DOI:
10.1039/C4RA09908G
(Paper)
RSC Adv., 2015,
5, 6160-6171
Gemini quaternary ammonium-incorporated biodegradable multiblock polyurethane micelles for brain drug delivery†
Received
8th September 2014
, Accepted 12th December 2014
First published on 12th December 2014
Abstract
Brain drug delivery is still facing significant challenges due to the low permeability of the blood–brain barrier (BBB). To overcome such an insurmountable obstacle, we developed gemini quaternary ammonium (GQA) as a cell penetrating molecule incorporated into biodegradable multiblock poly(ε-caprolactone urethanes)s (BMPUs) drug nanocarriers for improvement of drug accumulation in brain parenchyma. The zeta potential of Dox-loaded GQA-BMPUs micelles was around 26 mV with a mean particle size near 100 nm. It was found that GQA-BMPUs micelles achieved steadily time-dependent and concentration-dependent Dox accumulation in human brain microvascular endothelial cells (HBMECs) much higher than GQA-free BMPUs micelles and free Dox, as confirmed by flow cytometry and confocal laser scanning microscopy (CLSM) experiments. Meanwhile, no pronounced cytotoxicity was noticed in GQA-BMPUs micelles and GQA-free BMPUs micelles, and Dox associated cytotoxicity might be reduced once encapsulated into micelles. More importantly, CLSM of brain sections showed higher accumulation of Dox-loaded GQA-BMPUs micelles in the subcortical area after administrated intravenously, while no Dox accumulation was observed in either Dox-BMPUs micelles or free Dox formulation. Coupling with in vivo pharmacokinetics, biodistribution and histological toxicity studies, the results show that GQA introduced into drug nanocarriers is a promising avenue to transport therapeutic agents across BBB and improve brain drug accumulation.
1. Introduction
Brain drug delivery has long been a challenge in the treatment of various brain related diseases, including malignant primary brain tumors, Alzheimer's disease (AD), Parkinson's disease (PD), multiple sclerosis (MS), etc.1 This is because most therapeutic agents find it difficult to permeate across the blood–brain barrier (BBB) into brain parenchyma and lesions.2–4 BBB refers to continuous and nonfenestrated capillaries in normal brain tissue with tight junctions between endothelia,3 which have formed a mechanical barrier to reduce most harmful xenobiotics outside the brain. Additionally, the efflux pumps such as the well-known P-glycoprotein (P-gp) and multidrug resistance protein family (MDR) locating on BBB endothelial cell membranes have also formed a molecular barrier to extrude various drugs (i.e., doxorubicin) from the brain.2,5 Thus, brain disorders may hide behind these barriers to avoid being detected and eliminated.
Over the decades, attempts have been made to increase drug accumulation in brain parenchyma, such as invasive approaches via breaching the BBB, pharmacological approaches via chemical modification of drugs, and physiological approaches via attaching peptides, antibodies or small molecules directly to drugs.6 However, the improvement of drug accumulation in brain tissue is still a formidable challenge. Nanoparticles may serve as an effective vector to protect drugs from degradation throughout the circulation and also reduce systemic side effects caused by free drugs.7 With specific properties, nanoparticles can transport therapeutic agents into brain orientatively via surface modified effects like receptor mediated transcytosis (RMT), which could increase the percentage of injected dose (ID) per g brain tissue to 0.01–0.5% compared to an impermeable control.6,8 Micellar drug carriers have been particularly attractive in studies of BBB penetration and brain drug delivery for their controllable self-assembly, high drug loading capacity, good biocompatibility, enhanced brain drug accumulation and prolonged circulation half-life.9 Surface decorated with antibodies or ligands, micelles will actively target to the corresponding receptors on BBB endothelial cells and help improve drugs across BBB into brain lesions via RMT effects.10,11 Additionally, cell penetrating peptides (i.e., TAT with a basic isoelectric point shows cationic sites under neutral environment in vivo) modified micelles may bind to the negatively charged luminal plasma membrane of BBB endothelial cells through electrostatic interaction and trigger BBB penetration by a mechanism independent of RMT, somewhat called adsorptive mediated transcytosis (AMT).12,13 But the possibility exists that antibodies, ligands, or peptides on surface of micelles can be recognized by immune system and eliminated throughout the circulation, the drug accumulation in brain is still unsatisfactory.9 Thus, it is important to pursue new nanocarriers for brain drug delivery, nonprotein cationic moieties incorporated into nanoparticles may be an attempt to meet requirements.
Biodegradable multiblock polymers (BMP) may provide more controllable self-assembly and enlarge diversity of micellar structure, which will lead to incorporation of various functional groups on formed nanocarriers.14 Moreover, biodegradable segmented polyurethanes (SPUs) have been widely applied in biomedicine for their excellent structural tailorability as well as good biocompatibility and facile preparation.15,16 In our recent work, we developed a novel generation of cationic biodegradable multiblock poly(ε-caprolactone urethanes)s (BMPUs) containing gemini quaternary ammonium (GQA).17 GQAs as a class of cationic amphiphilic compounds with two quaternary ammonium positive head and two hydrophobic alkyl tails are ease of synthesis,18 which have shown good surface activity, low critical micelle concentration (CMC) and high solubilizing ability. Our work demonstrated that GQA-incorporated BMPUs could easily self-assembled into micelles in aqueous solution at extremely low concentrations.19 The micelles had shown highly controllable self-assembly, stable physicochemical properties, high drug loading capacity, good biocompatibility.19,20 More importantly, we found GQAs endowed BMPUs micelles with fast cell internalizing ability, which acted like cell penetrating peptide (i.e., TAT).21 To date, to the best of our knowledge, no work has been reported on the application of micelles based on either BMPUs or GQAs as brain drug carriers.
In this study, we mainly focus on the role of GQA-incorporated BMPUs micelles in transporting doxorubicin (Dox) across the BBB. Dox was selected due to a typical anti-tumor drug which could not permeate through BBB efficiently as well as the autofluorescent property. MTT assay, H&E assay, flow cytometry, confocal laser scanning microscopy (CLSM) studies and HPLC were carried out to evaluate the in vitro cellular uptake and cytotoxicity of BMPUs micelles as well as in vivo pharmacokinetics, histological distribution, histological toxicity, BBB penetration and brain accumulation of Dox-loaded BMPUs.
2. Materials and methods
2.1. Materials
A series of functional monomers, including L-lysine-derivatized GQA and L-lysine ethyl ester diisocyanate (LDI) were synthesized according to previous reports,17,19,20,22 and the BMPUs copolymers G0 and G70 were prepared via a procedure in our previous work21 (see the ESI† for detailed information). Doxorubicin hydrochloride (Dox·HCl) was from Tecoland Corporation, USA. 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was purchased from Sigma Aldrich, USA. 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) and chloroform of analytical grade were obtained from Shanghai Jianglai Bio-Technology Co., Ltd., China. Methanol and acetonitrile of HPLC grade were from Sigma Aldrich, USA. Animal vein catheter (hard tube BB31695-PE/3, soft tube BB518-20) was purchased from American Health & Medical Supply International Corp. Co., Ltd., Chengdu, China.
2.2. Preparation of Dox-loaded BMPUs micelles
Dox was loaded into BMPUs micelles via a dialysis method. First of all, Dox·HCl and polyurethanes with different weight ratios were codissolved in DMAc solution, and treated with triethylamine for 2 h under stirring condition to remove hydrochloride. After that, the solution was added dropwise (1 drop every 30 s) into deionized water. The resulting solution was then transferred to a dialysis tube (MWCO 3500) and dialyzed against deionized water for about 3 days to remove the organic solvent and unloaded free Dox at room temperature. The micellar solution was centrifugalized at 3000 rpm for 10 min and passed through a 0.45 μm pore-sized syringe filter (Milipore, Carrigtwohill, Co. Cork, Ireland). Before cell and animal experiment, the micellar solution was passed through 0.22 μm bacteria filter and the osmotic pressure was adjusted to physiological condition.
2.3. General measurements
The gel permeation chromatography (GPC), proton nuclear magnetic resonance spectra (1H NMR) and fluorescence measurements of BMPUs was finished in our previous work (see the ESI† for detailed structure information of BMPUs).21
Transmission electron microscopy (TEM) of Dox-loaded micelles was performed on a Hitachi model H-600-4 transmission electron microscope (Japan) with an accelerating voltage of 75 kV. A drop of Dox-micellar solution stained by 1% (w/v) phosphotungstic acid was placed on a copper grid with Formvar film, and then the liquid was blotted off and air-dried before measurement.
Sizes and zeta potentials of Dox-micelles were measured with a Zetasizer Nano ZS dynamic light-scattering (DLS) instrument (Malvern, UK) at 25 °C at an angle of 90°.
The amount of Dox loaded inside micelles was determined using a Hitachi F-7000 FL spectrophotometer (excitation at 480 nm). Drug loading content (LC) and encapsulation efficiency (EE) were calculated according to the following equations:
| LC (%) = weight of loaded drugs/weight of drug-loaded micelles × 100% |
| EE (%) = weight of loaded drugs/weight of feeding drugs × 100% |
2.4. In vitro release study
The release assay of Dox from G70-Dox and G0-Dox was conducted using a dialysis method in HEPES solution (10 mM, pH 7.0) and acetate buffer solution (10 mM, pH 5.0) at 37 °C with gentle shaking. The release media also contains 100 mM sodium salicylate to maintain a sink condition. The incubation media was replaced with fresh media at desired time points. The detection of released Dox was performed by determining absorbance at 485 nm using a spectrophotometer (M5, Molecular Devices, Corp.). The release experiments were repeated three times and the result was expressed as means ± standard deviation.
2.5. Cell culture
Human brain microvascular endothelial cells (HBMECs) were purchased from American Type Culture Collection (ATCC, Rockville, MD). The cell lines were cultivated in Dulbecco's Modified Eagle's Medium (DMEM, Gibco Life, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT) and 1% penicillin–streptomycin (Gibco Life, USA). The cell cultures were maintained in a 37 °C incubator (Hera Cell, Thermo Scientific) with a humidified 5% CO2 atmosphere.
2.6. Cytotoxicity assay
HBMECs were seeded in 96-well plates at 5 × 103 cell per well and incubated for 24 h. The culture media was removed and replaced with 100 mL medium containing various concentrations of drug-free and Dox-loaded micelles, sterile normal saline and free Dox solutions diluted to the same concentrations with culture medium were set as negative control and positive control, respectively. After 48 h of incubation, the cells were exposed to 20 μL of MTT solution (5 mg mL−1) and incubated for 4 h at 37 °C. Then, the MTT solution was removed and the insoluble formazan crystals were dissolved in 100 μL of dimethyl sulfoxide (DMSO). The plates were shaken for 10 min, and the absorbance of formazan product was measured at 490 nm on a microplate reader (DNM-9602, Nanjing Perlove Medical Equipment Co., Ltd., China). The cell viability was normalized to that of untreated cells.
2.7. Cellular uptake
2.7.1. Confocal laser scanning microscope (CLSM) study. HBMECs were seeded at a density of 1 × 105 cells per well in 6-well chamber slides for 24 h. The cells were incubated with free Dox and Dox-loaded micelles equivalent to 1 μg mL−1 of free Dox for different time intervals at 37 °C. After removal of the medium, the cells were washed three times with cold PBS (PH 7.4), fixed with 1 mL of 4% paraformaldehyde for 20 min at 37 °C and stained with DAPI (0.5 μg mL−1) for 10 min at room temperature. The slides were mounted with 10% glycerol solution and observed under CLSM (DM6000 CS, Leica, Germany).
2.7.2. Flow cytometry study. HBMECs were seeded at a density of 1 × 105 cells per well in 6-well culture plates for 24 h. The cells were then treated with free Dox and Dox-loaded micelles at various concentrations at 37 °C for 2 and 4 h. Then, culture medium was removed and the cells were washed with PBS (PH 7.4) and treated with trypsin. The cells were collected in flow tube, and washed with PBS (PH 7.4) three times by centrifugation (2000 rpm, 5 min). After discarding the supernatant, the cells were resuspended in 0.3 mL PBS (PH 7.4), and the intracellular Dox fluorescence was analyzed by BD FACSCalibur (BD, USA).
2.8. In vivo pharmacokinetics and biodistribution studies
2.8.1. Animals. The animal studies were carried out with the approval from the Ethics Committee of Sichuan University and in compliance with the Principles of Laboratory Animal Care of the National Institutes of Health, China. SPF adult female Sprague–Dawley (SD) rats (250 ± 25 g, 10 week-old) were purchased from Vital River Laboratory Animal Technology Co., Ltd., Beijing, China. Rats were housed at controlled temperature of 20–22 °C, relative humidity of 50–60% and 12 h light-dark cycles and provided with standard laboratory chow and tap water ad libitum. All animals would be in quarantine for a week before experiment.
2.8.2. Pharmacokinetics study. SD rats were anesthetized with 10% chloral hydrate (i.p., 0.3 mL/100 g body weight) and randomly assigned to four groups (n = 3). Each rat was received right external jugular vein catheterization,23 the distal end of hard tube was exteriorized and fixed on the back of rat neck. The catheter was filled with heparinized saline without any air bubbles. When ambulatory, the rats were transferred to a clean cage and well-kept overnight. Next day, each group of rats were received intravenous injection of free Dox and Dox-loaded micelles at a dose of 500 μg kg−1 and sterile normal saline as negative control, respectively. At different time intervals after administration, approximate 0.5 mL of blood was collected by heparinized tube from the vein catheter. Plasma samples were harvested by immediately centrifugation (3000 rpm, 5 min) and stored at −20 °C. Thereafter, 300 μL of plasma was extracted with 1.5 mL methanol/chloroform (1/4 v/v), the organic layer was separated by centrifugation (3000 rpm, 10 min) and collected into a clean tube. The drug residue was obtained by evaporation and resuspended in 1 mL methanol. The plasma concentrations of Dox were determined by HPLC with a mobile phase of acetonitrile/methanol/acetate (10/40/50 v/v/v).
2.8.3. Histological distribution. SD rats were randomly assigned to four groups (n = 3), each group received free Dox and Dox-loaded micelles at a dose of 500 μg kg−1 and sterile normal saline, respectively. Rats were anesthetized with 10% chloral hydrate (i.p., 0.3 mL/100 g body weight) 8 h post administration. Then, they were perfused with PBS (pH 7.4) followed by 4% paraformaldehyde (pH 7.4). Afterwards, various tissues, including heart, liver, spleen, lung and kidney were collected and frozen in O.C.T. embedding medium at −80 °C. Frozen sections of 7 μm thickness were prepared with a cryostat microtome (Thermo Fisher Scientific, USA) and stained with DAPI (0.5 μg mL−1) for 10 min at room temperature. The sections were washed with PBS (pH 7.4) three times, mounted with 10% glycerol solution and viewed under CLSM (DM6000 CS, Leica, Germany).
2.9. Transport across the BBB in vivo
2.9.1. Qualitative analysis of brain tissue uptake. For qualitative studies of brain tissue, rats were treated as described above. After perfusion, brains were collected, embedded in O.C.T., cut into section of 7 μm thickness, stained with DAPI and viewed under CLSM.
2.9.2. Quantitative analysis of brain tissue uptake. The brain tissue concentrations of Dox were determined by HPLC. For this, rats were treated as described above. After perfusion with PBS (pH 7.4), brains were collected, weighted and stored at −20 °C. Thereafter, 1 g of brain tissue was homogenized in 300 μL PBS (pH 7.4). The homogenate was extracted with 1.5 mL methanol/chloroform (1/4 v/v), the organic layer was separated by centrifugation (3000 rpm, 10 min) and collected into a clean tube. The drug residues were obtained and measured as described above.
2.10. Histological toxicity assay
SD rats were randomly assigned to six groups (n = 3), each group received routine doses of drug-free micelles, Dox-loaded micelles, sterile normal saline and free Dox solutions, respectively. Then, 24 h post administration, the rats were anesthetized and perfused as described above. Tissue samples (brain, heart, liver, spleen, lung and kidney) were collected and routinely stained with hematoxylin and eosin followed by microscopic examination of tissue toxicity.
2.11. Statistical analysis
Data were expressed as means ± standard deviations (SD). Statistical significance in cellular uptake, brain drug accumulation, pharmacokinetics, and cytotoxicity effects was determined using one-way ANOVA followed by a Student's t test for multiple comparison tests. Statistical analysis was analyzed using statistical software (SPSS 17.0, Chicago, USA). A difference of P < 0.05 was considered statistically significant, and P < 0.001 was considered high statistically significant.
3. Results and discussion
3.1. Preparation and characterization of Dox-loaded BMPUs micelles
To achieve excellently biocompatible GQA-BMPUs micelles for Dox delivery across BBB, a similar series of BMPUs were re-synthesized according to our previous work,19,22 the detailed process is provided in the ESI.† It was demonstrated in our previous research that higher concentration of incorporated GQA was associated with more cellular uptake of BMPUs micelles,21 yet high cationic density may raise safety concern. Therefore, in our ongoing work, polyurethane G70 with 70% of GQA molar fraction in chain extender was synthesized, G0 with no GQA in chain extender was prepared as negative control. In our previous study, it was found that longer poly(ethylene glycol) (PEG) chains could shield the hydrophobic poly(ε-caprolactone) (PCL) inner core and mask the cationic GQA shell as well,21 which could reduce drug or carrier toxicity as well as prolong blood half-life of the carrier.24 Thus, in this study, we readjusted the PEG end chains with mPEG (MW 1900) in both G70 and G0 polyurethanes. The detailed monomer feed ratios of BMPUs is shown in Table S1.† The molecular structure of polyurethanes is shown in Fig. S1.† The polyurethanes exhibited excellent micellization properties with very low CMC values (Table S2†). It was demonstrated that G70 micelles consist of PCL core, GQA shell and PEG corona, which displayed a core–shell–corona structure (Fig. S2†), while G0 micelles displayed a core–corona structure with no GQA shell.21 The characterization values are shown in Fig. S3 and Table S2.†
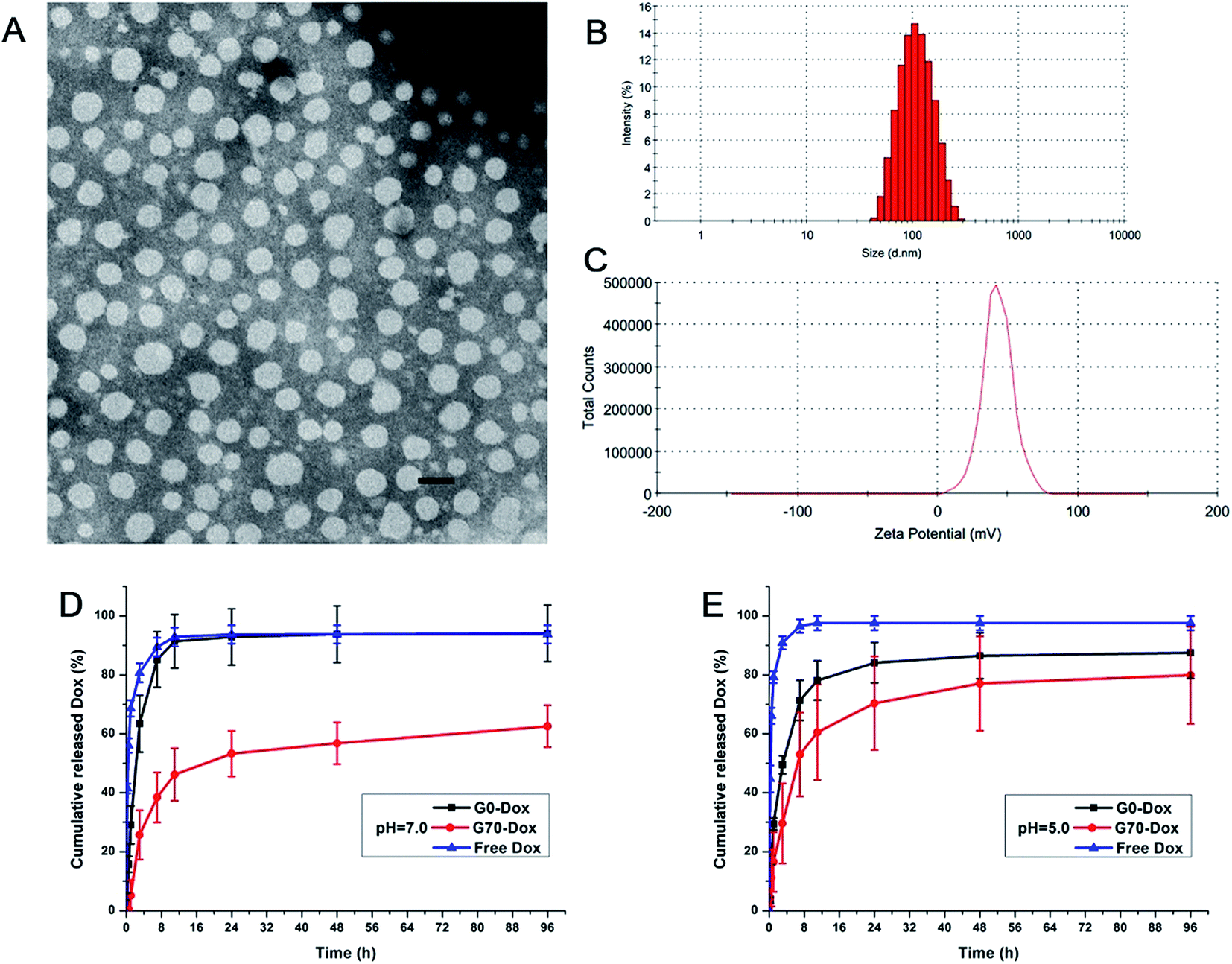
The TEM image of Dox-loaded G70 micelles is shown in Fig. 1A, the micellar particles were spherical with homogeneous size distribution. The average particle size of Dox-loaded G70 micelles determined by DLS was 102.6 ± 0.8 nm with a PDI of 0.13 at 25 °C (Fig. 1B and S3†). Both methods indicated that Dox-loaded G70 micelles were dispersed individual particles with regular spherical shapes and narrow size distribution. The particle size of G70 increased after loading Dox in the inner core while G0 particle size changed little after loading Dox (Fig. 1B and Table S2†). Since G70 micelles have higher drug loading capacity than G0 micelles, which has been described as follows. The zeta potential of Dox-loaded G70 micelles was 26.1 ± 0.3 mV (Fig. 1C and S3†), which was consistent with Dox free G70 micelles (Table S2†). It indicated that positive charge on surface of G70 was stable, which was beneficial to the interaction between Dox-loaded G70 micelles and BBB in vivo. Dox loading content and encapsulation efficiency of G70 were 9.8% and 47%, respectively, much higher than that of G0 (LC of 2.7% and EE of 20%, respectively). This is attributed to the excellent surfactant properties and solubilization capacity of GQA cationic groups.21 Thus, we successfully obtained Dox-loaded G70 micelles with more stable dispersivity, higher zeta potential and much higher Dox loading capacity.
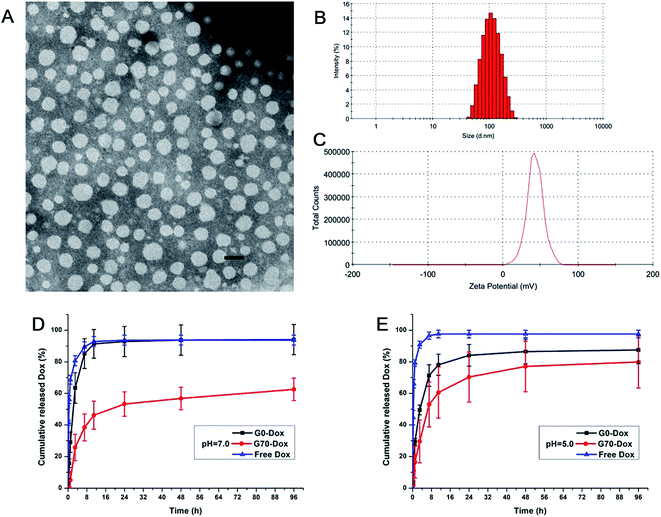 |
| | Fig. 1 Characterization of Dox-loaded G70 micelles. (A) TEM image, the scale bar is 150 nm; (B) particle size distribution measured by DLS; (C) zeta potential measured by DLS; in vitro release profile of G0-Dox, G70-Dox and free Dox at pH 7.0 (D) and pH 5.0 (E). | |
The in vitro release profiles of G0-Dox, G70-Dox and free Dox are presented in Fig. 1D and E. The results indicate that Dox could be released from G70-Dox micelles over an extended period at pH 7.0 and pH 5.0, much longer than that from G0-Dox, free Dox as a positive control. Dox release from G0-Dox micelles was quick in the first 24 h of incubation, over 90% of cumulative release similar to that of free Dox. This indicated that G0-Dox underwent a burst release in incubation media, while GQA incorporated G70-Dox was relatively stable throughout the observation time. Meanwhile, Dox release from G70-Dox micelles was faster at pH 5.0 than that at pH 7.0, indicating that G70-Dox might be stable in circulation (pH 7.0) before it reaches the interesting foci (pH 5.0) to achieve therapeutic efficacy with less side effects.
3.2. In vitro uptake of Dox-loaded BMPUs micelles by HBMECs
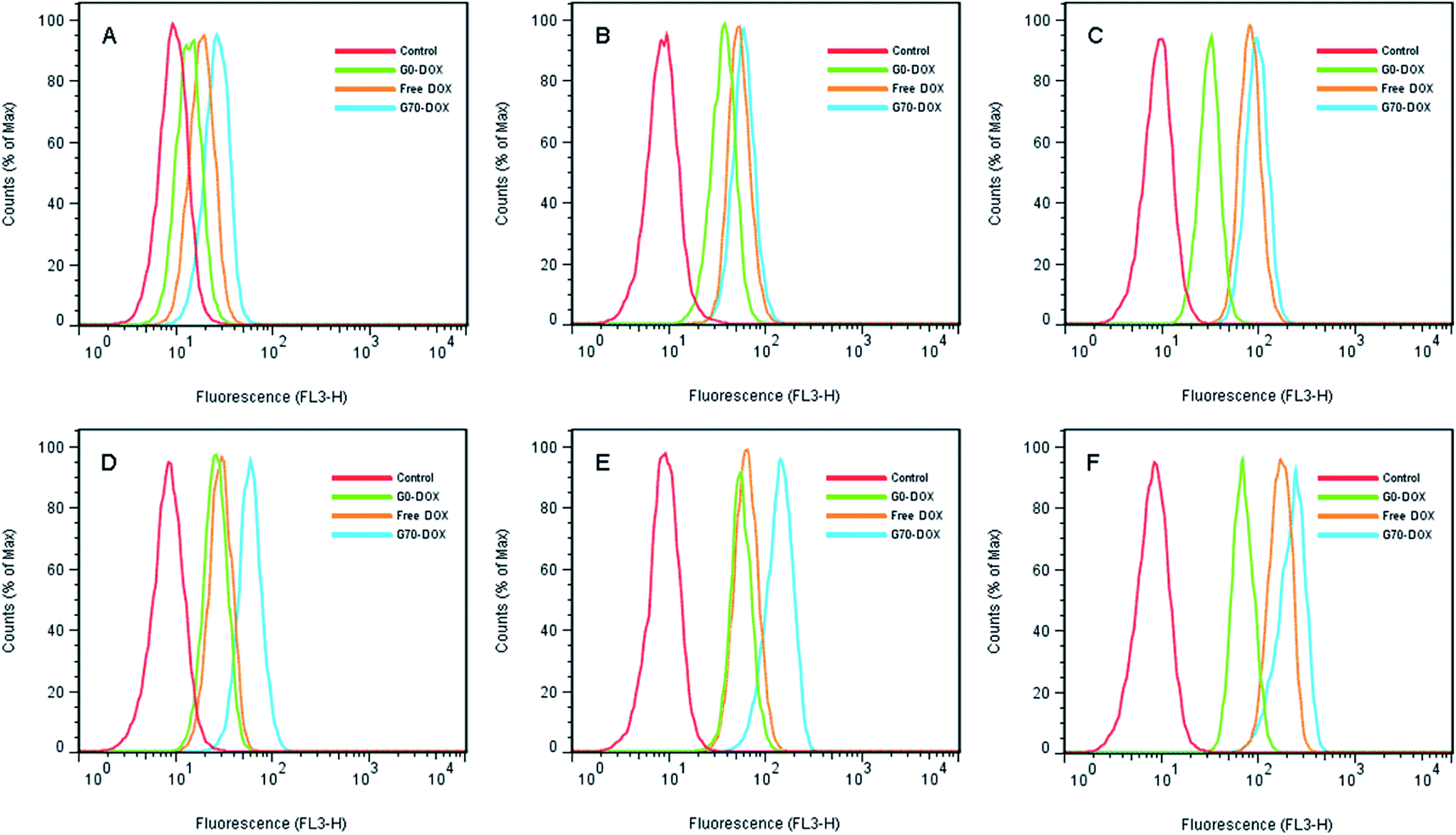
HBMECs are commonly applied as BBB penetrating models in vitro.25,26 In this study, the cellular uptake of Dox-loaded BMPUs micelles by HBMECs was evaluated using flow cytometry and CLSM experiments. The histogram profiles of Dox-associated fluorescence (FL3-H) for HBMECs incubated with free Dox and Dox-loaded BMPUs are shown in Fig. 2. Clearly, the uptake of G70-Dox, G0-Dox and free Dox by HBMECs was both concentration-dependent and time-dependent. The uptake of G70-Dox was the highest of all, while the uptake of G0-Dox was the lowest at various concentration point and each incubation time. This discriminative uptake indicated that the loading of Dox in G70 micelles increased the uptake of Dox due to incorporation of GQA on the surface of BMPUs micelles. Interestingly, the intracellular increment of mean fluorescence intensity (MFI) with time at each concentration point was unparallel for G70-Dox, G0-Dox and free Dox (Table 1). Briefly, different concentrations of G70-Dox improved the MFI by 2.6 fold more or less from 2 h to 4 h of incubation. Whereas, the similar phenomenon was only found in high concentration of free Dox (0.5 μg mL−1) treated HBMECs, low concentration of free Dox (0.1 μg mL−1 and 0.3 μg mL−1) just improved the MFI by 1.7 and 1.2 fold from 2 h to 4 h of incubation. We assumed that the unparallel fluorescent increase of HBMECs by different concentrations of free Dox with time was mainly due to the molecular efflux pumps (i.e., P-gp and MDR) locating on HBMECs membrane.2,5 Dox is a kind of anthracyclines antineoplastic agents, which belongs to the substrate of P-gp and MRP1 and will be transported out of BBB endothelial cells once it diffuses into cell membrane.2 Low concentrations of free Dox would be turned down by these efflux pumps, while high concentrations of free Dox might saturate the substrate site, increasing intracellular accumulation of Dox. On the other hand, once Dox hided in the inner core of G70 micelles, it would not be detected by P-gp efflux pump. Cationic GQA shell would stick to the negatively charged surface of HBMECs via two quaternary ammonium positive head groups, the long hydrophobic alkyl tails would promote endocytosis of G70-Dox, resulting in stable increase of MFI.21 This effect is the so-called adsorptive mediated endocytosis (AME) as was observed in other cationic nanocarriers.27,28 However, the exact mechanism of cellular uptake of G70-Dox by HBMECs was not very clear here, further studies are needed to interpret the internalization details.
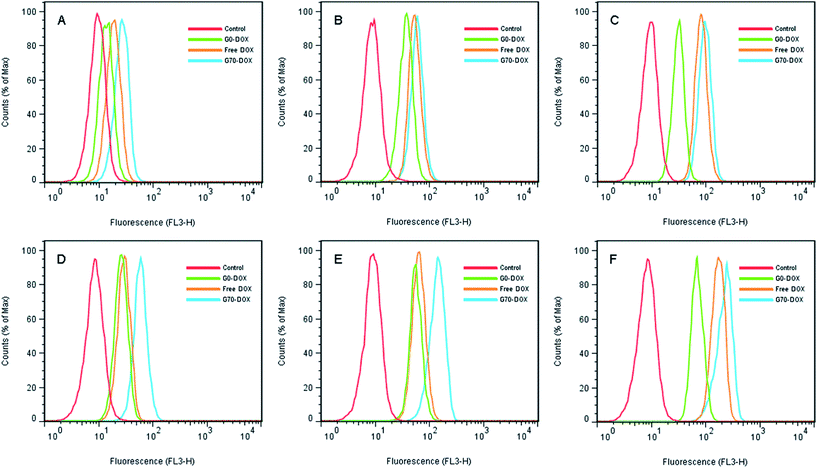 |
| | Fig. 2 Flow cytometry histogram profiles of HBMECs incubated with various concentration of free Dox and Dox-loaded BMPUs micelles for 2 h (A–C) and 4 h (D–F). (A and D) 0.1 μg mL−1 of G70-Dox, G0-Dox and free Dox. (B and E) 0.3 μg mL−1 of G70-Dox, G0-Dox and free Dox. (C and F) 0.5 μg mL−1 of G70-Dox, G0-Dox and free Dox. | |
Table 1 The intracellular increment of MFI with time at each concentration point (fold) measured by flow cytometrya
| Concentration (μg mL−1) |
Fluorescence increment with time |
Increment from 2 h to 4 h |
| 2 h |
4 h |
| Data was presented at means ± standard deviation for n = 3. |
| G70-Dox |
0.1 |
2.84 ± 0.23 |
6.93 ± 0.11 |
2.45 ± 0.15 |
| 0.3 |
6.36 ± 0.22 |
16.14 ± 0.44 |
2.54 ± 0.13 |
| 0.5 |
9.97 ± 0.40 |
29.53 ± 0.67 |
2.97 ± 0.18 |
| Free Dox |
0.1 |
2.05 ± 0.20 |
3.47 ± 0.10 |
1.71 ± 0.17 |
| 0.3 |
6.03 ± 0.01 |
7.64 ± 0.59 |
1.27 ± 0.10 |
| 0.5 |
8.73 ± 0.06 |
20.10 ± 0.48 |
2.30 ± 0.06 |
| G0-Dox |
0.1 |
1.45 ± 0.11 |
3.05 ± 0.03 |
2.11 ± 0.17 |
| 0.3 |
3.45 ± 0.17 |
5.92 ± 0.16 |
1.72 ± 0.08 |
| 0.5 |
3.80 ± 0.22 |
8.75 ± 0.22 |
2.31 ± 0.17 |
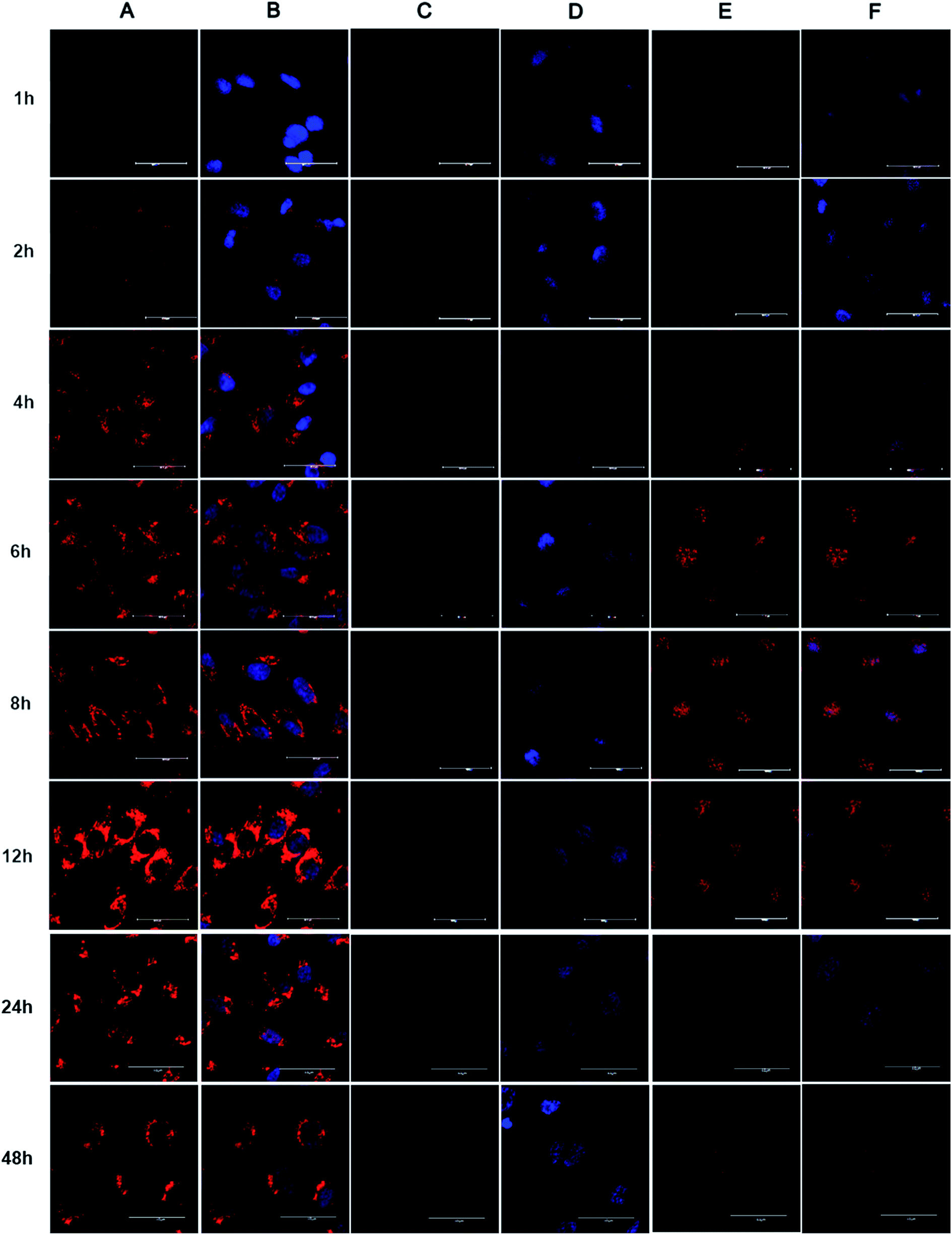
To investigate the intracellular distribution of Dox-loaded BMPUs and free Dox, CLSM experiment of HBMECs treated with equal concentration of G70-Dox, G0-Dox and free Dox at different time points were conducted. The results showed that cellular uptake of Dox was time-dependent (Fig. 3). G70-Dox treated HBMECs achieved the highest uptake of drug with Dox-associated fluorescence locating mainly in cytoplasm close to the nuclei, no sign of nucleic accumulation could be found throughout the 48 h of incubation (Fig. 3A and B). Whereas, free Dox treated HBMECs achieved lower fluorescence augmentation with absorbed Dox mainly distributing in the nuclei. Long time incubation of HBMECs with free Dox could cause unwanted cytotoxicity and cell damage, thus there was no cell observed at 48 h time point (Fig. 3E and F). Unfortunately, there was rare uptake of G0-Dox by HBMECs, only faintly visible Dox-associated fluorescence could be detected in the nuclei at 12 h of incubation. Again, these results demonstrated that incorporation of GQA into BMPUs could greatly improve the cellular uptake of the self-assembled micelles. Free Dox is nucleophilic, often accumulating in nucleus once into the cell. The CLSM results also indicated that G70-Dox was stable in cytoplasm of HBMECs, no early burst release in BBB endothelial cells was observed up to 48 h of incubation. Interestingly, although the intracellular fluorescence intensity increased with time up to 12 h, when incubation time was prolonged to 24 h and 48 h, the fluorescence intensity decreased. This suggested that G70-Dox might escape from HBMECs, the exact mechanism was not known by now and our next immediate study would emphasize on it. Such characteristics were good for G70 to deliver Dox across BBB with less toxicity on endothelial cells. As highly invasive malignant primary brain tumor cells will infiltrate normal brain parenchyma and hide behind intact BBB endothelia,5 it is of primary importance to search and eliminate these tumor cells as well as to protect the integrity of BBB. GQA-incorporated BMPUs micelles may meet such requirement by keeping stable with less drug release throughout the penetrating process. Since the fundamental aim of micellar anticarcinogen delivery is to reach tumor site and release loaded drugs to kill tumor cells, one may raise the question of whether the excellently stable of GQA-BMPUs micelles would result in poor drug release in tumor site and may not achieve any anticancer effect. To further address this issue, we have already developed environment sensitive polyurethane micelles to promote the micellar release of anticancer drugs in tumor site,29–31 which could serve as a promising nanocarrier for drug delivery across BBB treating high grade malignant primary brain tumors in our further studies. The main strategy of this study was to investigate the role of GQA as an enhancer to promote polyurethane micelle across BBB both in vitro and in vivo.
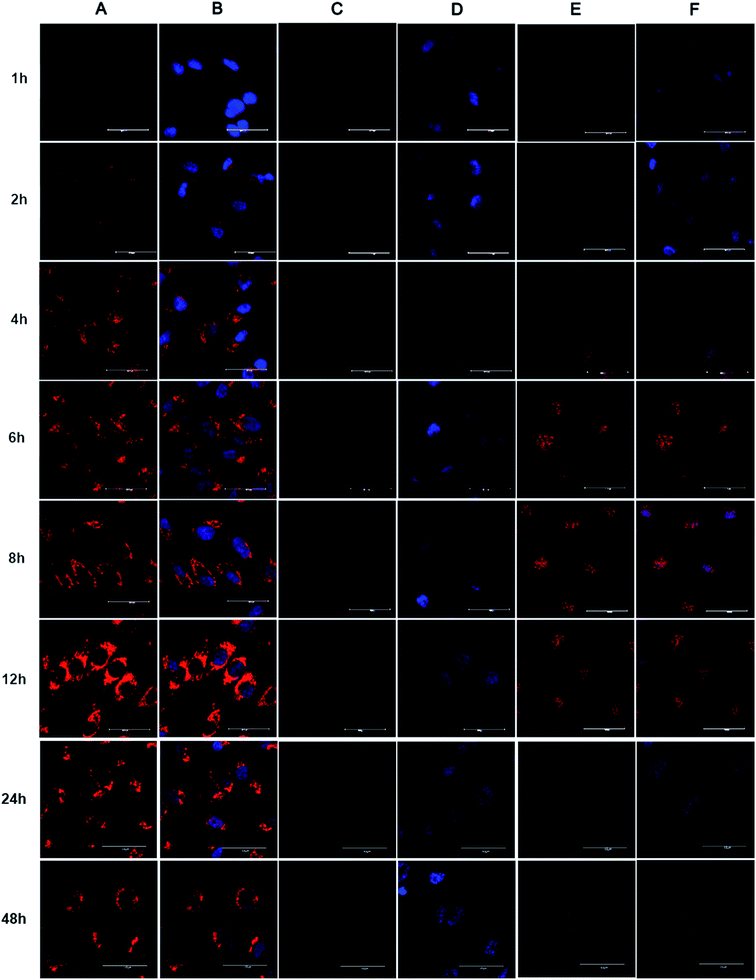 |
| | Fig. 3 CLSM images of HBMECs incubated with G70-Dox (A and B), G0-Dox (C and D) and free Dox (E and F) for different time intervals. The equal concentration of Dox was 1 μg mL−1. Scale bars: 40 μm. (A and B) G70-Dox located in cytoplasm close to nuclei, (C and D) G0-Dox was faintly visible, (E and F) free Dox accumulated in nuclei. Nuclei of cells were stained with DAPI (blue). (B, D and F) showed fluorescence overlays. | |
3.3. BBB penetration and brain accumulation of Dox-loaded BMPUs micelles
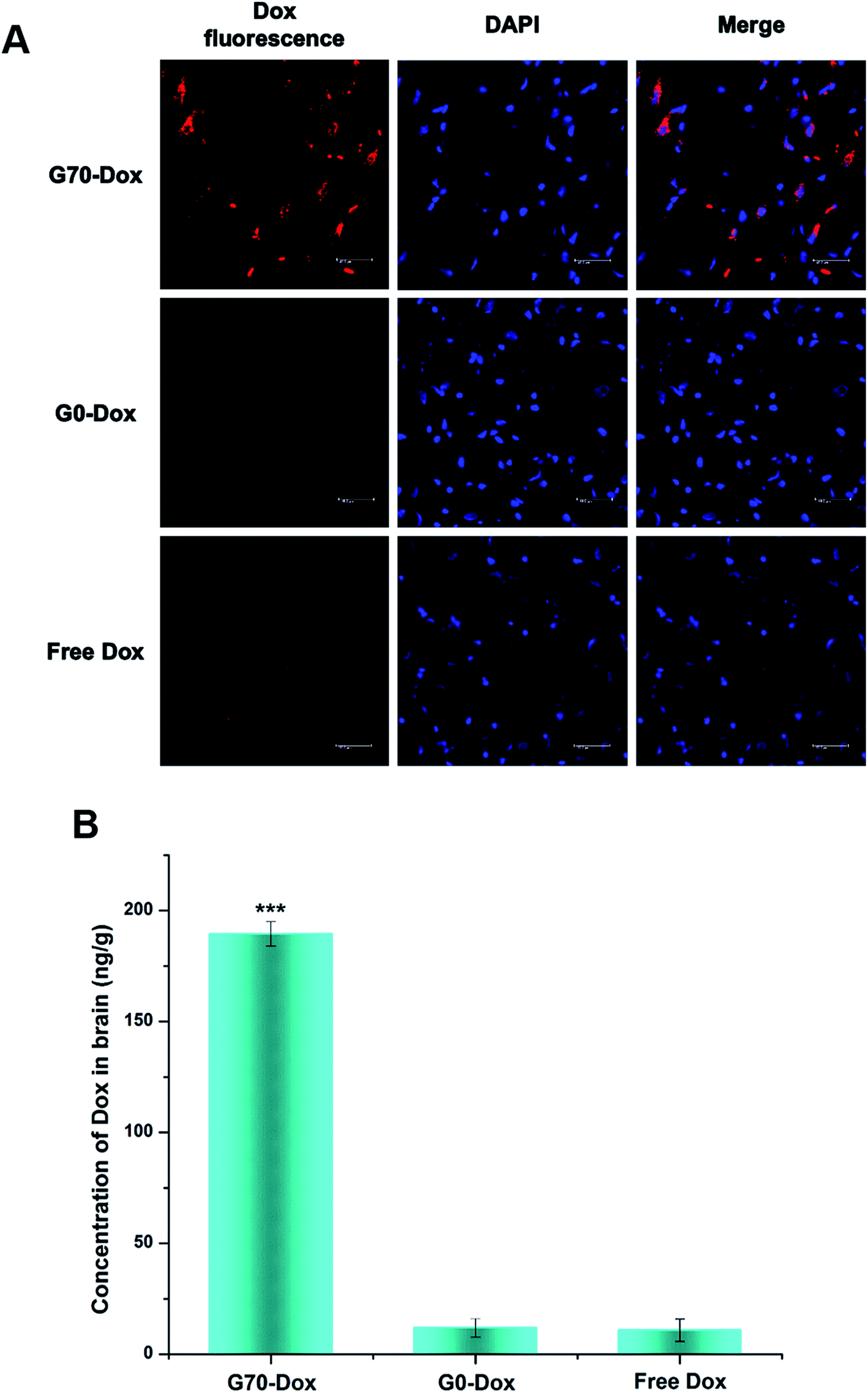
Since BBB is a complex physiological barrier between central nervous system and circulating blood, brain drug delivery in vivo is far more sophisticated than cellular uptake in vitro. Thus, to investigate the efficacy of BBB penetration by GQA-incorporated BMPUs in vivo, G70-Dox, G0-Dox and free Dox were intravenously administrated into the tail vein of female adult SD rats. G70-Dox treated rats exhibited remarkable Dox-related fluorescence extensively distributed in the subcortical area of the cerebrum, whereas G0-Dox and free Dox treated rats rarely displayed any Dox fluorescence throughout the brain parenchyma (Fig. 4A). In addition, G70-Dox treated rats showed 16 and 17 times higher Dox accumulation in brain tissue than G0-Dox and free Dox treated rats (P < 0.001, Fig. 4B), respectively, as confirmed by HPLC assay. G70-Dox increased the brain Dox up to 0.188% of the injected dose (ID) per g brain tissue, which was consistent with the previous published work.32 These results indicated that GQA incorporation into the BMPUs micelles indeed facilitate the transportation of Dox across the BBB, mainly locating at the subcortical area of the cerebrum. Interestingly, high grade malignant primary brain tumors (i.e., glioblastoma) are high invasive tumors which often infiltrate far away into normal brain parenchyma along the nerve fiber at subcortical area. Therefore, GQA-BMPUs micelles are suitable for antitumor drug delivery to such tumors and our next immediate studies will further investigate the efficacy of drug transportation to high grade brain tumors by modified GQA-polyurethane micellar drug carriers.
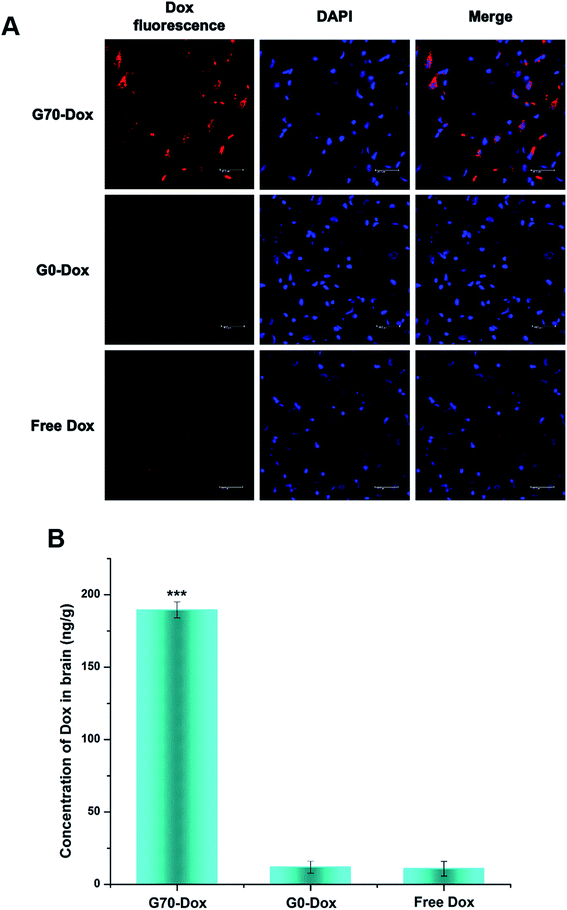 |
| | Fig. 4 Qualitative (A) and quantitative (B) uptake of free Dox and Dox-loaded BMPUs micelles by brain tissue of SD rats 8 h post administration, equal concentration of Dox (100 μg mL−1). (A) CLSM images of brain parenchyma in subcortical area, red (Dox), blue (DAPI). The scale bars: 40 μm. (B) Brain accumulation profile of G70-Dox, G0-Dox and free Dox. G70-Dox showed more accumulation in brain parenchyma. (***) G70-Dox vs. G0-Dox and free Dox, P < 0.001. | |
3.4. In vivo pharmacokinetics and biodistribution of Dox-loaded BMPUs micelles
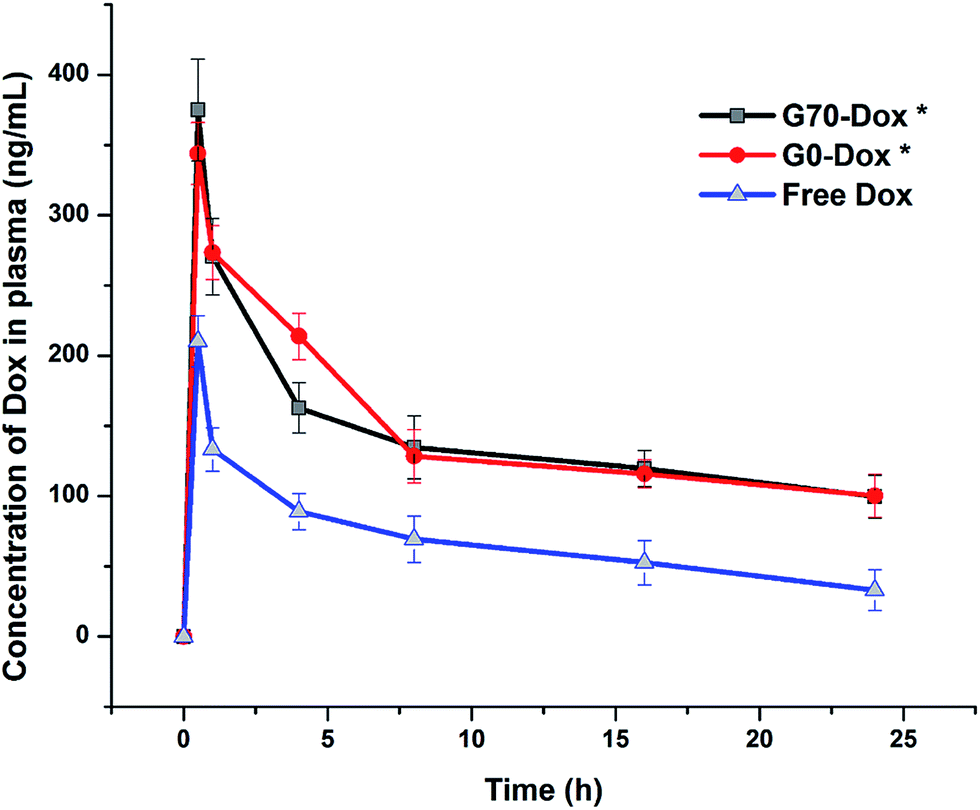
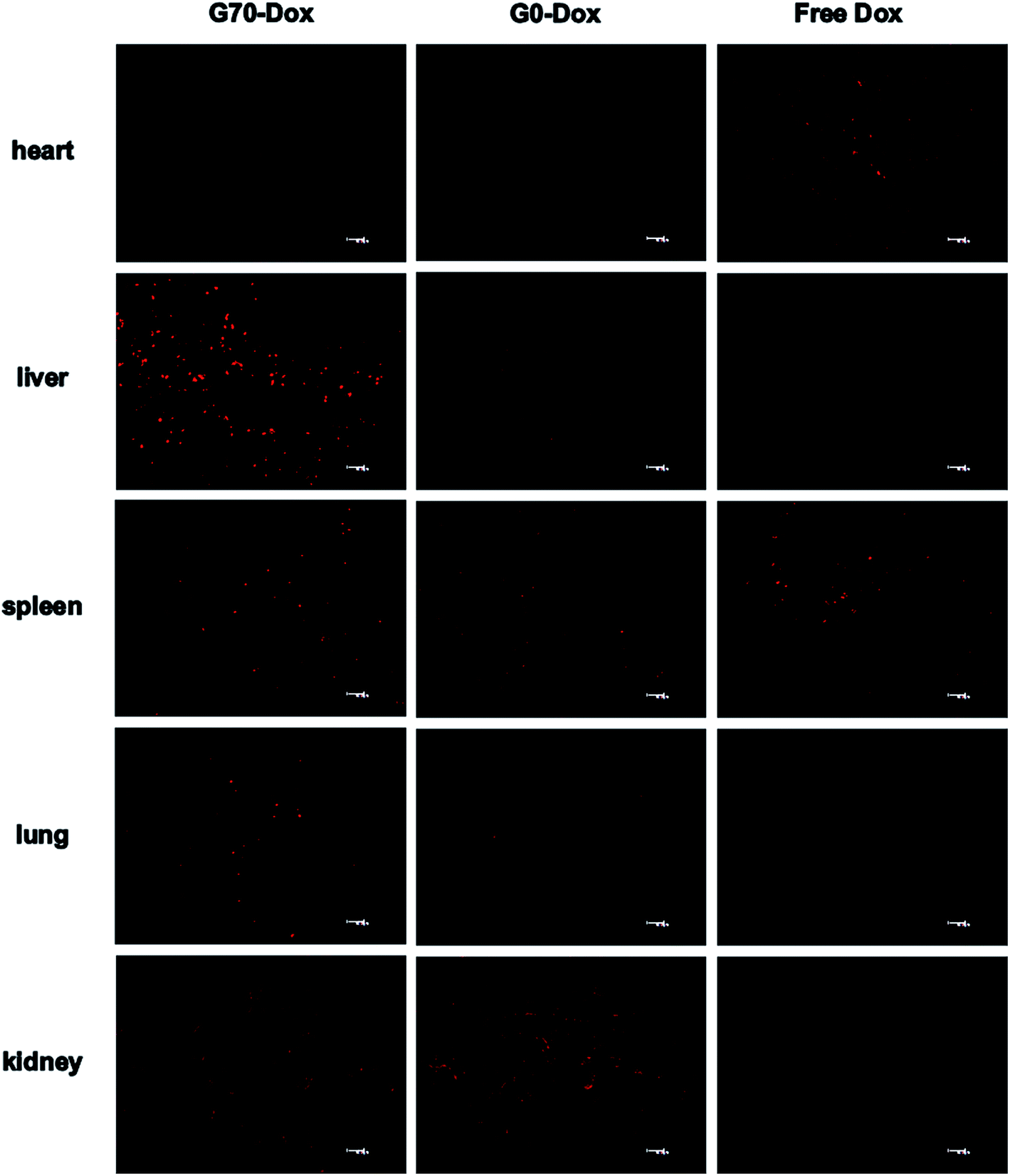
To better evaluate the situation of Dox-loaded BMPUs micelles in vivo, we examined the pharmacokinetics and peripheral tissue distribution of G70-Dox, G0-Dox and free Dox in SD rats. The plasma concentration-time profiles of Dox for the three formulations showed that G70-Dox and G0-Dox significantly increase the blood retention time of Dox, comparing to free Dox (P < 0.05, Fig. 5). Dox is a small molecule antineoplastic agent, which may easily extravasate to surrounding tissues from peripheral capillaries.33 BMPUs micelles could slow the extravasation of Dox which may reduce the systemic toxicity of G70-Dox and G0-Dox treatment. By the way, the prolonged circulation time of G70-Dox and G0-Dox may improve the content of drug carriers exposing to the BBB, which is beneficial to the following BBB penetrating process. Biodistribution results are presented in Fig. 6. Free Dox distributed equally to peripheral tissues except for heart (more intensive fluorescence signal in heart tissue), indicating the heart-philic feature of Dox.34–36 G70-Dox and G0-Dox had an accumulation in mono-phagocytic system of liver, spleen and lung tissues, demonstrating that micellar drug carriers are easily hijacked by reticuloendothelial system (RES). Charged surface of nanocarriers could improve such process, while surface coating of some hydrophilic polymers like PEG can reduce RES uptake. Thus, we redesigned G70 and G0 with long PEG chains (MW 1900) as described in previous section. In future studies, further efforts are still needed (i.e., introduction of longer PEG chains) to deeply reduce RES elimination and prolong circulation time of GQA-BMPUs micelles. Both G70-Dox and G0-Dox were hydrophilic systems, which could be excreted through urine. Therefore, they had an accumulation in kidney.
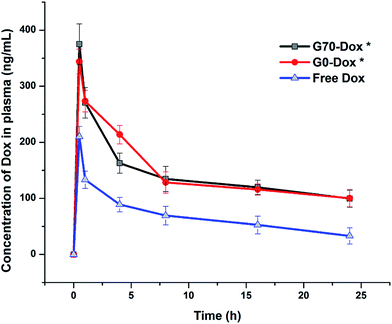 |
| | Fig. 5 Plasma concentration-time profiles of Dox after intravenous injection of G70-Dox, G0-Dox and free Dox at 0.5 mg kg−1 dose in SD rats. Error bars represent means ± standard deviation for n = 3. Dox loaded in BMPUs micelles showed longer circulation time than free Dox. (*) P < 0.05 vs. free Dox. | |
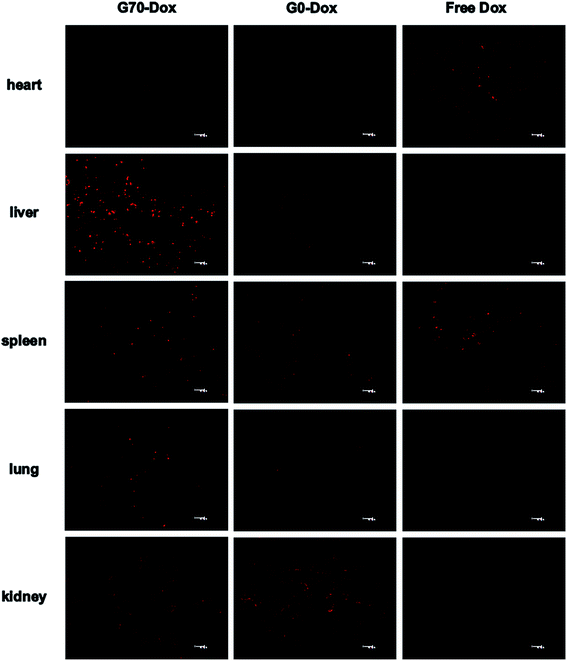 |
| | Fig. 6 Tissue distribution images of Dox 8 h post administration of G70-Dox, G0-Dox and free Dox at 0.5 mg kg−1 dose in SD rats, scale bars: 40 μm. G70-Dox accumulated more in RES, while free Dox was more in heart. | |
3.5. Toxicity evaluation
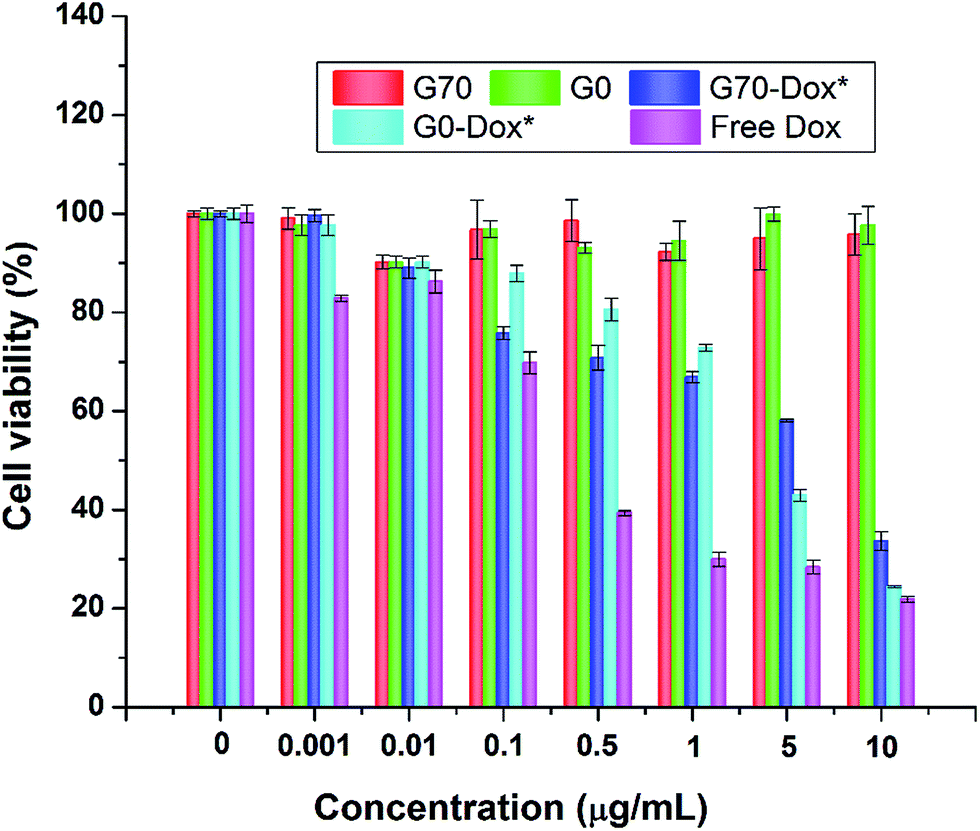
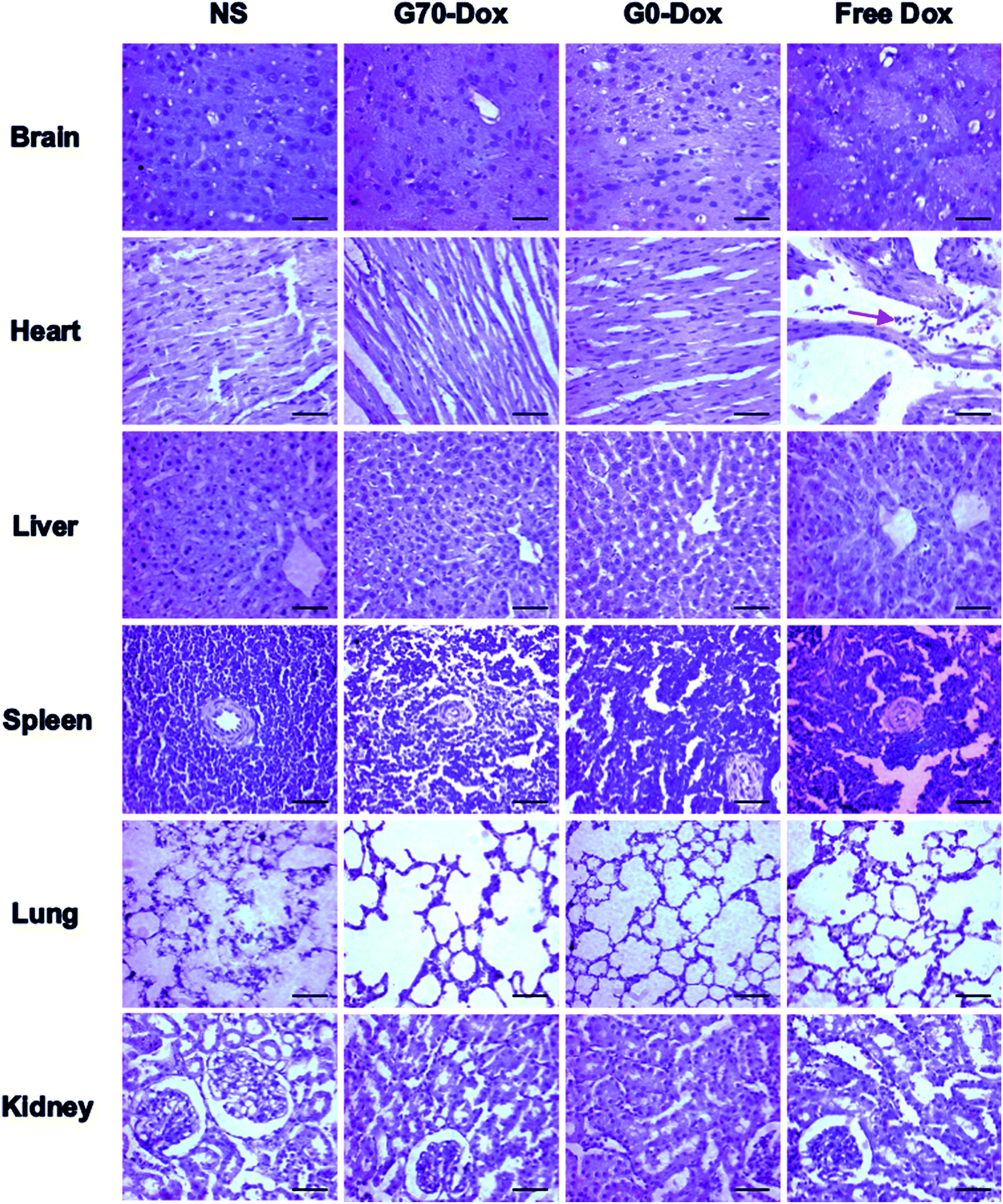
Since wide application of nanomaterials in drug delivery has raised toxicology concerns, herein, toxicity evaluation of blank BMPUs micelles and Dox-loaded BMPUs micelles was conducted both in vitro and in vivo. MTT assay of HBMECs treated with various concentrations of Dox-loaded and drug free micelles showed that blank micelles maintained over 90% cell viability at each concentration (Fig. 7). G70-Dox and G0-Dox could reduce cytotoxicity of Dox, comparing to free Dox (P < 0.05), while there was no statistical difference observed in cytotoxicity between G70-Dox and G0-Dox (P > 0.1). H&E stains showed that no noticeable toxicity and tissue damage could be detected in organs (brain, heart, liver, spleen, lung and kidney) of G70-Dox and G0-Dox treated SD rats (Fig. 8). Whereas, somewhat inflammatory exudation could be found in heart tissue of free Dox group. These results suggested that BMPUs micelles could reduce Dox toxicity both in vitro and in vivo. For further development of the polyurethane micelles, however, detailed toxicity evaluation as well as chronic in vivo toxic effect of polyurethane micelles are needed.
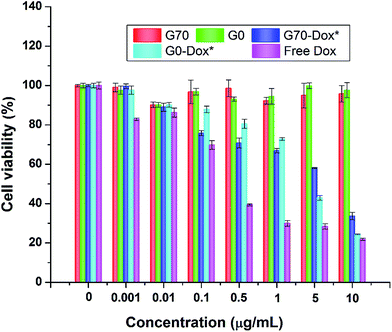 |
| | Fig. 7 Cytotoxicity profiles of HBMECs incubated with various concentrations of drug free and Dox-loaded BMPUs micelles. Blank G70 and G0 micelles were equally diluted with G70-Dox and G0-Dox. Cell viability was over 90% with blank micelles, cells treated with G70-Dox and G0-Dox showed higher viability than with free Dox of different concentrations. (*) P < 0.05 vs. free Dox. | |
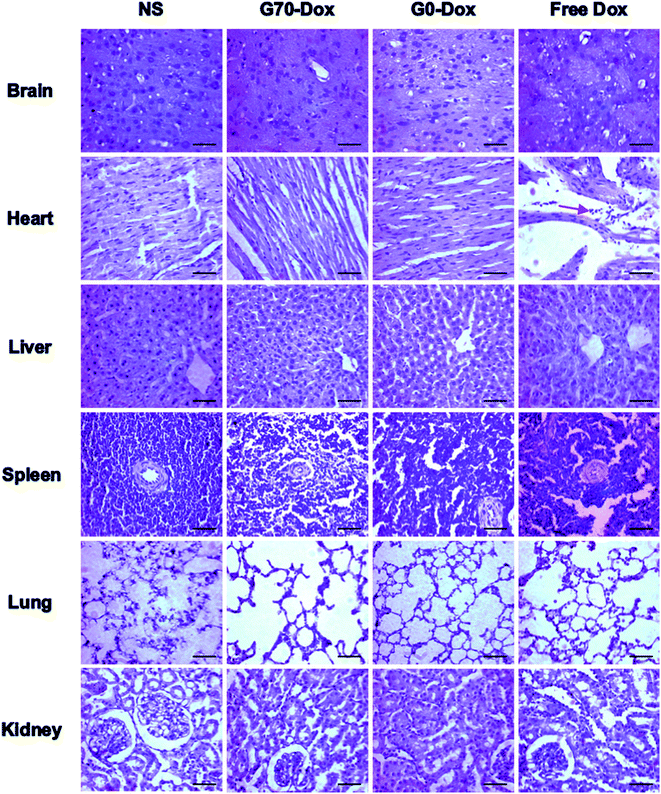 |
| | Fig. 8 H&E stains of various tissues collected from SD rats receiving different treatments, scale bars: 100 μm. Red arrow: inflammatory cells. Almost no tissue damage or inflammation was observed in NS, G70-Dox and G0-Dox treated rats, while inflammatory response was detected in heart of free Dox treated rats. | |
4. Conclusions
In summary, we firstly introduced GQA into BMPUs micelles in the delivery of antitumor drug across BBB. The GQA not only acted as a micellization enhancer with stable zeta potential and high drug loading capacity, but also played an important role in BBB penetration, as demonstrated by flow cytometry and CLSM experiments. In vitro cellular uptake and in vivo brain uptake of Dox-loaded GQA-BMPUs micelles demonstrated great advantage over bare BMPUs micelles and free Dox formulations. Cytotoxicity assay and histological toxicity examination showed great biocompatibility of the cationic GQA-BMPUs micelles. Moreover, pharmacokinetics and biodistribution studies provided a better understanding of the metabolism of such gemini cationic micelles in vivo, and would help us design more suitable GQA-BMPUs micelles in further work. Our findings would provide new prospective in brain drug delivery and encourage further studies using GQA-based polymers as micellar drug carriers to transport therapeutic/imaging agents across BBB.
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
We would like to thank the financial supports by the projects of National Natural Science Foundation of China (contract/grant number: 81100925, 81472361, 51073104, 51273126 and 51121001).
References
- J. R. Kanwar, X. Sun, V. Punj, B. Sriramoju, R. R. Mohan, S. F. Zhou, A. Chauhan and R. K. Kanwar, Nanomedicine, 2012, 8, 399–414 CrossRef CAS PubMed.
- G. Lee, S. Dallas, M. Hong and R. Bendayan, Pharmacol. Rev., 2001, 53, 569–596 CrossRef CAS PubMed.
- D. R. Groothuis, Neuro-Oncology, 2000, 2, 45–59 CAS.
- M. Srikanth and J. A. Kessler, Nat. Rev. Neurol., 2012, 8, 307–318 CrossRef CAS PubMed.
- H. Wolburg, S. Noell, P. Fallier-Becker, A. F. Mack and K. Wolburg-Buchholz, Mol. Aspects Med., 2012, 33, 579–589 CrossRef CAS PubMed.
- R. Gabathuler, Neurobiol. Dis., 2010, 37, 48–57 CrossRef CAS PubMed.
- G. A. Silva, BMC Neurosci., 2008, 9(suppl 3), S4 CrossRef PubMed.
- J. D. Meyers, T. Doane, C. Burda and J. P. Basilion, Nanomedicine, 2013, 8, 123–143 CrossRef CAS PubMed.
- L. Costantino and D. Boraschi, Drug Discovery Today, 2012, 17, 367–378 CrossRef CAS PubMed.
- H. Xin, X. Sha, X. Jiang, W. Zhang, L. Chen and X. Fang, Biomaterials, 2012, 33, 8167–8176 CrossRef CAS PubMed.
- H. Gao, J. Qian, S. Cao, Z. Yang, Z. Pang, S. Pan, L. Fan, Z. Xi, X. Jiang and Q. Zhang, Biomaterials, 2012, 33, 5115–5123 CrossRef CAS PubMed.
- K. S. Rao, M. K. Reddy, J. L. Horning and V. Labhasetwar, Biomaterials, 2008, 29, 4429–4438 CrossRef CAS PubMed.
- L. Liu, K. Guo, J. Lu, S. S. Venkatraman, D. Luo, K. C. Ng, E. A. Ling, S. Moochhala and Y. Y. Yang, Biomaterials, 2008, 29, 1509–1517 CrossRef CAS PubMed.
- D. G. Bucknall and H. L. Anderson, Science, 2003, 302, 1904–1905 CrossRef CAS PubMed.
- S. Grad, L. Kupcsik, K. Gorna, S. Gogolewski and M. Alini, Biomaterials, 2003, 24, 5163–5171 CrossRef CAS.
- G. Jianjun, K. L. Fujimoto, M. S. Sacks and W. R. Wagner, Biomaterials, 2005, 26, 3961–3971 CrossRef PubMed.
- M. Ding, J. Li, X. Fu, J. Zhou, H. Tan, Q. Gu and Q. Fu, Biomacromolecules, 2009, 10, 2857–2865 CrossRef CAS PubMed.
- F. M. Menger and J. S. Keiper, Angew. Chem., Int. Ed., 2000, 39, 1906–1920 CrossRef.
- M. Ding, L. Zhou, X. Fu, H. Tan, J. Li and Q. Fu, Soft Matter, 2010, 6, 2087 RSC.
- M. Ding, X. He, L. Zhou, J. Li, H. Tan, X. Fu and Q. Fu, J. Controlled Release, 2011, 152, E87–E89 CrossRef CAS PubMed.
- M. Ding, X. He, Z. Wang, J. Li, H. Tan, H. Deng, Q. Fu and Q. Gu, Biomaterials, 2011, 32, 9515–9524 CrossRef CAS PubMed.
- D. Mingming, Q. Zongzheng, W. Jin, L. Jiehua, T. Hong, G. Qun and F. Qiang, Polym. Chem., 2011, 2, 885–891 RSC.
- K. V. Thrivikraman, R. L. Huot and P. M. Plotsky, Brain Res. Protoc., 2002, 10, 84–94 CrossRef CAS.
- E. Gullotti and Y. Yeo, Mol. Pharm., 2009, 6, 1041–1051 CrossRef CAS PubMed.
- Y. C. Kuo and C. T. Liang, Biomaterials, 2011, 32, 3340–3350 CrossRef CAS PubMed.
- Y. C. Kuo and H. W. Yu, Int. J. Pharm., 2011, 416, 365–375 CrossRef CAS PubMed.
- W. Lu, J. Wan, Z. She and X. Jiang, J. Controlled Release, 2007, 118, 38–53 CrossRef CAS PubMed.
- T. Parikh, M. M. Bommana and E. Squillante III, Eur. J. Pharm. Biopharm., 2010, 74, 442–450 CrossRef CAS PubMed.
- L. Zhou, L. Yu, M. Ding, J. Li, H. Tan, Z. Wang and Q. Fu, Macromolecules, 2011, 44, 857–864 CrossRef CAS.
- L. Zhou, D. Liang, X. He, J. Li, H. Tan, Q. Fu and Q. Gu, Biomaterials, 2012, 33, 2734–2745 CrossRef CAS PubMed.
- M. Ding, N. Song, X. He, J. Li, L. Zhou, H. Tan, Q. Fu and Q. Gu, ACS Nano, 2013, 7, 1918–1928 CrossRef CAS PubMed.
- I. van Rooy, S. Cakir-Tascioglu, W. E. Hennink, G. Storm, R. M. Schiffelers and E. Mastrobattista, Pharm. Res., 2011, 28, 456–471 CrossRef CAS PubMed.
- M. Gou, H. Shi, G. Guo, K. Men, J. Zhang, L. Zheng, Z. Li, F. Luo, Z. Qian, X. Zhao and Y. Wei, Nanotechnology, 2011, 22, 095102 CrossRef PubMed.
- F. S. Carvalho, A. Burgeiro, R. Garcia, A. J. Moreno, R. A. Carvalho and P. J. Oliveira, Med. Res. Rev., 2014, 34, 106–135 CrossRef CAS PubMed.
- T. H. Tran, C. T. Nguyen, L. Gonzalez-Fajardo, D. Hargrove, D. Song, P. Deshmukh, L. Mahajan, D. Ndaya, L. Lai, R. M. Kasi and X. Lu, Biomacromolecules, 2014, 15, 4363–4375 CrossRef CAS PubMed.
- S. D. Russell, K. L. Blackwell, J. Lawrence, J. E. Pippen Jr, M. T. Roe, F. Wood, V. Paton, E. Holmgren and K. W. Mahaffey, J. Clin. Oncol., 2010, 28, 3416–3421 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09908g |
| ‡ Fang Fang contributed equally to this work with Rui-Chao Liang, and is the co-first author for this paper. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.