products 1, 2, 3 and 6. The yields of by-products 3 and 5 increased while the yields of by-products 1, 2, and 4 decreased with the increase of the reaction temperature. The by-product 6 occurred when the temperature was above 498 K, which indicates that the further hydrogenation becomes easy at high temperature. It can be concluded that 428 K is the optimal reaction temperature for the hydrogenation of BHET to BHCD.
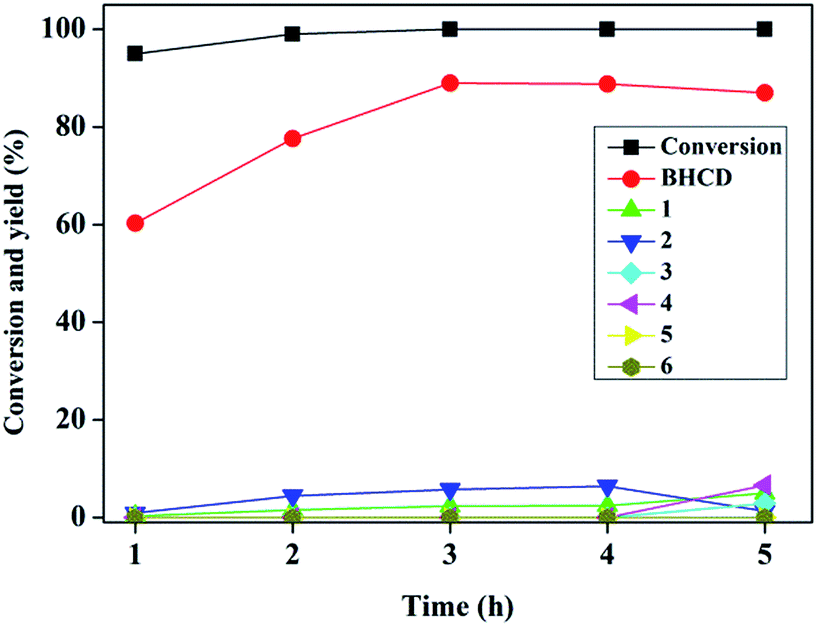
3.1.2. Influence of reaction time. Fig. 2 presents the influence of reaction time on the conversion of BHET and yields of BHCD and main by-products. It is shown that the conversion of BHET increased with the increase of the reaction time. However, the yield of the BHCD increased rapidly in reaction time before 3 h but decreased slightly with the further increase of the reaction time, which implies that the further hydrogenation of BHCD leads to more side reactions. The yield of the by-product 1 increased, while the yield of the by-product 2 increased initially and then decreased with the increase of the reaction time. Moreover, the by-products 3 and 4 occurred when the reaction time was longer than 4 h, indicating that the terminal hydroxyl group is easier to be reduced than the ester group catalyzed by Pd/C, and the further increase of the reaction time would not only reduce the yield of the main product but also not distinctly affect the conversion. Based on the analysis, a reaction time of 3 h gave the highest product yield and lowest side products and therefore it is regarded as the best reaction time.
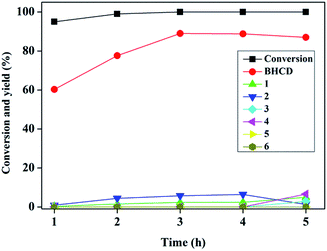 |
| | Fig. 2 Influence of reaction time on the conversion of BHET and yields of BHCD and main by-products. Reaction conditions: 2.0 g BHET, 0.1 g 10% Pd/C, 3.5 MPa, and 428 K. | |
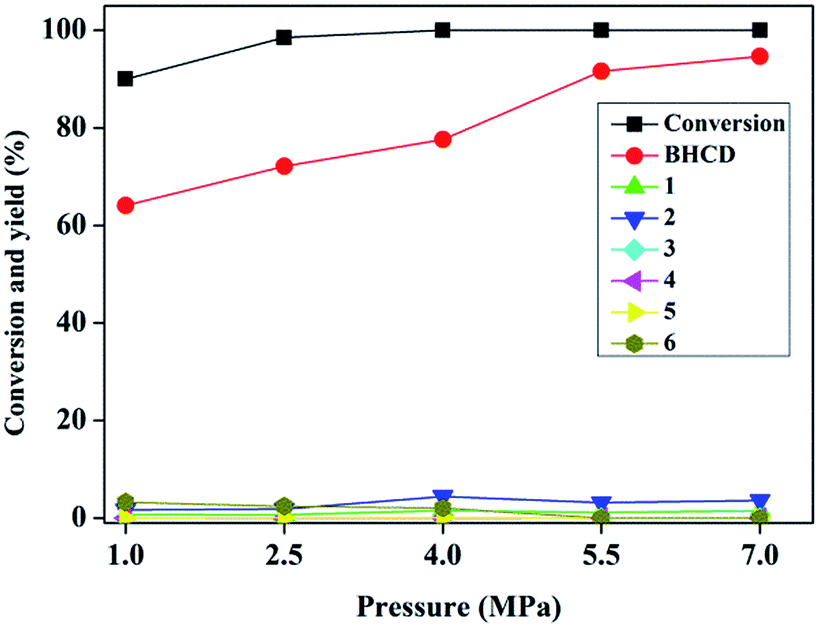
3.1.3. Influence of reaction pressure. Fig. 3 shows the influence of reaction pressure on the conversion of BHET and yields of BHCD and main by-products. It is observed that with an increase of the reaction pressure, the conversion of BHET increased and the yield of the BHCD increased and the rate of the increase in the yield of BHCD became smaller. The by-products 5 and 6 disappeared when the reaction pressure was above 5.5 MPa. The by-products 1 and 2 increased and then decreased with the increase of the reaction pressure. However, the by-products 3 and 4 were not detected in the products. It indicates that the high pressure improves the selectivity of the hydrogenation of phenyl to cyclohexane, but could not remove the further hydrogenation of ester and hydroxyl groups. It can be concluded that a certain range under 7 MPa of the high pressure can not only drive the equilibrium to move to the desired direction but also prevent the catalysts from losing activity.
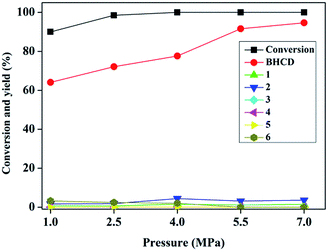 |
| | Fig. 3 Influence of reaction pressure on the conversion of BHET and yields of BHCD and main by-products. Reaction conditions: 2.0 g BHET, 0.1 g 10% Pd/C, 2 h, and 428 K. | |
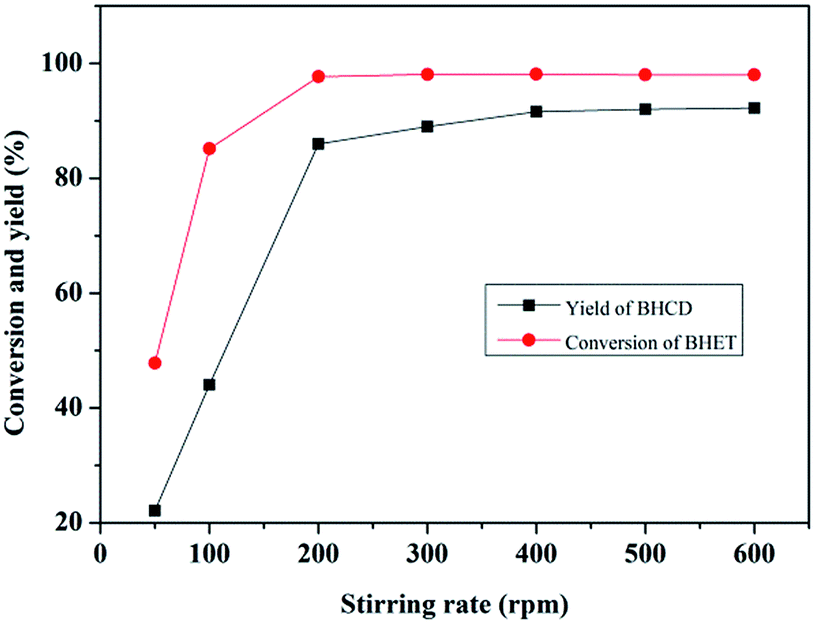
3.1.4. Influence of stirring rate. Fig. 4 shows the influence of stirring rate on the conversion of BHET and the yield of BHCD. As can be seen, the conversion of BHET and yield of BHCD firstly increased with the increase of the stirring rate from 50 to 300 rpm, and then kept stable from 300 to 600 rpm. Thus, there should be no mass or heat transfer limitation with a stirring rate higher than 300 rpm.
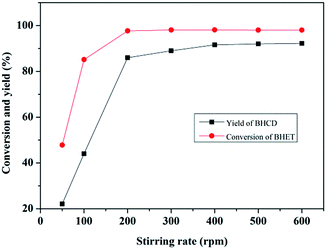 |
| | Fig. 4 Influence of stirring rate on the conversion of BHET and the yield of BHCD. Reaction conditions: 2.0 g BHET, 0.1 g Pd/C, 5.5 MPa, 428 K, and 2 h. | |
As a consequence, the Pd/C catalyst shows high activity and selectivity of the hydrogenation of BHET to BHCD without solvent under respective mild reaction conditions. Under the optimal reaction conditions of 428 K, 3 h, and 7 MPa, the conversion of BHET and the yield of BHCD have reached 100% and 95%, respectively.
3.2. Hydrogenation of BHCD to CHDM
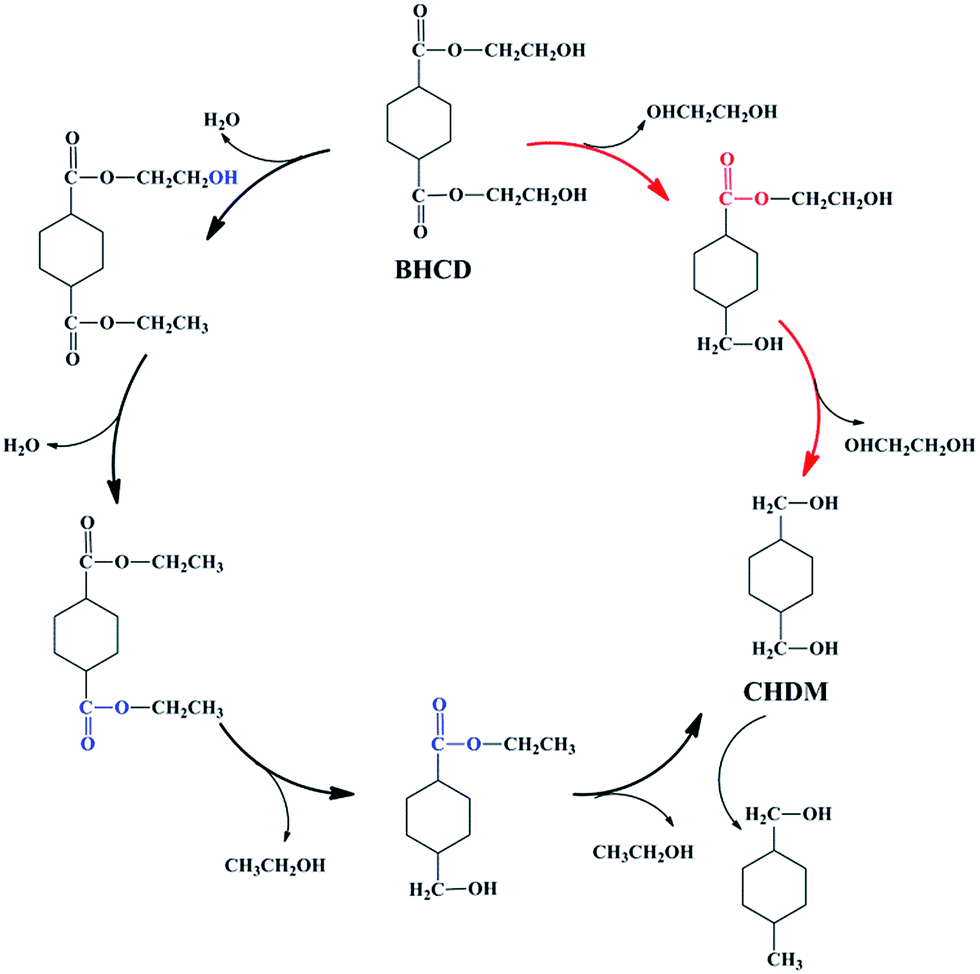
In the second step, BHCD with a certain purity (90%) obtained from the first step was converted to CHDM by the selective hydrogenation of ester group. However, it is well known that the hydroxyl group can be hydrogenated by the Cu-based catalysts,32,33 which make the second step difficult because of the hydroxyl groups in BHET. As shown in Scheme 4, we suggest the possible reaction pathway for the hydrogenation of BHCD to CHDM. The hydrogenation reaction follows two pathways: (i) the ester group in one side chain is firstly reduced to hydroxyl group, and then the other ester group is reduced, (ii) the terminal hydroxyl groups are firstly reduced and then the ester groups are reduced. The reaction pathway (i) and (ii) produce ethylene glycol and ethanol, respectively. The amount of ethylene glycol is much higher than ethanol, which implies that the reaction mainly follows pathway (i). We used Cu–Cr and Cu–Zn/Al2O3 as catalysts in the second step. In order to get the maximum yield of CHDM, we investigated the influences of different reaction conditions on the hydrogenation reaction of BHCD to CHDM.
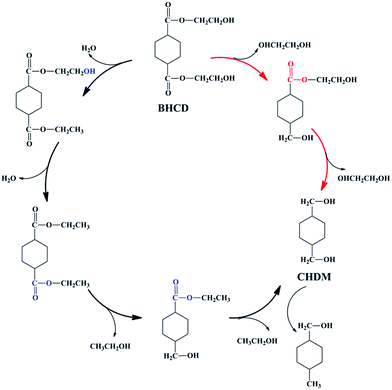 |
| | Scheme 4 The possible reaction pathway for the hydrogenation of BHCD to CHDM. | |
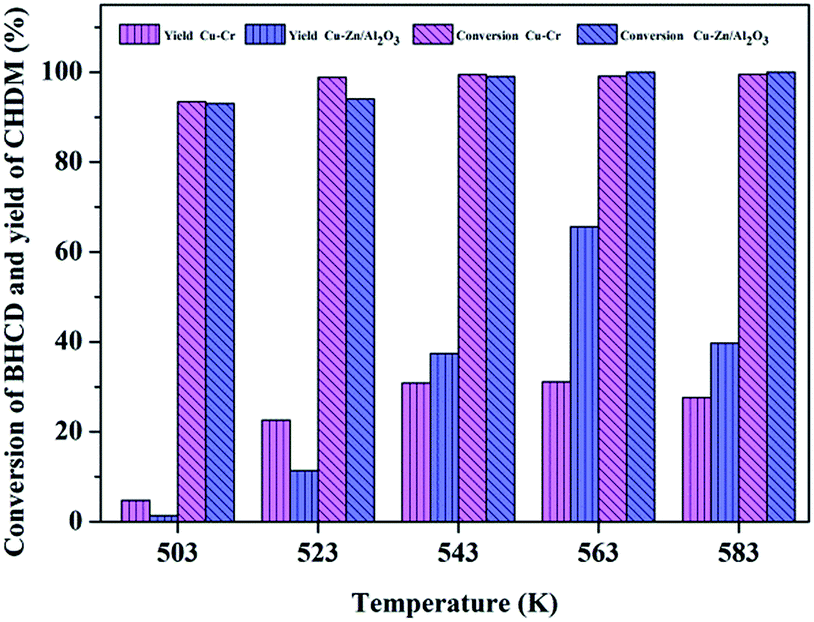
3.2.1. Influence of reaction temperature. Fig. 5 shows the influence of reaction temperature on the conversion of BHCD and yield of CHDM catalyzed by Cu–Cr and Cu–Zn/Al2O3 catalysts. The conversion of BHCD reached almost 100% at the temperature of 543 K and 523 K catalyzed by Cu–Cr and Cu–Zn/Al2O3 catalysts, respectively. However, the yield of CHDM initially increased and then decreased with the increase of the reaction temperature, due to the excessive hydrogenation of BHCD. A reaction temperature of 563 K is the best for the hydrogenation reaction, affording about 32% and 66% CHDM yield catalyzed by Cu–Cr and Cu–Zn/Al2O3 catalysts, respectively. When the temperature was higher than 563 K, the hydroxyl groups in CHDM would be further reduced, resulting in the increase of the by-products. Moreover, it is apparent that the yield of CHDM catalyzed by Cu–Zn/Al2O3 is much higher than that catalyzed by Cu–Cr catalyst.
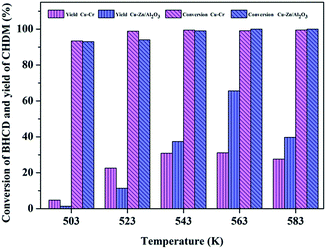 |
| | Fig. 5 Influence of reaction temperature on the conversion of BHCD and yield of CHDM. Reaction conditions: Cu–Cr: 2.0 g intermediate products (90% BHCD), 0.1 g Cu–Cr, 10 h, and 10 MPa; Cu–Zn/Al2O3: 2.0 g intermediate products (90% BHCD), 0.1 g Cu–Zn/Al2O3, 10 h, and 6.5 MPa. | |
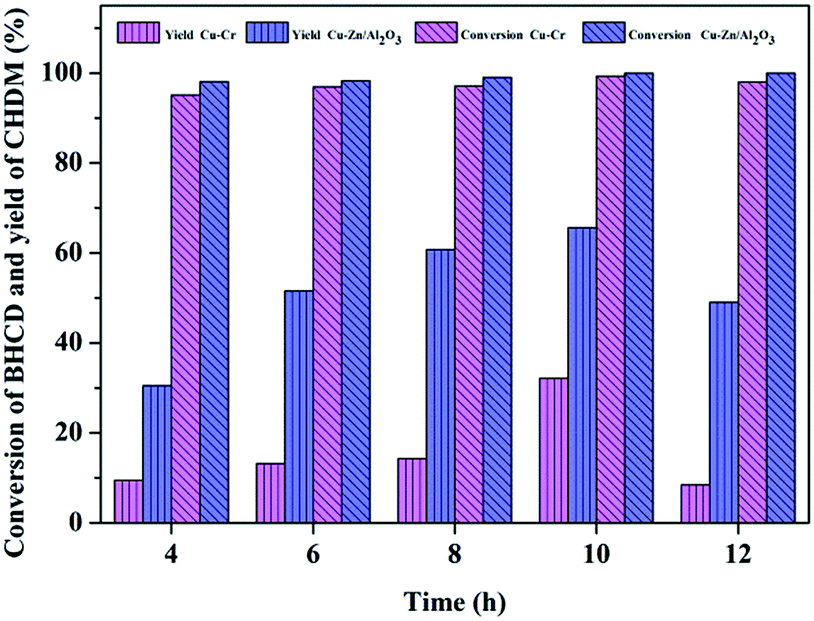
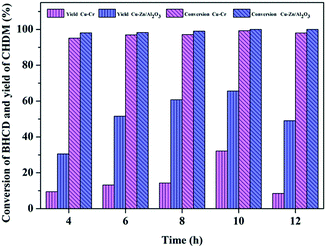
3.2.2. Influence of reaction time. The influence of reaction time on the conversion of BHCD and yield of CHDM catalyzed by Cu–Cr and Cu–Zn/Al2O3 catalysts is shown in Fig. 6. It displays that the reaction time is a dominant parameter affecting the ultimate catalyst performance. The conversion of BHCD increased when the reaction time increased, whereas the yield of CHDM initially increased when the reaction time increased to 10 h and then decreased when the reaction time increased prolonged from 10 h to 12 h, which is attributed to the further hydrogenation of the hydroxyl group of CHDM. As a consequence, the preferred reaction time is 10 h, affording about 32% and 65% CHDM yield catalyzed by Cu–Cr and Cu–Zn/Al2O3 catalysts, respectively. It displays that the excessive increase of reaction time has no effect on the improvement of the yield of CHDM.
 |
| | Fig. 6 Influence of reaction time on the conversion of BHCD and yield of CHDM. Reaction conditions: Cu–Cr: 2.0 g intermediate products (90% BHCD), 0.1 g Cu–Cr, 543 K, and 10 MPa; Cu–Zn/Al2O3: 2.0 g intermediate products (90% BHCD), 0.1 g Cu–Zn/Al2O3, 563 K, and 6.5 MPa. | |
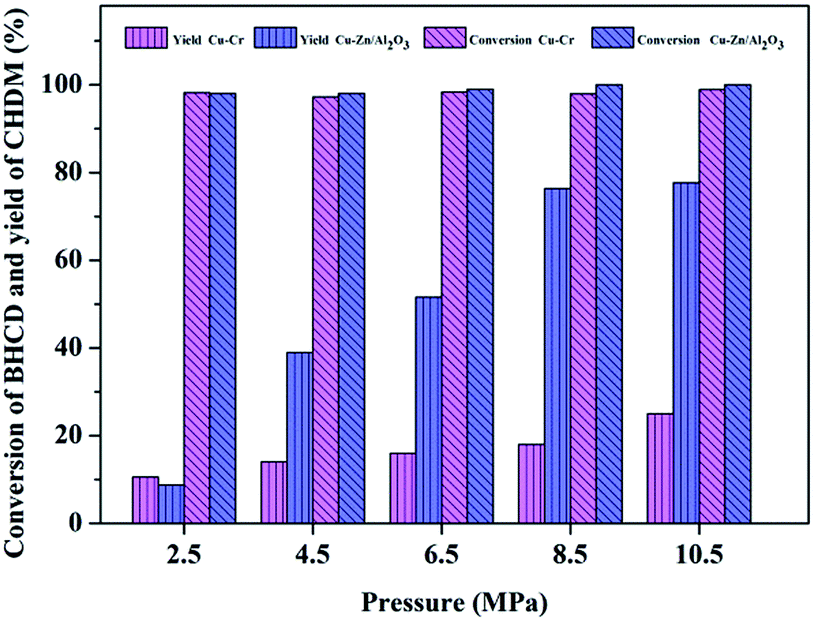
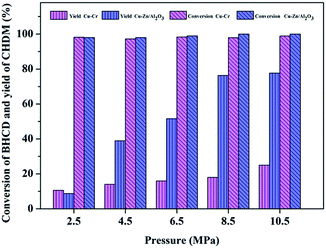
3.2.3. Influence of reaction pressure. Fig. 7 shows the influence of reaction pressure on the conversion of BHCD and yield of CHDM catalyzed by Cu–Cr and Cu–Zn/Al2O3 catalysts. The conversion of BHCD reached almost 100% when the reaction pressure reached 2.5 MPa. It could be found that with the increase of the reaction pressure from 2.5 to 10.5 MPa, the yield of CHDM increased and then became unchanged without affecting the conversion of BHCD. It reveals that the high pressure greatly improves the selective hydrogenation of the ester groups in BHCD and decreases the by-products which resulted in the high yield of CHDM. Under the optimal reaction pressure of 10.5 MPa, the yield of CHDM reached 26% and 78% catalyzed by Cu–Cr and Cu–Zn/Al2O3 catalysts respectively.
 |
| | Fig. 7 Influence of reaction pressure on the conversion of BHCD and yield of CHDM. Reaction conditions: Cu–Cr: 2.0 g intermediate products (90% BHCD), 0.1 g Cu–Cr, 543 K, and 10 h; Cu–Zn/Al2O3: 2.0 g intermediate products (90% BHCD), 0.1 g Cu–Zn/Al2O3, 563 K, and 6 h. | |
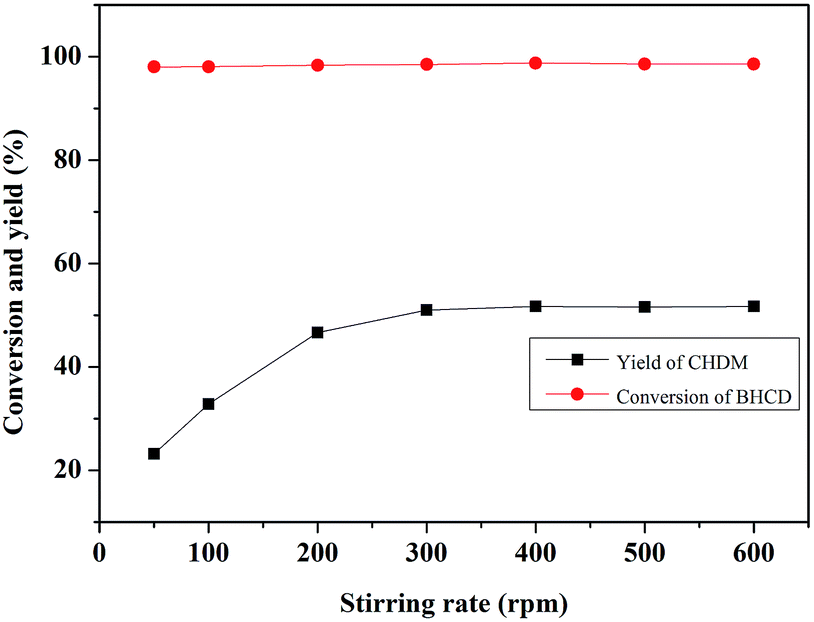
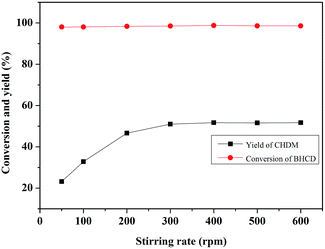
3.2.4. Influence of stirring rate. Fig. 8 shows the influence of stirring rate on the conversion of BHCD and the yield of CHDM. As can be seen, the yield of BHCD firstly increased with the increase of the stirring rate from 50 to 300 rpm, and then kept about 52% from 300 to 600 rpm. However, the conversion of BHCD reached about 98% when the stirring rate was 50 rpm. The hydrogenation of BHCD to CHDM is a multi-step reaction, the previous reaction steps were performed with low stirring rate, however, the high stirring rate promoted the followed reaction steps, resulting in the increase of the yield of CHDM with the increase of the stirring rate. There should be no mass or heat transfer limitation with a stirring rate higher than 300 rpm in the hydrogenation.
 |
| | Fig. 8 Influence of stirring rate on the conversion of BHCD and the yield of CHDM. Reaction conditions: 2.0 g intermediate products (90% BHCD), 0.1 g Cu–Zn/Al2O3, 6.5 MPa, 563 K, and 6 h. | |
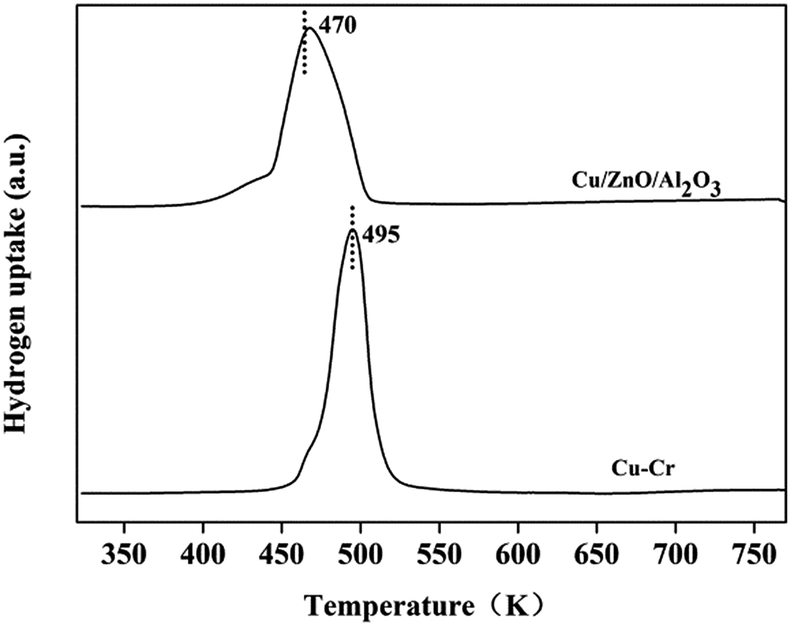
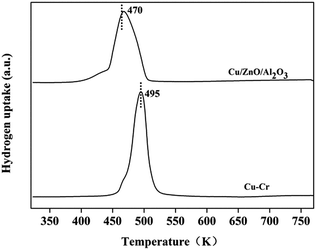
3.2.5. Characterization of Cu-based catalysts. The reducibility of the Cu–Cr and Cu–Zn/Al2O3 catalysts is shown in Fig. 9. The reduction peaks of the Cu–Cr and Cu–Zn/Al2O3 catalysts are 495 K and 470 K, respectively. It reveals that the Cu2+ species can be reduced in situ to Cu+ or/and Cu0 because the reduction temperature of Cu–Cr and Cu–Zn/Al2O3 catalysts in the experiment are of 623 K and 573 K which are higher than the reduction peaks of Cu–Cr and Cu–Zn/Al2O3 catalysts, respectively.
 |
| | Fig. 9 TPR profiles of the Cu–Cr and Cu–Zn/Al2O3 catalysts before reduction. | |
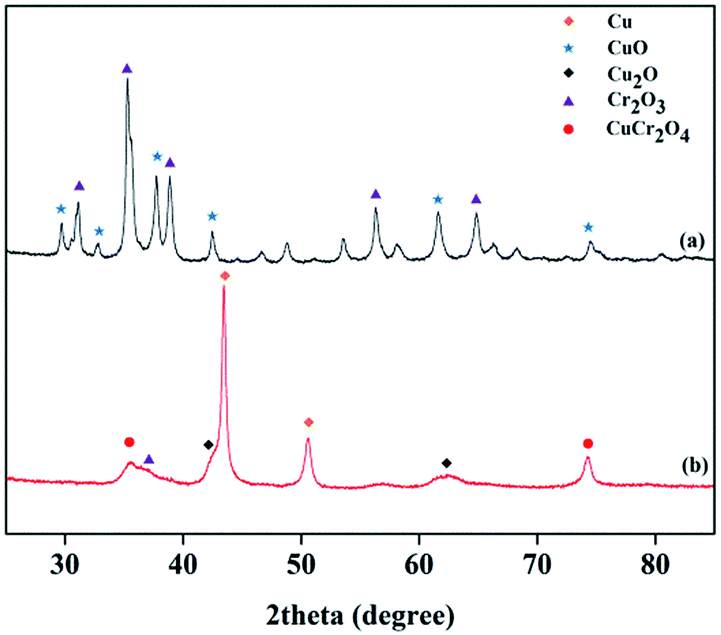
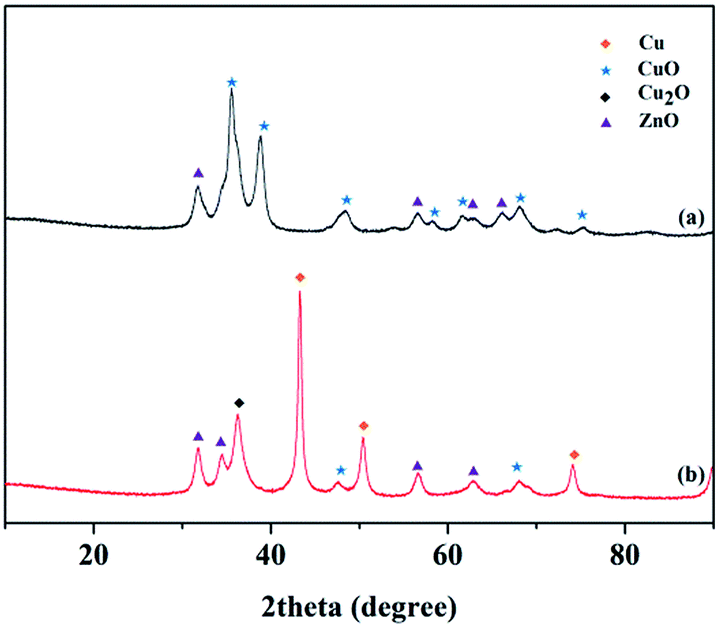
The XRD patterns of the Cu–Cr and Cu–Zn/Al2O3 catalysts before and after reduction are shown in Fig. 10 and 11, respectively. The typical diffraction peaks with 2θ indexed with approximately at 32.3, 36.2, 42.2, 53.6 and 58.1° in the Cu–Cr and Cu–Zn/Al2O3 catalysts before reduction which can be assigned to the crystalline phase of CuO. The appearance of Cu0 crystallite diffraction peaks at 43.29, 50.42 and 74.03° in the XRD patterns of the Cu–Cr and Cu–Zn/Al2O3 catalysts after reduction, clearly demonstrates that the Cu2+ is reduced to Cu0. While, the Cu+ diffraction peaks are also found. Therefore, the Cu+/Cu0 species are the active sites in our present catalytic system. Additionally, diffraction peaks belonging to Cr2O3 are detectable in Fig. 10, it is suggested that Cr2O3 serve as supports in Cu–Cr catalyst. Moreover, there are no diffractions related to Al2O3 in Fig. 11 are observed, indicative of the presence of amorphous Al2O3, the ZnO and amorphous Al2O3 phases may function as supports or dispering agents in the Cu–Zn/Al2O3 catalyst.
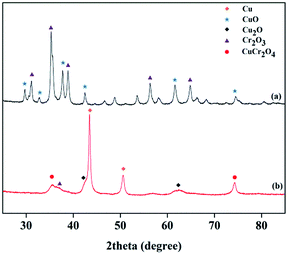 |
| | Fig. 10 XRD patterns of the Cu–Cr catalyst (a) before reduction (b) after reduction. | |
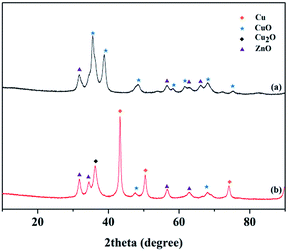 |
| | Fig. 11 XRD patterns of the Cu–Zn/Al2O3 catalyst (a) before reduction (b) after reduction. | |
The Cu-based catalysts were characterized with low temperature N2 adsorption–desorption to elucidate the structural features. As shown in Table 1, it displays that the surface area of the Cu–Zn/Al2O3 catalyst is larger than the Cu–Cr catalyst, which may be contributing to the higher activity of the Cu–Zn/Al2O3 catalyst than the Cu–Cr catalyst.
Table 1 BET surface area, pore volume and mean pore size of the Cu-based catalysts
| Sample |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Mean pore size (nm) |
| Cu–Cr |
10.88 |
0.09 |
33.8 |
| Cu–Zn/Al2O3 |
33.52 |
0.15 |
16.4 |
The optimal reaction conditions of the hydrogenation of BHCD to CHDM are 543 K, 10 h, and 10.5 MPa. The yield of CHDM and the conversion of BHCD have reached 32%, 78%, and 100% catalyzed by Cu–Cr and Cu–Zn/Al2O3 catalysts, respectively. Cu–Zn/Al2O3 catalyst shows much higher activity and selectivity of the hydrogenation of ester to alcohol than Cu–Cr catalyst, it is deduced that the addition of Zn and Al can improve the activity and selectivity of the Cu-based catalysts in the hydrogenation of ester group resulting in the increase of the yield of CHDM. On the contrary, the addition of Cr can improve the activity and selectivity of the Cu-based catalyst in the hydrogenation of hydroxyl group resulting in the decrease of the yield of CHDM. In addition, Cu–Zn/Al2O3 catalyst is more environmentally friendly than Cu–Cr catalyst. Therefore, we choose Cu–Zn/Al2O3 as the excellent catalyst in the hydrogenation reaction of BHCD to CHDM.
4. Conclusions
On the basis of the above discussion, it is shown that the waste PET monomer BHET can be efficiently converted into CHDM by a two-step hydrogenation reaction using Pd/C and Cu-based catalysts without solvent. The conversion of BHET and the yield of CHDM have reached as high as 100% and 78%, respectively. The phenyl group can be efficiently hydrogenated to cyclohexane group catalyzed by Pd/C catalyst under relative mild conditions in the first step. However, the competition of hydroxyl and ester groups in the reaction with hydrogen increases the difficulty of the selective hydrogenation of ester groups in the second step. The Cu–Zn/Al2O3 catalyst shows much higher activity and selectivity of the hydrogenation of ester group to hydroxyl group than Cu–Cr catalyst. Moreover, it is suggested that the Cu+/Cu0 species are the active sites in the Cu-based catalysts. In summary, it is successfully converted the waste PET monomer BHET to high-valued CHDM, thus providing not only a new method for the production of CHDM, but also another direction for the chemical recycling of waste PET.
Acknowledgements
This work was supported financially by the Program of National High Technology Research and Development Program of China (863 Program) (no. 2012AA063001), National Natural Science Foundation of China (no. 21276255) and Beijing Natural Science Foundation of China (no. 2131005, no. 2132055, no. 21276260).
Notes and references
- S. R. Turner, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5847–5852 CrossRef CAS.
- H. J. Bang, H. Y. Kim, F. L. Jin and S. J. Park, J. Ind. Eng. Chem., 2011, 17, 805–810 CrossRef CAS PubMed.
- L. H. Zhu, L. Zheng, K. Q. Du, H. Fu, Y. H. Li, G. R. You and B. H. Chen, RSC Adv., 2013, 3, 713–719 RSC.
- Y. J. Zhao, J. Zhou, J. G. Zhang, D. Y. Li and S. D. Wang, Ind. Eng. Chem. Res., 2008, 47, 4641–4647 CrossRef CAS.
- S. K. Sharma, K. B. Sidhpuria and R. V. Jasra, J. Mol. Catal. A: Chem., 2011, 335, 65–70 CrossRef CAS PubMed.
- M. Saeys, M. F. Reyniers, M. Neurock and G. B. Marin, J. Phys. Chem. B, 2005, 109, 2064–2073 CrossRef CAS PubMed.
- Y. Q. Huang, Y. Ma, Y. W. Cheng, L. J. Wang and X. Li, Ind. Eng. Chem. Res., 2014, 53, 4604–4613 CrossRef CAS.
- P. Kasinathan, D. W. Hwang, U. H. Lee, Y. K. Hwang and J.-S. Chang, Catal. Commun., 2013, 41, 17–20 CrossRef CAS PubMed.
- J. L. Gong, H. R. Yue, Y. J. Zhao, S. Zhao, L. Zhao, J. Lu, S. P. Wang and X. B. Ma, J. Am. Chem. Soc., 2012, 134, 13922–13925 CrossRef CAS PubMed.
- P. Yuan, Z. Y. Liu, W. Q. Zhang, H. J. Sun and S. C. Liu, Chin. J. Catal., 2010, 31, 769–775 CrossRef CAS.
- M. Toba, S.-i. Tanaka, S.-i. Niwa, F. Mizukami, Z. Koppány, L. Guczi, K.-Y. Cheah and T.-S. Tang, Appl. Catal., A, 1999, 189, 243–250 CrossRef CAS.
- K. Tahara, H. Tsuji, H. Kimura, T. Okazaki, Y. Itoi, S. Nishiyama, S. Tsuruya and M. Masai, Catal. Today, 1996, 28, 267–272 CrossRef CAS.
- M. J. Hanton, S. Tin, B. Boardman and P. Miller, J. Mol. Catal. A: Chem., 2011, 346, 70–78 CrossRef CAS PubMed.
- S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 7869–7872 CrossRef CAS PubMed.
- Z. Q. Zhu, Z. H. Lu, B. Li and S. Z. Guo, Appl. Catal., A, 2006, 302, 208–214 CrossRef CAS PubMed.
- R. Raja, T. Khimyak, J. M. Thomas, S. Hermans and B. F. G. Johnson, Angew. Chem., Int. Ed., 2001, 40, 4638–4642 CrossRef CAS.
- A. B. Hungria, R. Raja, R. D. Adams, B. Captain, J. M. Thomas, P. A. Midgley, V. Golovko and B. F. G. Johnson, Angew. Chem., Int. Ed., 2006, 45, 4782–4785 CrossRef CAS PubMed.
- X. F. Li, Z. Y. Sun, J. L. Chen, Y. Zhu and F. Z. Zhang, Ind. Eng. Chem. Res., 2014, 53, 619–625 CrossRef CAS.
- M. L. Zhu, Z. X. Li, Q. Wang, X. Y. Zhou and X. M. Lu, Ind. Eng. Chem. Res., 2012, 51, 11659–11666 CrossRef CAS.
- M. L. Zhu, S. Li, Z. X. Li, X. M. Lu and S. J. Zhang, Chem. Eng. J., 2012, 185, 168–177 CrossRef PubMed.
- G. X. Xi, M. X. Lu and C. Sun, Polym. Degrad. Stab., 2005, 87, 117–120 CrossRef CAS PubMed.
- M. Ghaemy and K. Mossaddegh, Polym. Degrad. Stab., 2005, 90, 570–576 CrossRef CAS PubMed.
- Q. Wang, X. Q. Yao, S. F. Tang, X. M. Lu, X. P. Zhang and S. J. Zhang, Green Chem., 2012, 14, 2559–2566 RSC.
- H. Wang, Y. Q. Liu, Z. X. Li, X. P. Zhang, S. J. Zhang and Y. Q. Zhang, Eur. Polym. J., 2009, 45, 1535–1544 CrossRef CAS PubMed.
- H. Wang, Z. X. Li, Y. Q. Liu, X. P. Zhang and S. J. Zhang, Green Chem., 2009, 11, 1568–1575 RSC.
- S. R. Shukla, A. M. Harad and L. S. Jawale, Waste Manag., 2008, 28, 51–56 CrossRef CAS PubMed.
- N. M. Abdullah and I. Ahmad, Fibers Polym., 2013, 14, 584–590 CrossRef CAS.
- S. R. Shukla, A. M. Harad and L. S. Jawale, Polym. Degrad. Stab., 2009, 94, 604–609 CrossRef CAS PubMed.
- Y. Kai and A. C. Chen, Energy, 2013, 58, 357–363 CrossRef PubMed.
- L. M. He, H. Y. Cheng, G. F. Liang, Y. C. Yu and F. Y. Zhao, Appl. Catal., A, 2013, 452, 88–93 CrossRef CAS PubMed.
- H. Z. Liu, T. Jiang, B. X. Han, S. G. Liang and Y. X. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS PubMed.
- Z. H. Xiao, X. K. Wang, J. H. Xiu, Y. M. Wang, C. T. Williams and C. H. Liang, Catal. Today, 2014, 234, 200–207 CrossRef CAS PubMed.
- R. V. Sharma, P. Kumar and A. K. Dalai, Appl. Catal., A, 2014, 477, 147–156 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.