DOI:
10.1039/C4RA12075B
(Paper)
RSC Adv., 2015,
5, 6151-6159
Detection of NaCN in aqueous media using a calixarene-based fluoroionophore containing ruthenium(II)-bipyridine as the fluorogenic unit†
Received
9th October 2014
, Accepted 20th November 2014
First published on 2nd December 2014
Abstract
A new molecular sensor containing calixarene and ruthenium(II)-bipyridine as the fluorophore bridged by an amide moiety has been synthesised and characterized, and its anion binding properties have been investigated. It selectively detected cyanide in 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 water:acetonitrile when sodium salts of various anions such as F−, Cl−, Br−, I−, PO42−, IO4−, BO3−, CH3COO−, CN− and SO42− were investigated. The recognition event was monitored by fluorescence spectroscopy and the lower detection limit found was 70 ppb. However, when tetrabutylammonium salts of the same anions were used then, in addition to CN−, CH3COO− was also detected under similar experimental conditions. Interestingly, CN− exhibited substantial quenching, whereas CH3COO− showed an enhancement in emission intensity. The interaction of anions with the fluoroionophore was also monitored by electrochemical technique and the result obtained is consistent to that found by fluorogenic method. Binding constants were determined from emission titration, composition of the anion-complexes formed were determined from mass and emission titration data, mechanistic aspects of the interaction have been discussed with the aid of NMR data and the role of cations in the contrast fluorescent off and on behaviour has also been discussed. This sensor has also been used to estimate cyanide in real samples and the result obtained is satisfactory.
:5 water:acetonitrile when sodium salts of various anions such as F−, Cl−, Br−, I−, PO42−, IO4−, BO3−, CH3COO−, CN− and SO42− were investigated. The recognition event was monitored by fluorescence spectroscopy and the lower detection limit found was 70 ppb. However, when tetrabutylammonium salts of the same anions were used then, in addition to CN−, CH3COO− was also detected under similar experimental conditions. Interestingly, CN− exhibited substantial quenching, whereas CH3COO− showed an enhancement in emission intensity. The interaction of anions with the fluoroionophore was also monitored by electrochemical technique and the result obtained is consistent to that found by fluorogenic method. Binding constants were determined from emission titration, composition of the anion-complexes formed were determined from mass and emission titration data, mechanistic aspects of the interaction have been discussed with the aid of NMR data and the role of cations in the contrast fluorescent off and on behaviour has also been discussed. This sensor has also been used to estimate cyanide in real samples and the result obtained is satisfactory.
Introduction
Selective detection of CN− has received considerable interest because of its toxic effect towards mammals resulting from its ability to interfere with the electron transfer process.1 Cyanide toxicity occurs via inactivation of cytochrome oxidase and inhibition of cellular respiration and consequent cellular anoxia.2 Hence, humans' and animals' cardiovascular, respiratory and central nervous systems are highly prone to being affected by acute cyanide poisoning.1 Despite its toxic nature, cyanides are industrially made in large quantities and used in electroplating, as raw materials for synthetic fibres, in resins, in herbicides and for gold extraction.3,4 Various industries produce as much as 1400000 tons of toxic cyanide per year worldwide.5 Because of its acute toxicity, human beings as well as aquatic life have very low tolerance limits for CN−. The U.S. EPA regulates cyanide content at very low levels of 0.2 ppm and 0.005 mg L−1 for drinking water and environmental primary standards, respectively.6
Several techniques have been developed for the detection of CN− in water as well as non-aqueous media. These techniques include chromatography, potentiometric methods, and amperometry.7–10 However, these techniques are not user friendly and are difficult to apply for physiological monitoring or for monitoring of drinking water. Significant work has been done on the development of fluorometric and colorimetric based chemosensors for the detection of CN−.11 However, simple and efficient methods to monitor cyanide contamination in water, soil and biological fluids (blood, urine, saliva) are still needed.
When designing molecular sensors for anions, especially for CN−, metal complex based sensors have been extensively used as the positive charge on the metal ion facilitates interaction with negatively charged ions.11–19 Apart from metal complexes, other chemosensors based on quantum dots, organic dyes and other protic chromophores have also been reported for detection of CN−.20–25 Chromophores containing NH and OH are advantageous as they can form strong hydrogen-bonding interactions with the anions.26–28 However, some of the CN− receptors reported also exhibit limitations, such as interference from other anions, especially from F− and CH3CO2−, basic pH, non-aqueous medium, high detection limit, and risk of releasing HCN during experiments.11,12,20,22,24,29,30
Keeping the reported limitations in mind, we have designed a fluoroionophore containing a photoactive Ru(II)-bipyridine unit as the fluorogenic unit and amide containing a calixarene moiety as the binding site for anions. As receptors, calixarenes are found to be very attractive because modifications of calixarene give rise to a large variety of derivatives with various functional groups, which provide a highly preorganized architecture for the assembly of converging binding sites.31–35 As mentioned above, incorporation of a metal ion has some advantages: the positive charge on it is expected to assist strong hydrogen bonding interactions between anions and the NH and OH protons of the calix moiety. Moreover, the spectroscopic and electrochemical properties of the metal ion can also be used for monitoring of the recognition event.36–38 This new fluoroionophore selectively detects CN− from NaCN in aqueous media in the presence of various other anions including the most interfering anions, F− and CH3COO−, and the binding of CN− is also reversible. The performance of this CN− sensor has also been tested successfully in real samples. Herein we report synthesis and characterization of this new fluoroionophore and details of its performance as an anion sensor in aqueous media.
Results and discussion
Synthesis and characterization of the fluoroionophore (1)
The route followed for the synthesis of L and its Ru(II) complex (1) is shown in Scheme 1 and details of the experimental procedure are given in the Experimental section. Compound L was synthesized from compounds C and F (Scheme 1) in dry acetonitrile using Et3N as the base under inert atmosphere. The acid chloride (F), obtained from the corresponding acid (E), was directly used for the next step without isolation. Elemental analysis (C, H and N), IR, ESMS and 1H NMR spectral data for the intermediate compounds, ligand L and complex 1 are given in the Experimental section. The elemental analysis and mass data are in excellent agreement with the calculated values. The mass spectra for L and 1 are presented in the ESI (Fig. S1 and S2†). It should be noted that the m/z value of the compound L corresponds to the H+ adduct, which is a well-known phenomenon when LC-MS is used for the measurement of mass.32–34,36
 |
| | Scheme 1 The route followed for synthesis of L and 1 is shown. Reagents and conditions: (I) CH2BrCO2Me, K2CO3, ACN; (II) NH2CH2CH2NH2, MeOH; (III) SeO2, dioxane; Ag2O, dioxane/EtOH; (IV) SOCl2, toluene; (V) ACN, Et3N; (VI) Ru(bpy)2Cl2, EtOH/H2O. | |
The IR spectrum of L exhibited bands of moderate intensity at 3545 cm−1 and 3049 cm−1, which are due to ν(OH) of phenolic group of calixarene and ν(NH) of the amide moiety, respectively. The strong band that appeared at 1677 cm−1 is assigned to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). In the 1H NMR spectrum of L (Fig. S3, ESI†), the meta-protons (with respect to the OH substituent) of the calixarene moiety appeared as three very closely spaced singlets in the region δ 7.13–6.95, which is expected due to the partial cone conformation of the calix moiety, as shown in Scheme 1.39 Three phenolic –OH appeared at δ 10.17, 9.58 and 9.48, as confirmed by D2O exchange experiment. The bipyridine moiety exhibited five distinct signals in the region δ 8.72–7.61, and another signal probably overlapped with the signals from the calix moiety. The protons of the amide moieties –CONH– are overlapped with the multiplets due to the aromatic moiety around δ 8.5. The methylene protons of ArO–CH2–CO– appeared as a singlet at δ 4.63 and the Ar–CH2–Ar protons of the calixarene moiety appeared as two closely spaced doublets in the region δ 3.49–3.31 and one singlet at δ 3.86, which suggest that the calixarene moiety exists in a paco conformation.39 The –CH2 of the ethylene chain appeared as a triplet at δ 4.12. The Ru(II) complex of L was synthesized by the reaction of cis-[Ru(bpy)2Cl2]2H2O and L in refluxing ethanol–water, isolated with a PF6− counter anion and purified by column chromatography. This complex gave satisfactory C, H and N analysis and the mass data (m/z) (Fig. S2, ESI†) are in excellent agreement with the calculated value of 1503.55. The IR spectrum of 1 exhibited a broad band at 3433 cm−1, which is due to ν(O–H). The bands at 1676 and 842 cm−1 are assigned to ν(CO) and PF6−, respectively. The NMR spectrum (Fig. S4†), the data from which are given in the Experimental section, is consistent with the structure shown in Scheme 1.
O). In the 1H NMR spectrum of L (Fig. S3, ESI†), the meta-protons (with respect to the OH substituent) of the calixarene moiety appeared as three very closely spaced singlets in the region δ 7.13–6.95, which is expected due to the partial cone conformation of the calix moiety, as shown in Scheme 1.39 Three phenolic –OH appeared at δ 10.17, 9.58 and 9.48, as confirmed by D2O exchange experiment. The bipyridine moiety exhibited five distinct signals in the region δ 8.72–7.61, and another signal probably overlapped with the signals from the calix moiety. The protons of the amide moieties –CONH– are overlapped with the multiplets due to the aromatic moiety around δ 8.5. The methylene protons of ArO–CH2–CO– appeared as a singlet at δ 4.63 and the Ar–CH2–Ar protons of the calixarene moiety appeared as two closely spaced doublets in the region δ 3.49–3.31 and one singlet at δ 3.86, which suggest that the calixarene moiety exists in a paco conformation.39 The –CH2 of the ethylene chain appeared as a triplet at δ 4.12. The Ru(II) complex of L was synthesized by the reaction of cis-[Ru(bpy)2Cl2]2H2O and L in refluxing ethanol–water, isolated with a PF6− counter anion and purified by column chromatography. This complex gave satisfactory C, H and N analysis and the mass data (m/z) (Fig. S2, ESI†) are in excellent agreement with the calculated value of 1503.55. The IR spectrum of 1 exhibited a broad band at 3433 cm−1, which is due to ν(O–H). The bands at 1676 and 842 cm−1 are assigned to ν(CO) and PF6−, respectively. The NMR spectrum (Fig. S4†), the data from which are given in the Experimental section, is consistent with the structure shown in Scheme 1.
Anion binding study using fluorescence spectroscopy
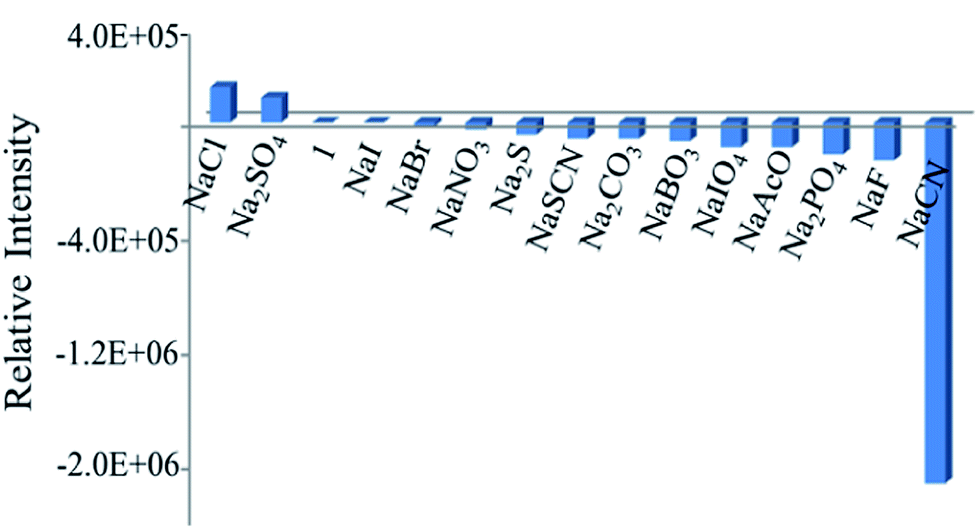
The photophysical and electrochemical properties of the ruthenium(II)-bipyridine complexes are well known and these properties can be used as a tool to monitor ion-recognition events. For complex 1, upon excitation the MLCT absorption band at 460 nm exhibited a strong emission band at 634 nm, which has been used to monitor its interaction with anions. Upon addition of the Na+ salts of anions such as F−, Cl−, Br−, I−, PO42−, IO4−, BO3−, CH3COO−, CN− and SO42− in 95:5H2O–CH3CN medium (details of the procedure are given in the Experimental section), the intensity of the emission band for the CN− anion was substantially quenched with a slight blue shift, whereas for other anions no significant change was noted (Fig. S5†). A bar diagram showing the plot of the relative change in the intensity of the emission band with respect to various anions used in this study is shown in Fig. 1. The diagram suggests that the fluoroionophore 1 selectively interacts with CN− in aqueous media.
 |
| | Fig. 1 Bar diagram showing the plot of the relative change in emission intensity for 1 (2 × 10−6 M) in the presence of sodium salts of various anions (2 × 10−4 M) in H2O:CH3CN (95:5). Excitation wavelength: 461 nm. | |
Effect of pH
The emission spectrum of 1 was recorded with and without the addition of CN− in aqueous media with different pHs. It should be noted that complex 1 exhibits its luminescence property in the pH range 2 to 12; however, the intensity of the emission band increased at the lower pH (Fig. 2). After addition of NaCN, the intensity of the emission band is quenched but the extent of quenching is low at lower pH, similar to that found in the absence of CN− (Fig. 2). In acidic pH, the –CONH can easily get protonated, which is expected to lead to a reverse PET effect, resulting in an increase in the emission intensity of the Ru-bipyridine moiety.40
 |
| | Fig. 2 Bar diagram showing the relative change in emission intensity of complex 1 before (blue bar) and after (red bar) addition of NaCN (25 equivalent) at different pHs in water–acetonitrile (95:5). | |
Interference study
The interferences of other anions for the detection of CN− were also investigated by recording emission spectra of 1 upon addition of a mixture of 10% of cyanide and 90% of other anions and the plot of the fraction of quenching in emission intensity as a function of mixture of different anions is shown in Fig. 3. It may be noted that no significant interference from any other anion is observed.
 |
| | Fig. 3 Bar diagram showing the fraction of quenching in emission intensity for 1 at 642 nm upon addition of CN− and a mixture of anions containing 10% of CN− (1.2 × 10−6 M) and 90% of other anions (1.08 × 10−5 M) recorded in H2O–CH3CN (95:5) solvent. | |
Reversible binding of CN−
The reversible binding of CN− with 1 was also investigated using Cu(ClO4)2, as it can remove CN− from the complex forming insoluble CuCN.34 It should be noted that after addition of 1 equivalent of Cu(ClO4)2 with respect to NaCN added into the solution of 1, the emission intensity is regained and it is close to that observed for 1 before the addition of CN−. To this solution, after further addition of the same equivalent amount of NaCN, similar quenching as noted earlier was observed (Fig. S6, ESI†). The observation thus indicates that the binding of CN− with complex 1 is reversible in aqueous medium.
Determination of binding constant
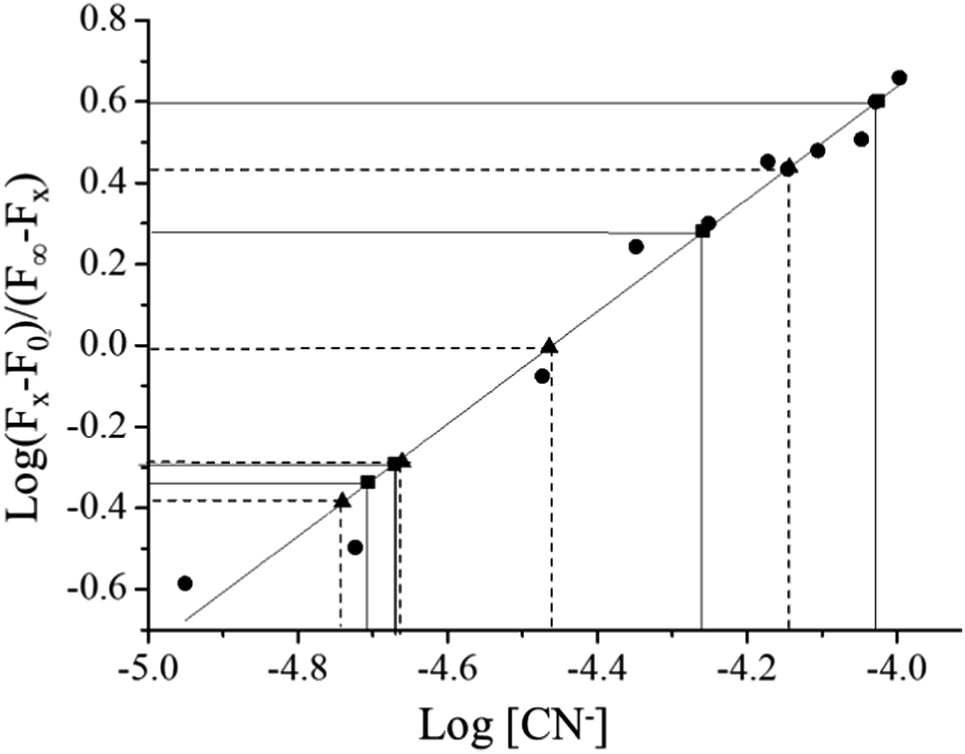
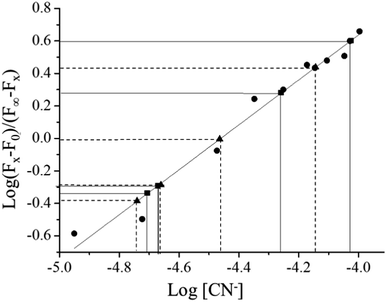
The binding constant of CN− with complex 1 in aqueous solution (H2O:CH3CN, 95:5) was determined by fluorescence titration method.36,41 Details of the method are given in the Experimental section and the emission spectral change upon incremental addition of CN− is shown in Fig. 4. The binding constant was calculated from the titration data using the equation (F0 − Fx)/(Fx − F) = ([M]/Kdiss)n. The binding constant (Ks) was obtained by plotting log[(F0 − F)/(F − F∞)] vs. log[M], the double logarithm plot, which exhibits linearity in the range 5.03 × 10−5 to 1.12 × 10−4 M (R2 = 0.969), shown as the inset in Fig. 4. The slope of this line (1.86115) gave the binding constant, 2.5 × 104 M−1, and the value of n (2.2), which suggests 1:2 stoichiometry. The lower limit of detection for NaCN was also calculated from the emission spectral change and it was found to be 70 ppb in the same solvent system.
 |
| | Fig. 4 Emission spectral changes of 1 (1 × 10−6 M) upon addition of increasing concentrations of NaCN. Excitation wavelength: 458 nm. Inset: linear regression fit (double-logarithmic plot) of the titration data as a function of concentration of CN− ion. | |
Anion-sensing study using tetrabutylammonium salts
When the anion-binding study was conducted with tetrabutylammonium (TBA) salts under similar experimental conditions as used for NaCN, then the selectivity pattern changed. Like NaCN, the TBA salt of CN− exhibited substantial quenching; however, fluoride also showed some quenching but CH3COO− exhibited a significant enhancement in emission intensity, whereas other anions did not show any considerable change (Fig. 5). The observations suggest that the cation also plays an important role in sensing and in the presence of the TBA ion, like CN−, CH3COO− also forms a strong interaction with 1 and F− also shows a weak interaction. This cation dependent selectivity is probably related to the interaction of the cations with the calix moiety and also its conformation in solution. It is well established that alkali metal ions form complexes with calixarene and that the metal–calix interaction takes place at the lower rim of the calix moiety because of the presence of OH groups and also due to the enhanced π electron delocalization at that site.42 Anions can interact with the hydrogen atoms of the amide moiety or the OH groups of the calix; however, in this case NMR study showed that (discussed in the mechanism section) anions formed interactions with the amide hydrogen atoms. Therefore, if Na+ is in the vicinity of the lower rim of the calix, then due to steric crowding it is difficult for the large anions to enter the loop made by the calix unit and the Ru-bipyridine unit (structure of 1 in Scheme 1) to form an interaction with the amide hydrogen atom. Due to the TBA cation being large, it is difficult for it to enter in the loop to form any interaction and therefore anions are free to enter in the cage like space to interact with the amide. This is probably the reason why acetate interacts strongly when the TBA cation is used but not in the presence of Na+ salts. We also carried out a study with 1-butyl-3-methylimmidazolium acetate (ionic liquid), which contains a large cation, and observed that, like TBA+, it also exhibits a significant enhancement in the emission intensity of 1 (Fig. S7†), which supports the speculation that large cations do not enter in the loop to form any interactions and it facilitates entry of only anions into the loop to interact with the amide moiety. For a particular cation, the ability of anions to form interactions depends on the competitive hydrogen bonding/electron donating ability of the anions in a given solvent. In other words, the basicity of the anions and the decreasing order of the pKb values of anions in aqueous medium is CN− > AcO− > F− > other halides.43 This is qualitatively consistent with the observed selectivity. The mode of binding of anions and the consequent effects on emission spectra are discussed below in the mechanism section.
 |
| | Fig. 5 Emission spectral changes of 1 (1 × 10−6 M) in the presence of various anions in H2O:ACN (95:5) solvent. Excitation wavelength 461 nm. | |
Binding constants for AcO− and CN− with the TBA cation have also been calculated from emission titration data following a similar procedure as described above for NaCN. The spectral change for acetate is shown in Fig. 6 and that of CN− is presented in the ESI (Fig. S8†) and the binding constants obtained are 8.4 × 104 M−1 and 2.4 × 103 M−1 for CN− and AcO−, respectively. The slight difference in the binding constant for CN− with Na+ and TBA+ ions is probably due to the involvement of cation(s) in forming interactions with the calix moiety.
 |
| | Fig. 6 Emission spectral change of complex 1 (1 × 10−6 M) upon addition of increasing amounts of AcO− in H2O–CH3CN (95:5). Excitation wavelength: 458 nm. Inset: linear regression fit (double-logarithmic plot) of the titration data as a function of concentration of the AcO− ion. | |
Anion sensing by electrochemical method
For Ru(II)-bipyridine based molecular sensors, the redox property of the Ru(II) can be used for monitoring the interaction of the analyte with the sensor molecule.36–38 The redox property of the Ru(II) in 1 was investigated by recording cyclic voltammetry (CV) as well as differential pulse voltammetry (DPV) in acetonitrile and the CV and DPV for the region 0.0 to +2.0 V are shown in Fig. S9.† It should be noted that the CV of the complex 1 exhibited a quasireversible redox wave at the potential 1.35 V and the DPV also showed an oxidation wave at the same potential, which is attributed to Ru(II) → Ru(III) oxidation.36,44,45 The CV and DPV of 1 were again recorded under similar experimental conditions in the presence of excess amounts (50 molar equivalent) of TBA salts of CN− and AcO−. The DPV of 1 before and after addition of CN− and AcO− are shown in Fig. 7. It is interesting to note that in the presence of CN−, the oxidation potential of Ru(II) is cathodically shifted from 1.35 to 1.1 V and in the presence of AcO− the same oxidation potential has anodically shifted from 1.35 to 1.8 V. The observation suggests that both the CN− and the AcO− anions interacted with the molecule and altered the redox potential of the metal ion significantly. However, the opposite shift in the redox potential indicates that the interaction with anions resulted in an opposite electronic effect on the metal ion. The CN− ion's strong back bonding ability enhanced the electron density on the metal ion resulting in oxidation of it at lower potential whereas the AcO− ion reduced the electron density on Ru(II) making it difficult to oxidize and hence oxidation occurred at a higher potential. The observation is consistent with the finding noted in the luminescence study; the CN− with higher electron density on the metal ion promoted intramolecular quenching whereas AcO− with lower electron density on the metal ion reduced intramolecular quenching, which in turn enhanced the intensity of the emission band.
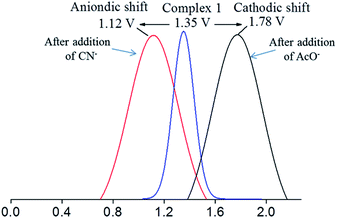 |
| | Fig. 7 Differential pulse voltammogram DPV of 1 before and after addition of TBA salts of CN− and AcO− (50 molar equivalent excess) recorded in acetonitrile. | |
Mass spectrometry
In order to find out the compositions of the complexes formed, mass spectra of 1 were recorded in acetonitrile–water upon addition of excess (10 molar equivalent) amounts of tetrabutylammonium salts of AcO− and CN−. The relevant portions of the mass spectra of 1 recorded with CN− and AcO− are shown in Fig. 8 and S10 (ESI†). In the case of CN−, the e/m value 1429.74 corresponds to the species [1–2PF6− + 2CN− + H2O–H+]− (calculated value 1429.72), in which one of the OH is deprotonated to generate an anionic species and the composition is consistent with the formation of a 1:2 complex. For acetate ions, the observed e/m value of 1764.43 corresponds to the composition [1 + AcO− + ACN + H2O]− (calculated value 1767.92), which is in agreement with the 1:1 complex formation, as found from emission titration.
 |
| | Fig. 8 Relevant portion of the mass spectra for 1 in the presence of TBACN (10 equivalents) recorded in acetonitrile. | |
Mechanistic aspects of anion binding
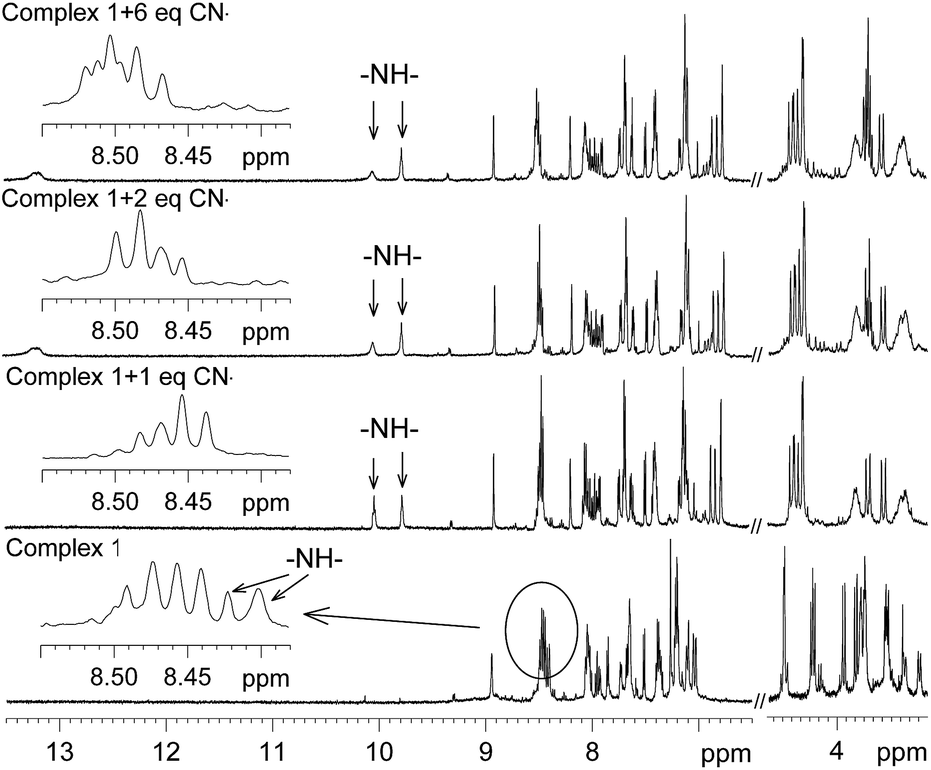
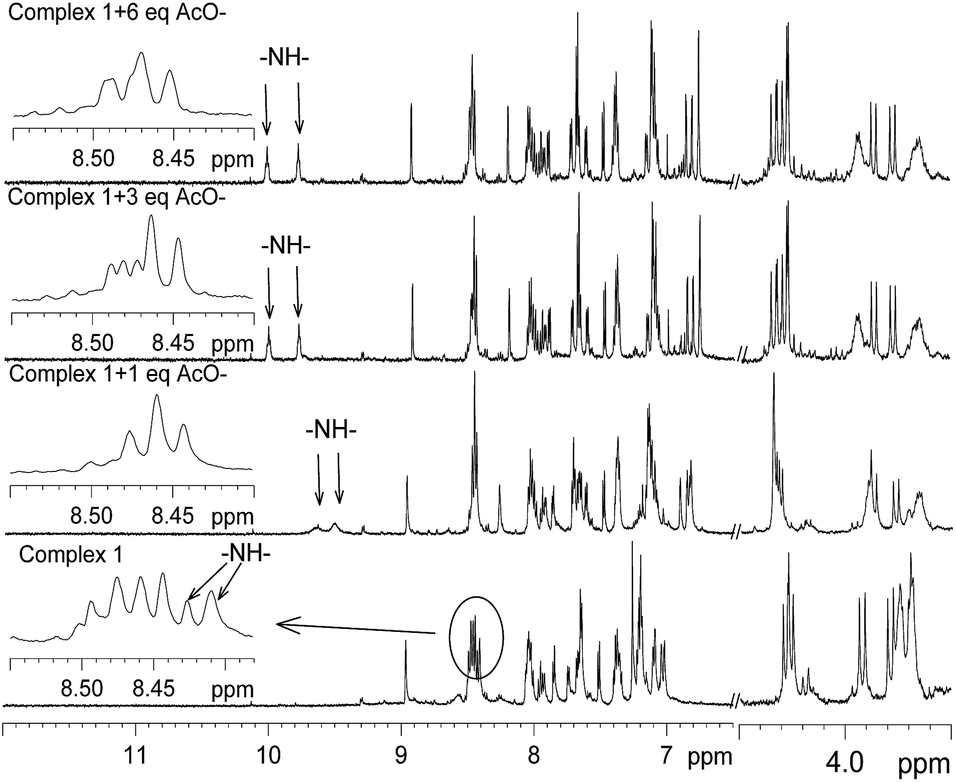
To investigate the site of interaction with anions, 1H NMR study was carried out. The 1H NMR spectrum of 1 was recorded in CD3CN before the addition of the anions and then after incremental addition of the anions up to six molar equivalents. The spectra thus obtained for CN− and AcO− are shown in Fig. 9 and 10. It should be noted that two N–H signals, which were overlapped with the aromatic protons in the range δ 8.39–8.46, appeared as two distinct singlets and the significant down field shifts of the signals are due to strong interactions with the anions. It should be noted that the intensity of one of these two signals reduced with the formation of a new broad signal at δ 13.20 upon addition of more than equimolar amounts of CN−. This broad signal observed at δ 13.2 suggests that one of the protons behaves differently and it may be due to a very strong interaction with CN− and also the formation of HCN by deprotonation of amide N–H may not be ruled out.24 For the AcO− anion, almost a similar observation was noted (Fig. 10); however, the intensity of the new signal for N–H did not diminish and no new broad signal, as observed for CN−, was noted. On the basis of this information, it is proposed that the two oxygen atoms of the AcO− anion interacted with two N–H protons, as shown in Fig. 11. The electron delocalization at the bidentate acetate anion probably pulls electron density towards itself from the adjacent amide groups, reducing the intramolecular luminescence quenching, which consequently enhances the intensity of the emission band. For CN−, the push of electron density from two anions to the bipyridine ligand enhanced the electron density, causing enhancement of the intramolecular quenching. It is also consistent with the observation noted in the electrochemical oxidation of the metal ion in the presence of CN− and AcO−.
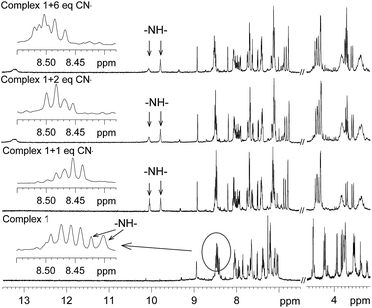 |
| | Fig. 9 Selected portion of the 1H NMR spectra of complex 1 in CD3CN with the addition of increasing amounts of CN−. | |
 |
| | Fig. 10 Selected portion of the 1H NMR spectra of complex 1 in CD3CN with the addition of increasing amounts of AcO−. | |
 |
| | Fig. 11 Proposed structural drawing of the CH3COO− bound complex 1. | |
Application in real sample
Complex 1 has been used for quantitative detection of cyanide in human saliva and drinking water. As the human saliva and drinking water were free from CN−, known concentrations (given in Table 1) of sodium cyanide were spiked. At first the collected human saliva and drinking water samples were filtered through 0.45 μm GNWP nylon membranes to remove insoluble materials, if any, and 0.5 mL samples of both of them were spiked with a suitable amount (2 × 10−4 M) of sodium cyanide stock solution. Then the sensor was used to estimate the concentration of NaCN (recovery) in human saliva and drinking water by using the standard double logarithm curve generated by estimating CN− in various solutions of known concentration (Fig. 12). The results obtained are summarized in Table 1, which shows the satisfactory recovery of NaCN compared to the amount added. These experimental results thus demonstrate that this fluorometric assay has potential for the detection of NaCN in various aqueous samples.
Table 1 Determination of CN− of NaCN in real samples
| |
Added (ppm) |
Found (ppm) |
Recovery (%) |
RSD% (n = 3) |
| Not detectable. |
| Drinking water |
| D0 |
0 |
nda |
— |
— |
| D1 |
0.40 |
0.42 |
105 |
±4 |
| D2 |
0.50 |
0.48 |
96 |
±7 |
| D3 |
1.50 |
1.60 |
106 |
±2 |
| D4 |
2.50 |
2.44 |
98 |
±6 |
|
| Human saliva |
| S0 |
0 |
nda |
— |
— |
| S1 |
0.40 |
0.38 |
95 |
±5 |
| S2 |
0.50 |
0.47 |
94 |
±3 |
| S3 |
1.00 |
1.10 |
110 |
±6 |
| S4 |
2.00 |
2.10 |
104 |
±7 |
 |
| | Fig. 12 Calibration line generated from standard solution of NaCN (●) in aqueous media and it has been used for estimation of CN− in real sample. The values for spike of NaCN in drinking water (■) and human saliva (▲) are shown and corresponding concentrations are calculated. | |
Conclusions
A molecular sensor comprising calix[4]arene as the receptor and ruthenium(II)-bipyridine moiety as the fluorophore connected by an amide moiety has been synthesised and its anon binding properties have been investigated. It selectively detects CN− out of a large number of Na+ salts of various anions in aqueous media with a lower detection limit of 70 ppb, whereas with tetrabutylammonim salts, it detects CN− as well as AcO− under similar experimental conditions. The anion recognition event was monitored by fluorescence spectroscopy and electrochemical study. The compositions of the anion bound complexes and their binding constants were determined. The mechanism of interaction and the possible energy transfer process involved in the recognition event has been discussed with the aid of spectroscopic data. The sensor has also been tested successfully to estimate cyanide in real samples, such as water from lake and saliva.
Experimental section
Instrumentation
Elemental analyses (C, H, and N) were performed on a model Vario Micro CUBE elemental analyzer. Mass spectra were recorded on a Q-TOF MicroTM LC-MS instrument. Infrared spectra were recorded on a PerkinElmer spectrum GX FT-system as KBr pellets. NMR spectra were recorded on models DPX 200 and Avance II 500 Bruker FT-NMR instruments. The UV/vis and luminescence spectra were recorded on a CARY 500 scan Varian spectrophotometer and model Fluorolog Horiba Jobin Yvon spectrofluorimeter at room temperature. Electrochemical measurements were carried out using PARASTA 2273 equipment.
Chemicals and reagents
The compounds 2,2′-bipyridine, 4,4′-dimethyl-2,2′-bipyridine, selenium dioxide, ammonium hexafluorophosphate, and tetrabutylammonium salts of anions used in this study were purchased from Alfa Aesar (Johnson Matthey Company). Hydrated ruthenium trichloride was purchased from Arora Matthey. Neutral alumina and silica gel were obtained from the National Chemical Co. All other reagents including the sodium salts of anions used in this study were purchased from S.D. Fine Chemicals. All organic solvents were of analytical grade and were used as received for synthetic purposes. Solvents for spectral studies were freshly purified by standard procedures. The starting compounds p-tert-butylcalix[4]arene,46 calix[4]arene,47 calix[4]arene monoester,48 4′-ethyl-2,2′-bipyridine-4-caboxylic acid, and cis-[Ru(bpy)2Cl2]·2H2O49 were synthesized following literature procedures.
Synthesis of compound C
A mixture of compound B (0.72 g, 1 mmol) and an excess amount of ethylene diamine (0.5 mL, 7.5 mmol) was refluxed in chloroform–methanol (1:3, 60 mL) for 12 h. Upon addition of water (1 mL) to this solution, a white precipitate was formed. It was filtered off and washed with water and methanol. The white product was then kept in high vacuum overnight. Yield: 0.83 g (80%). IR, νmax (KBr pellet)/cm−1 3398 ν(OH), 1688 ν(CO); 1H NMR (200 MHz, CDCl3): δ 9.25 (br, s, 1H, Ar–OH), 7.08–7.02 (overlapped signals, 8H, Ar–Hm), 4.6 (s, 2H, –OCH2CO), 4.26–4.16 (m, 4H, ArCH2Ar), 3.61–3.47 (m, 8H, ArCH2Ar, –CONHCH2CH2–NH–), 1.22–1.18 (overlapped signals, 36H, J = 13.4 Hz, –C(CH3)3). ESMS (m/z): found 748.31 (100%) calcd for [C − H+]− 748.02. Anal. calcd for C48H64O5N2; C, 76.96; H, 8.86; N, 3.73. Found: C, 75.58; H, 8.61; N, 3.78.
Synthesis of compounds F and L
A mixture of E (0.428 g, 2 mmol) and thionyl chloride (3 mL) in 30 mL dry toluene was stirred and refluxed under nitrogen for 6 h. The excess SOCl2 and toluene were then removed by rotary evaporation. The resulting greenish-yellow solid mass (F) was used directly in the next step for the synthesis of L. In the next step, compound C (1.50 g, 2 mmol) was dissolved in dry ACN (50 mL) and triethylamine (1 mL) was added to this solution. Then the ACN solution (30 mL) of the acid chloride (F) obtained in the previous step was added dropwise to the reaction mixture over a period of 1 h and the solution was then stirred at room temperature for another 2 h. Next, the reaction mixture was refluxed for 24 h under nitrogen atmosphere. The solution was then allowed to cool to room temperature and evaporated to dryness by rotary evaporation. The residue was dissolved in CHCl3 (100 mL), and the organic layer was washed three times with water (70 mL each time), dried over Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography on activated neutral alumina by using 1% (v/v) MeOH/CHCl3 as the eluent. Yield: 0.592 g, (45%). IR, νmax (KBr pellet)/cm−1 3545 (OH), 3049 (NH), 1677 (CO). 1H NMR (500 MHz, CDCl3): δ = 10.13 (br, s, 1H, –CONH), 9.58–9.48 (m, 3H, Ar–OH), 8.72 (s, 1H, ligand bipy-H), 8.62 (d, 1H, J = 5 Hz, ligand bipy-H), 8.46 (d, 1H, J = 4.8, ligand bipy-H), 8.17 (s, 1H, ligand bipy-H), 7.62 (d, J = 3.6, 2H, ligand bipy-H), 7.00–7.05 (m, 6H, Ar–Hm), 6.95 (s, 2H, Ar–Hm), 4.63 (s, ArO–CH2–CO–), 4.12 (t, 4H, J = 12.6 Hz, CONH–CH2–CH2–CONH–), 3.86 (s, 4H, Ar–CH2–Ar), 3.49–3.31 (m, 4H, Ar–CH2–Ar), 2.43 (s, 3H, bipy-CH3), 1.19 (br, s, 27H, –C(CH3)3), 1.15 (br, s, 9H, –C(CH3)3). ESMS (m/z): found 946.38 (100%) calcd for [L + H+]+ 946.23. Anal. calcd for C60H72N4O6: C, 76.24; H, 7.868; N, 5.93. Found: C, 76.04; H, 7.43; N, 5.77.
Synthesis of complex [Ru(bpy)2(L)](PF6)2 (1)
A mixture of cis-[Ru(bpy)2Cl2]·2H2O (0.13 g, 0.25 mmol) and compound L (0.24 g, 0.25 mmol) in ethanol–water (2:1, 60 mL) was refluxed for 10 h. The reaction mixture was then allowed to cool to room temperature; the volume was reduced to ca. 20 mL by rotary evaporation then filtered and to the filtrate was added solid NH4PF6 (0.82 g, 5 mmol). The precipitate thus separated was collected by filtration, then washed with water and diethyl ether. The complex was purified by column chromatography using a column packed with deactivated (2% water) alumina and acetonitrile–toluene (1:1) as eluent. The small first fraction was discarded; the large orange-red colored second fraction gave the desired complex. After removing the solvent, the residue was again dissolved in acetonitrile and was precipitated by vapor diffusion method using diethyl ether. Yield: 60–70%. IR, νmax (KBr pellet)/cm−1 3433 (OH), 1653 (CO). 1H NMR [CD3CN] δ = 8.90 (s, 3H, Ar–OH), 8.39–8.49 (m, 6H, bipy-H, –CONH), 8.06–8.08 (m, 3H, bipy-H), 7.94 (t, J = 4.0, 2H, bipy-H), 7.86 (1H, J = 6, ligand bipy-H), 7.71 (d, J = 6.0, 1H, ligand bipy-H), 7.64–7.68 (m, 4H, bipy-H), 7.51 (d, J = 5.5, 1H, ligand bipy-H), 7.40–7.38 (m, 3H, ligand bipy-H, bipy-H), 7.27 (s, 2H, ligand bipy-H), 7.20–7.25 (m, 4H, Ar–Hm), 7.11 (d, 2H, J = 5.5, Ar–Hm), 7.05 (d, J = 7.75, 2H, Ar–Hm), 4.59 (s, 2H, ArO–CH2–CO–), 3.91 (t, J = 11.5, –CH2–CH2–CONH–), 3.44–3.48 (m, 4H, CONH–CH2–CH2–CONH–), 3.80 (d, J = 13.5, Ar–CH2–Ar), 3.75 (d, J = 20.5, 1H), 3.72–3.62 (m, 4H, Ar–CH2–Ar), 3.28 (d, J = 13.5, 1H, Ar–CH2–Ar), 3.12 (d, J = 13.5, 1H, Ar–CH2–Ar), 2.34 (s, 3H, bipy-CH3), 1.15–1.17 (m, 36H, –C(CH3)3), MS (m/z): found, 1503.57 (65%) calcd for [1 − PF6−]+, 1503.55. Anal. calcd for C80H90RuN8O6P2F12·2H2O; C, 57.03; H, 5.50; N, 6.66. Found: C, 56.83; H, 5.63; N, 6.66. UV-vis: λmax (CH3CN)/nm (ε/dm3 mol−1 cm−1): 461 (10730), 289 (25600).
Ion-binding study
The selectivity of 1 for anions was examined by fluorescence study. The luminescence spectrum of complex 1 (1 × 10−6 M) was recorded with excitation at the absorption maxima (λmax) of the MLCT band, which is 461 nm, then the luminescence spectra of 1 upon addition of anions (100 fold excess, 1 × 10−4 M) were recorded. These spectra were compared with that of the compound 1 to ascertain the interactions of the anions with the ionophore. For the determination of binding constants with strongly interacting anions, emission titration was performed. For this purpose, the same stock solution of the complex was used and the solutions of the anions with desired concentrations were prepared by diluting the concentrated standard stock solution (2 × 10−3 M). Then, 2 mL of each solution was mixed in a 5 mL volumetric flask and the luminescence spectra of the resulting solutions were recorded. The binding constants were calculated following the literature procedure.41,50 The binding constant and stoichiometry of the complex formation were calculated following the literature procedure describe in the results and discussion section. The stock solution of 1 × 10−4 M was also used to study UV/vis spectral changes. For the NMR study, 2 mg of the complex was dissolved in 0.5 mL of [D3] acetonitrile and the 1H spectrum of the resulting solution was recorded. The tetra butyl ammonium salt of cyanide and acetated (1–10 equivalent) was added to the solution and the spectra of the solutions were recorded.
Electrochemical study
Cyclic and differential pulse voltammetry (CV and DPV) studies were carried out in a three-electrode cell consisting of a platinum working electrode, a platinum-wire auxiliary electrode, and an SCE reference electrode. Solutions of the complexes in purified acetonitrile containing 0.1 M tetrabutylammonium tetrafluoroborate as the supporting electrolyte were deaerated by bubbling nitrogen for 10 min prior to each experiment.
Acknowledgements
CSIR-CSMCRI Registration no. 173/2014. We gratefully acknowledge CSIR, New Delhi for financial assistance for this work under the project CSC 0134 (M2D) and for generous support towards infrastructure and core competency development at CSIR-CSMCRI. D.M. gratefully acknowledges the CSIR for awarding Senior Research Fellowship (SRF). We thank Dr V. P. Boricha, A. K. Das, V. Agrawal and Anirban Pal for recording NMR, mass, IR spectra and for assistance with electrochemistry measurement, respectively.
Notes and references
- S. I. Baskin and T. G. Brewer, Medical Aspects of Chemical and Biological Warfare, ed. F. R. Sidell, E. T. Takafuji and D. R. Franz, TMM Publications, Washington, DC, 1997, ch. 10, pp. 271–286 and references therein Search PubMed.
- T. T. Margaret, W. Riley, H. L. Liang, J. T. Eells, M. M. Henry, E. Buchmann, M. Kane and H. T. Whelan, J. Biol. Chem., 2005, 280, 4761 CrossRef PubMed.
- J. E. Bailey, J. Brinder and M. Bohnet, Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH, New York, 6th edn, 1999 Search PubMed.
- G. C. Miller and C. A. Pritsos, Proceedings of the TMS Annual Meeting, 2001, p. 73 Search PubMed.
- H. Sun, Y. Y. Zhang, S. H. Si, D. R. Zhu and Y. S. Fung, Sens. Actuators, B, 2005, 108, 925 CrossRef CAS PubMed.
- U. Epa, K. Ikebukuro, H. Nakamusa and I. Karube, in Handbook of Water Analysis, Fed. Regist., 1998, vol. 63, FR 44511 Search PubMed.
- C. Giuriati, S. Cavalli, A. Gorni, D. Badocco and P. J. Pastore, J. Chromatogr. A, 2004, 1023, 105 CrossRef CAS PubMed.
- H. P. Beck, B. Zhang and A. Bordeanu, Anal. Lett., 2003, 36, 2211 CrossRef CAS PubMed.
- M. T. Fernandez-Arguelles, J. M. Costa-Fernandez, R. Pereiro and A. Sanz-Medel, Anal. Chim. Acta, 2003, 491, 27 CrossRef CAS.
- B. Deep, N. Balasubramanian and K. S. Nagaraja, Anal. Lett., 2003, 36, 2865 CrossRef PubMed.
- F. Wang, L. Wang, X. Chen and J. Yoon, Chem. Soc. Rev., 2014, 43, 4312 RSC.
- J. L. Worlinsky, S. Halepas and C. Brückner, Org. Biomol. Chem., 2014, 12, 3991 CAS.
- G. J. Park, I. H. Hwang, E. J. Song, H. Kim and C. Kim, Tetrahedron, 2014, 70, 2822 CrossRef CAS PubMed.
- W. Cao, X. J. Zheng, D. C. Fang and L. P. Jin, Dalton Trans., 2014, 43, 7298 RSC.
- M. J. Li, Z. Lin, X. Chen and G. Chen, Dalton Trans., 2014, 43, 11745 RSC.
- K. K. W. Lo and S. P. Y. Li, RSC Adv., 2014, 4, 10560 RSC.
- A. E. J. Broomsgrove, D. A. Addy, C. Bresner, I. A. Fallis, A. L. Thompson and S. Aldridge, Chem.–Eur. J., 2008, 14, 7525 CrossRef CAS PubMed.
- P. Chinna Ayya Swamy, S. Mukherjee and P. Thilagar, Anal. Chem., 2014, 86, 3616 CrossRef PubMed.
- P. Anzenbacher, D. S. Tyson, K. Jursikova and F. N. Castellano, J. Am. Chem. Soc., 2002, 124, 6232 CrossRef CAS PubMed.
- Y. Dong, R. Wang, W. Tian, Y. Chi and G. Chen, RSC Adv., 2014, 4, 3701 RSC.
- S. Han, J. Wang and S. Jia, Luminescence, 2014 DOI:10.1002/bio.2687.
- T. Noipa, T. Tuntulani and W. Ngeontae, Talanta, 2013, 105, 320 CrossRef CAS PubMed.
- Y. K. Tsui, S. Devaraj and Y. P. Yen, Sens. Actuators, B, 2012, 161, 510 CrossRef CAS PubMed.
- V. Kumar, H. Rana and M. P. Kaushik, Analyst, 2011, 136, 1873 RSC.
- U. G. Reddy, P. Das, S. Saha, M. Baidya, S. K. Ghosh and A. Das, Chem. Commun., 2013, 49, 255 RSC.
- S. Li, C. Zhang, S. Huang, F. Hu, J. Yin and S. H. Liu, RSC Adv., 2012, 2, 4215 RSC.
- V. Amendola, D. E. Omez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343 CrossRef CAS PubMed.
- J. L. Sessler and J. M. Davis, Acc. Chem. Res., 2001, 34, 989 CrossRef CAS PubMed.
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2014, 43, 10.1039/c4cs00179f.
- N. S. Shifrin, B. D. Beck, T. D. Gauthier, S. D. Chapnick and G. Goodman, Regul. Toxicol. Pharmacol., 1996, 23, 106 CrossRef CAS PubMed.
- R. Joseph and C. P. Rao, Chem. Rev., 2011, 111, 4658 CrossRef CAS PubMed.
- S. Patra, D. Maity, A. Sen, E. Suresh, B. Ganguly and P. Paul, New J. Chem., 2010, 34, 2796 RSC.
- D. Maity, A. Chakraborty, R. Gunupuru and P. Paul, Inorg. Chim. Acta, 2011, 372, 126 CrossRef CAS PubMed.
- S. Patra, R. Gunupuru, R. Lo, E. Suresh, B. Ganguly and P. Paul, New J. Chem., 2012, 36, 988 RSC.
- H. Tomiyasu, C. C. Jin, X. L. Ni, X. Zeng, C. Redshaw and T. Yamato, Org. Biomol. Chem., 2014, 12, 4917 CAS.
- S. Patra and P. Paul, Dalton Trans., 2009, 8683 RSC.
- H. J. Mo, Y. L. Niu, M. Zhang, Z. P. Qiao and B. H. Ye, Dalton Trans., 2011, 40, 8218 RSC.
- A. Mattiuzzi, L. Marcélis, I. Jabin, C. Moucheron and A. K. De Mesmaeker, Inorg. Chem., 2013, 52, 11228 CrossRef CAS PubMed.
- L. C. Croenen, B. H. M. Ruel, A. Casnati, W. Verboom, A. Pochini, R. Ungaro and D. N. Reinhoud, Tetrahedron, 1991, 47, 8379 CrossRef.
- G. Ajayakumar and K. R. Gopidas, Photochem. Photobiol. Sci., 2008, 7, 826 CAS.
- V. P. Boricha, S. Patra, Y. S. Chouhan, P. Sanavada, E. Suresh and P. Paul, Eur. J. Inorg. Chem., 2009, 1256 CrossRef CAS.
- A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713 CrossRef CAS PubMed.
- V. Amendola, D. Esteban-G`omez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343 CrossRef CAS PubMed.
- M. J. Li, B. W. K. Chu, N. Zhu and V. W. W. Yam, Inorg. Chem., 2007, 46, 720 CrossRef CAS PubMed.
- A. K. Bilakhiya, B. Tyagi, P. Paul and P. Natarajan, Inorg. Chem., 2002, 41, 3830 CrossRef CAS PubMed.
- C. D. Gutsche and M. Iqbal, Org. Synth., 1990, 68, 234 CrossRef CAS.
- C. D. Gutsche, J. A. Levine and P. K. Sujeeth, J. Org. Chem., 1985, 50, 5802 CrossRef CAS.
- L. C. Groenen, B. H. M. Ruel, A. Casnati, W. Verboom, A. Pochini, R. Ungaro and D. N. Reinhoudt, Tetrahedron, 1991, 47, 8379 CrossRef CAS.
- S.-S. Sun, A. J. Lees and Y. Zavalij, Inorg. Chem., 2003, 42, 3445 CrossRef CAS PubMed.
- A. C. Tedesco, D. M. Oliveria, Z. J. M. Lacava, R. B. Azevedo, E. C. D. Lima and P. C. Morais, J. Magn. Magn. Mater., 2004, 272–276, 2404 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S10 (mass, NMR, fluorescence spectra and CV and DPV). See DOI: 10.1039/c4ra12075b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.