
Water soluble well-defined acidic graft copolymers based on a poly(propylene glycol) macromonomer
Paulina Maksym-Bębeneka,
Tadeusz Bielab and
Dorota Neugebauer*a
aDepartment of Physical Chemistry and Technology of Polymers, Faculty of Chemistry, Silesian University of Technology, M. Strzody 9, 44-100 Gliwice, Poland. E-mail: dorota.neugebauer@polsl.pl
bCentre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Łódź, Poland
First published on 1st December 2014
Abstract
A series of well-defined amphiphilic graft copolymers based on poly(propylene glycol)methacrylate (PPGMA) extended with a polymethacrylate segment containing methacrylic acid units was successfully synthesized, combining atom transfer radical polymerization (ATRP), esterification, “grafting from” polymerization, and acidolysis. The multistep procedure yielded hydroxy-functionalized polymers (6–47 mol% of PPGMA), bromoester-functionalized copolymers as multifunctional macroinitiators (nBr = 8–25), PPGMA-based copolymers with poly(tert-butyl methacrylate) (PtBMA) segments (DPtBMA = 18–59), and copolymers varying in hydrophilic fraction content (FMAA = 0.28–0.54). After the generation of methacrylic acid units the PPGMA-based graft polymers attained a good solubility in polar solvents such as methanol and water. The particles, which were formed in aqueous solution, achieved sizes in the nanoscale range (165–230 nm) and negative zeta potentials.
Introduction
The ability to synthesize (co)polymers functionalized by hydroxyl groups with variable composition and topology leads to macromolecules with unique properties.1 However, the living polymerization of hydroxyl-functionalized monomers is possible via protected hydroxyl groups, due to the interaction of the reactive hydrogen with a catalyst or initiator in ionic or group transfer polymerizations.2,3 Fortunately, controlled radical polymerization (CRP) methods, including atom transfer radical polymerization (ATRP),4 allow the synthesis of tailor-made linear polymers from various hydroxyl monomers, such as 2-hydroxyethyl methacrylate (HEMA),5 2-hydroxyethyl acrylate (HEA),6 N-(2-hydroxyethyl acrylamide) (HEAA),7 and N-(2-hydroxypropyl)methacrylamide (HPMA).8 In the case of hydroxyl-functionalized macromonomers, that is, poly(D-lactide)9 and poly(ethylene glycol)methacrylate (PEGMA),10,11 grafted macromolecules have been obtained. It is worth noting that PEGMA requires purification to remove the dimethacrylate fraction and unreacted glycol by extraction prior to polymerization.10 The macromonomer functionalized with methacrylate and hydroxyl groups corresponding to other polyether is poly(propylene glycol)methacrylate (PPGMA). PPGMA homopolymers with linear and star-shaped structures,12 as well as statistical and block copolymers with perfluoroalkyl ethyl methacrylate13 or poly(dimethylaminoethyl methacrylate),14 have been reported in the literature. Moreover, PPGMA has been copolymerized with PEGMA by conventional free radical polymerization and ATRP for coating poly(lactide-co-glycolide) (PLGA) particles15 or using a PLGA macroininitiator to form self-assembled semigrafted block copolymers16 with biodegradable and thermoresponsive properties. In another case, thermoresponsive copolymers of PPGMA with poly(ethylene glycol) methyl ether methacrylate and ethylene glycol dimethacrylate have been obtained to form photo-cross-linked hydrogels.17,18The presence of pendant reactive –OH groups in polymers as substituents or at the end of side chains is an advantage for their further modification by various types of reactions; hence, these groups are useful for changing the physicochemical properties of macromolecules. One possibility is to use them directly for grafting reactions via ring-opening polymerization of cyclic monomers (for example, ε-caprolactone19,20 or lactide). Another strategy is based on the transformation of hydroxyl groups in linear polymers to halogenoesters. For example, PHEMA can be converted to poly(2-(2-bromoisobutyryloxy)ethyl methacrylate) (PBIEM), and then used as a multifunctional macroinitiator in “grafting from” polymerization.21 This strategy makes it possible to combine polymeric segments with different natures in branched structures which, similarly to linear block copolymers, can exhibit good phase separation and can be used in a variety of applications.22 However, because of their nonlinear topology they generally have lower melt viscosities, which is especially useful for processing.23 Recently, our group has reported the synthesis of well-defined amphiphilic and biocompatible polymers containing 5 mol% of PEGMA grafts extended by extra hydrophilic segments of poly(methacrylic acid) (PMAA), which significantly improved the solubility in polar solvents, including water.11 Solubility in aqueous solutions, as well as the composition and topology of polymers, is strongly recommended for biomedical applications, for example in the field of drug carriers.
Herein, we describe a versatile multistep procedure for the synthesis of another type of amphiphilic graft polymethacrylate with segments of PPG (5 units) and MAA units in the side chains, as shown in Scheme 1. ATRP provides well-defined PPGMA based copolymers with a high polymerization degree in the backbone (DPn = 100–225, including nPPGMA = 10–75) and relatively long PtBMA side chains (DPtBMA = 20–60), whereas esterification and acidolysis reactions guarantee the selective modification of hydroxyl groups to bromoester groups and the removal of tert-butoxy groups to introduce carboxyl groups, respectively. According to the literature, in the late 80’s PPGMA/MMA copolymers were prepared by conventional radical polymerization,24,25 whereas the other PPGMA copolymers mentioned above were applied as surface-engineered particles or hydrogels. Our work is focused on the first-time use of modified short-grafted PPGMA copolymers as new multifunctional macroinitiators in “side chain extension” polymerization to prepare biocompatible branched weak polyacids, which have the potential for self-assembly to form nanoparticles with drug entrapment capabilities.
 | ||
| Scheme 1 Synthetic route for the preparation of amphiphilic graft copolymers by a four-step procedure: copolymerization of PPGMA (a), esterification of hydroxyl groups (b), “side chain extension” polymerization (c), and acidolysis to form carboxyl groups (d). | ||
Experimental
Materials
Poly(propylene glycol)methacrylate (PPGMA, Sigma-Aldrich, MW = 375 g mol−1), methyl methacrylate (MMA, Sigma-Aldrich, 99%), tert-butyl methacrylate (tBMA, Sigma-Aldrich, 98%), anisole (Fluka, 99%), heptane (POCH) and toluene (POCH) were dried over molecular sieves and stored in a freezer under nitrogen. Copper(I) bromide (CuBr, Fluka 98%) and copper(I) chloride (CuCl, Fluka 97%) were purified by stirring with glacial acetic acid, followed by filtration and washing the solid with ethanol (three times) and diethyl ether (two times). Then, both CuBr and CuCl were dried under vacuum for 2 days. Ion-Exchange Resin Dowex® Marathon™ MSC hydrogen form (Sigma-Aldrich) was activated by stirring with nitric acid and water, then dried at 50 °C. 4,4′-Dinonyl-2,2′-dipyridyl (dNdpy, Sigma-Aldrich, 97%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, Sigma-Aldrich, 99%) ethyl 2-bromoisobutyrate (EtBriBu, Sigma-Aldrich, 98%), triethylamine (TEA, Sigma-Aldrich, ≥99%), α-bromoisobutyryl bromide (BriBuBr, Sigma-Aldrich) and trifluoroacetic acid (TFA, Sigma-Aldrich) were used as received. All other solvents were applied without purification.Synthesis
Characterization
Gel permeation chromatography
Molecular weights and dyspersities were determined by gel permeation chromatography (GPC) using a 1100 Agilent isocratic pump, autosampler, degasser, thermostatic box for columns, and differential refractometer Optilab Rex. ASTRA 4.90.07 software (Wyatt Technology Corporation) was used for data collecting and processing. Two PLGel 5 μm MIXD-C columns were used for separation. The samples were injected as a solution in methylene chloride. The volume of the injection loop was 100 μL. Methylene chloride was used as the mobile phase at a flow rate of 0.8 mL min−1. The calibration of the DAWN EOS was carried out using p.a. grade toluene and normalization with a polystyrene standard of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. The measurements were carried out at room temperature. In the case of the selected PPGMA based copolymers, a known amount of the sample was introduced for measurement to calculate the refractive index increment of the copolymer (dn/dc), so as to determine the absolute molecular weight using a MALLS DAWN EOS detector (Wyatt Technology Corporation, Santa Barbara, CA). dn/dc = 0.059–0.070.
000 g mol−1. The measurements were carried out at room temperature. In the case of the selected PPGMA based copolymers, a known amount of the sample was introduced for measurement to calculate the refractive index increment of the copolymer (dn/dc), so as to determine the absolute molecular weight using a MALLS DAWN EOS detector (Wyatt Technology Corporation, Santa Barbara, CA). dn/dc = 0.059–0.070.
Nuclear magnetic resonance spectroscopy
1H nuclear magnetic resonance (NMR) spectra for structure analysis were recorded on a UNITY/INOVA (Varian) 300 MHz spectrometer using CDCl3 or MeOD as solvents and tetramethylsilane or 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt as internal standards, respectively. Calculations based on 1H NMR: x = 1 − Ivt/Iv0, where x is the total monomer conversion, and Iv0 and Ivt are the peak areas of the vinyl protons of PPGMA and MMA (6.15 and 5.51 ppm) in relation to the peak of anisole protons (3.81 ppm) for the initial sample and for the sample taken during polymerization at time t, respectively. FPPGMA = [(IB,E − IA/2)/(IA/2)]/15, where FPPGMA is the PPGMA content in the copolymer, IB,E is the peak area of CH3 protons in the backbone (B) overlapped with CH3 protons in the PPG side chains (E) (0.65–1.3 ppm), IA is the peak area of CH2 protons in the backbone (1.81 ppm), and IA/2 = IB/3. FtBMA = (ItB/9)/[(ICH3 − IE)/3], where FtBMA is the tBMA content in the graft copolymer, ItB is the peak area of tert-butyl protons (1.42 ppm), ICH3 is the peak area of CH3 protons in the backbone and the PtBMA and PPG segments in the side chains (0.65–1.3 ppm), and IE is the peak area of CH3 protons in the PPG side chains estimated with respect to IA before grafting and used as a constant after grafting.Gas chromatography
The consumption of MMA and tBMA in the polymerizations was determined by gas chromatography (GC). The chromatograph (6850 Network GC System, Agilent Technologies) was equipped with a flame ionization detector. The injector and detector temperatures were kept constant at 250 °C (conditions: anisole as internal standard, column initial temperature 40 °C, column final temperature 200 °C). The conversions were determined by detecting the decrease in the MMA or tBMA peak area relative to the anisole peak as a standard.Fourier transform infrared spectroscopy
Fourier transform infrared spectroscopy (FT-IR) measurements were conducted on a PerkinElmer Spectrum Two Spectrometer (Attenuated Total Reflection method).Dynamic light scattering
The hydrodynamic diameters (dh) of particles were measured using a Malvern Zetasizer Nano-S90 equipped with a 4 mW He–Ne ion laser operating at λ = 633 nm. Samples placed in a PMMA cell after dilution with deionized water were put into the thermostatted cell compartment of the instrument at 25 ± 0.1 °C. All of the sample measurements were performed at a fixed scattering angle of 90°. At least three correlation functions were analyzed per sample in order to obtain an average value.ζ (Zeta)-potential measurements
The surface charge of the polymer matrix was measured in deionized water. Diluted samples (0.04 g L−1) were placed in a disposable folded capillary cell and put into the thermostatted cell compartment of the instrument (Malvern Zetasizer Nano-Z) at 25 ± 0.1 °C. Measurements were carried out on each sample from three independent runs.Results and discussion
Hydrophobic short-grafted copolymers P(MMA-co-PPGMA) with pendant hydroxyl groups (Scheme 1a) were prepared by ATRP of PPGMA (DPPPG = 5) with MMA in the presence of EtBriBu and CuBr/PMDETA or CuBr/dNbPy as initiating and catalytic systems, respectively. All reactions were performed in anisole at 60 °C. The molar mass of the polymers was controlled by the relative ratio of comonomers and initiator ([PPGMA]0+[MMA]0/[EtBrBu]0 = 500/1), while the content of incorporated hydroxyl-functionalized segments was controlled by changing the initial feed of PPGMA monomer (5–50 mol%). The basic characteristics of the copolymers were obtained from a combination of GC, NMR, and GPC data (Table 1).| No. | fPPGMA [mol%] | Time [h] | GC | NMR | GPCb | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| xMMA [%] | nMMA | x [%] | DPn | FPPGMA [mol%] | Mn,NMR × 10−3 [g mol−1] | Mn,GPC × 10−3 [g mol−1] | Mw/Mn | |||
| a CuBr/dNbPy = 0.75/1.5 as the catalytic system.b RI detector, CH2Cl2, PS standards; fPPGMA: initial PPGMA molar content in the reaction mixture; xMMA: conversion of MMA; x: overall monomer conversion; nMMA = xMMA[MMA]0/[EtBriBu]0 means the number of MMA units incorporated into the polymer; DPn = x([PPGMA]0 + [MMA]0)/[EtBriBu]0; FPPGMA: molar content of PPGMA units in the copolymer; Mn,NMR = x(375[PPGMA]0 + 100[MMA]0)/[EtBriBu]0.c PPGMA + MMA/EtBriBu/CuBr/PMDETA = 500/1/1/1; anisole = 30 vol%; T = 60 °C. | ||||||||||
| I | 5 | 1 | 23 | 109 | 31 | 155 | 6.6 | 18.5 | 24.5 | 1.26 |
| II | 10 | 0.2 | 21 | 93 | 23 | 116 | 13.0 | 15.7 | 18.6 | 1.25 |
| III | 10 | 1 | 34 | 153 | 33 | 167 | 12.9 | 22.6 | 29.6 | 1.18 |
| IVa | 10 | 0.5 | 19 | 86 | 23 | 115 | 12.8 | 15.4 | 16.0 | 1.36 |
| Va | 10 | 1 | 23 | 104 | 25 | 125 | 9.3 | 16.7 | 18.5 | 1.31 |
| VIa | 10 | 2 | 28 | 126 | 27 | 132 | 12.0 | 18.0 | 17.3 | 1.39 |
| VII | 15 | 1 | 35 | 149 | 36 | 180 | 14.8 | 25.4 | 30.4 | 1.27 |
| VIII | 25 | 0.3 | 15 | 56 | 21 | 105 | 35.9 | 21.0 | 21.5 | 1.18 |
| IX | 25 | 0.6 | 36 | 135 | 34 | 170 | 23.1 | 27.8 | 34.4 | 1.22 |
| X | 25 | 1 | 41 | 154 | 45 | 225 | 24.0 | 37.4 | 45.6 | 1.29 |
| XIa | 50 | 1 | 45 | 113 | 32 | 160 | 47.4 | 36.7 | 28.0 | 1.19 |
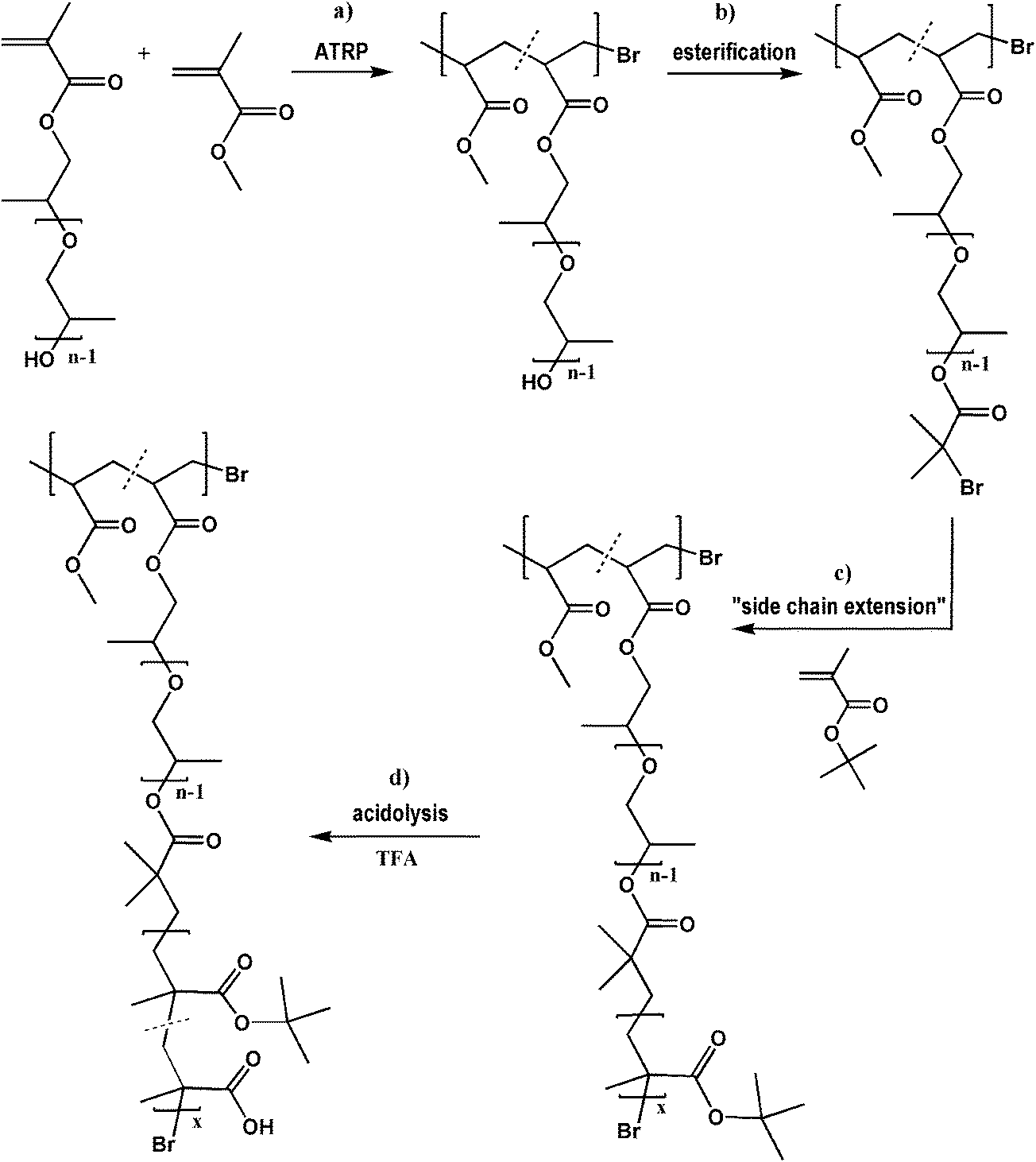
The chemical structures of the PPGMA-based copolymers after purification by repeated precipitation were characterized by 1H NMR spectroscopy. As shown in Fig. 1, the characteristic peaks corresponding to the methylene (A) and methyl protons (B) in the backbone are observed at 1.81 ppm and 0.65–1.3 ppm, respectively. Additionally, the protons of the methyl groups in the PPG side chains (1.20 ppm; E) are overlapped with these signals in the latter range of chemical shifts. The content of PPGMA units in the copolymer (FPPGMA) was estimated using these signals. The other signals were assigned to the methoxy protons of the MMA units (3.60 ppm; F) as well as the methylene and methine protons in the PPG chains of PPGMA units (3.14–4.22 ppm; C, D). 1H NMR was also used for analysis of reaction mixture samples to monitor the progress of polymerization via total monomer conversion (x), whereas conversion of MMA (xMMA) was determined by GC. The conversion parameters allowed calculation of the polymerization degree of polymethacrylate chains (DPn) and the number of MMA units (nMMA) introduced into the copolymer.
 | ||
| Fig. 1 1H NMR spectra of P(MMA-co-PPGMA) copolymers with varying content of PPGMA units obtained with an initial macromonomer feed in the range of 5–50 mol%. | ||
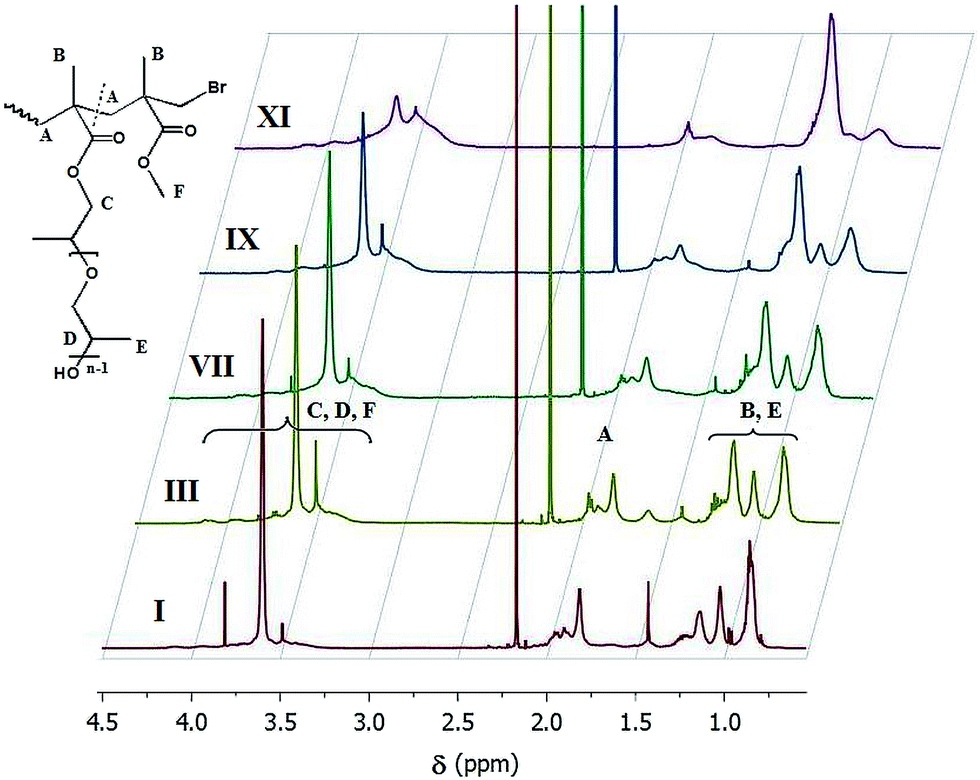
The P(MMA-co-PPGMA) copolymers were prepared in total monomer conversions of 21–45%. Various compositions were correlated with 10–75 units of PPGMA distributed along the polymethacrylate backbone with DPn in the range of 105–225, which yielded different grafting degrees of the short PPG chains as indicated by the content of PPGMA units (7–47 mol%). ATR copolymerization of PPGMA and MMA in the presence of 10 mol% initial feed of PPGMA was performed in two catalyst systems with different activity, CuBr/PMDETA vs. CuBr/dNbPy (II and III vs. IV–VI). A lower conversion within 1 h was obtained using the system with the latter catalyst (25% of V vs. 33% of III), indicating a reduced polymerization rate and broader molecular weight distribution (1.31 vs. 1.18). The kinetic plots in Fig. 2 confirm this observation and show some deviation from a linear relationship in the first-order time–conversion dependence. With CuBr/PMDETA, the process was well-controlled for the initial 20 min (21% conversion) and then proceeded more slowly, whereas in the second system, after 20 min the consumption of monomer was linear, but much lower.
 | ||
| Fig. 2 Kinetic plots of the semilogarithmic dependence of conversion vs. time for reactions III and V performed in various catalytic systems ([MMA]0/[PPGMA]0 = 90/10 mol%). | ||
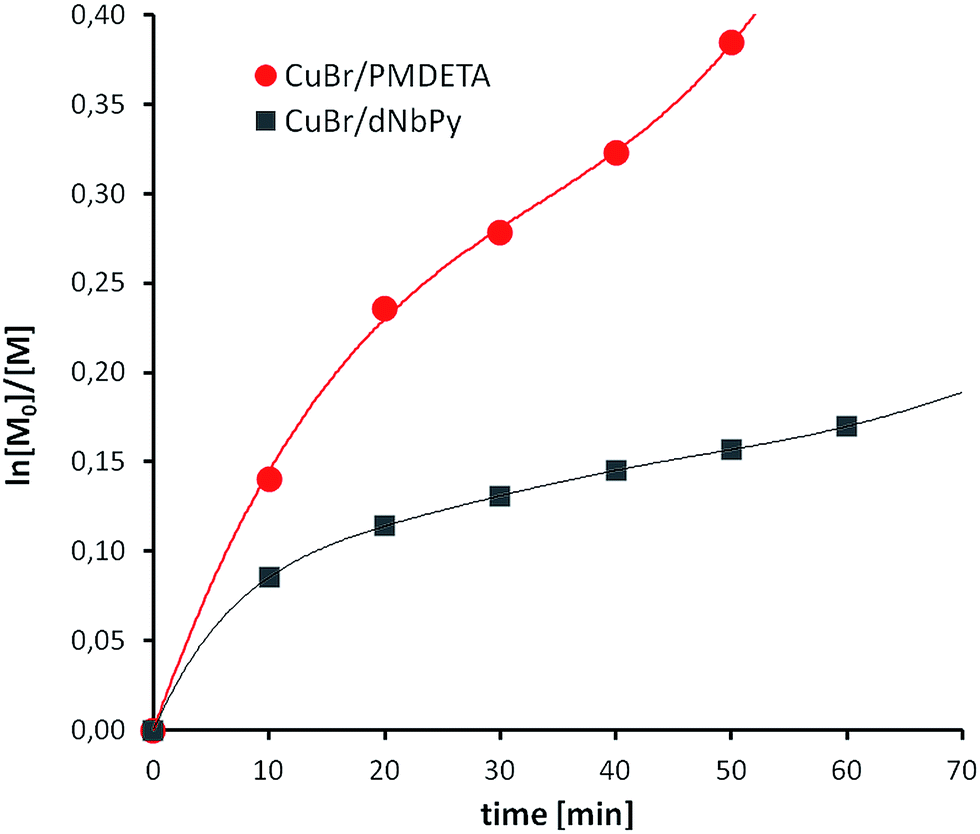
On the other hand, polymerizations IV–VI (fPPGMA = 10 mol%) performed under the same conditions but stopped at different times (0.5–2 h) indicated insignificant progress of conversion from 23 to 27% with the change in dispersity index (1.31–1.39). Similar behavior was also observed for polymerizations VIII–X (fPPGMA = 25 mol%). However, with a larger initial feed of PPGMA the change in conversion with reaction time was more spectacular, increasing from 21–45% in 0.3–1 h. The tendency of the conversion to increase on increasing the initial feed of PPGMA, which was monitored under comparable conditions (31% at fPPGMA = 5 mol%, 33% at 10 mol%, 36% at 15 mol%, and 45% at 25 mol% after 1 h), caused us to perform the next polymerization with 50 mol% of PPGMA with the less active catalyst complex CuBr/dNbPy, to avoid too fast a reaction and to minimize the participation of side reactions. In this case, 32% of the comonomers were converted to polymer XI containing 160 repeating units in the backbone, including 75 units of PPGMA with short side chains. As can be seen in Fig. 3a, the GPC traces display unimodal and narrow peaks without any tailing. The values of Mn,GPC obtained from conventional GPC in comparison with those from 1H NMR analysis showed some discrepancies in the method error range (Fig. 4a). The additional GPC analysis with a MALLS detector confirmed good agreement of the absolute Mn,MALLS with Mn,NMR, with one significant exception for copolymer XI obtained at the highest feed of PPGMA. In that case, Mn,MALLS ∼ 2xMn,NMR (58200 vs. 36500 g mol−1), whereas the molecular weight distribution was narrow (Mw/Mn,MALLS = 1.19), suggesting chain coupling termination.
 | ||
| Fig. 3 GPC traces of copolymers with varying content of PPGMA (13–46 mol%) (a) and a series of hydroxyl-functionalized copolymers, PPGMA-based MI, and tBMA modified copolymers (b). | ||
 | ||
| Fig. 4 Dependence of molecular weight on PPGMA content in hydroxyl-functionalized copolymers (a), polymerization degree of tBMA (b), and grafting degree (c) in graft copolymers. | ||
The diagram of instantaneous monomer-copolymer composition in Fig. 5 is related to the dependence of the content of PPGMA units in the copolymer (FPPGMA) vs. the initial feed of PPGMA in the polymerization mixture (fPPGMA). It shows that the composition of the copolymer is proportional to the monomer mixture, showing good agreement with a typical dependence for a “random copolymer” (solid line).
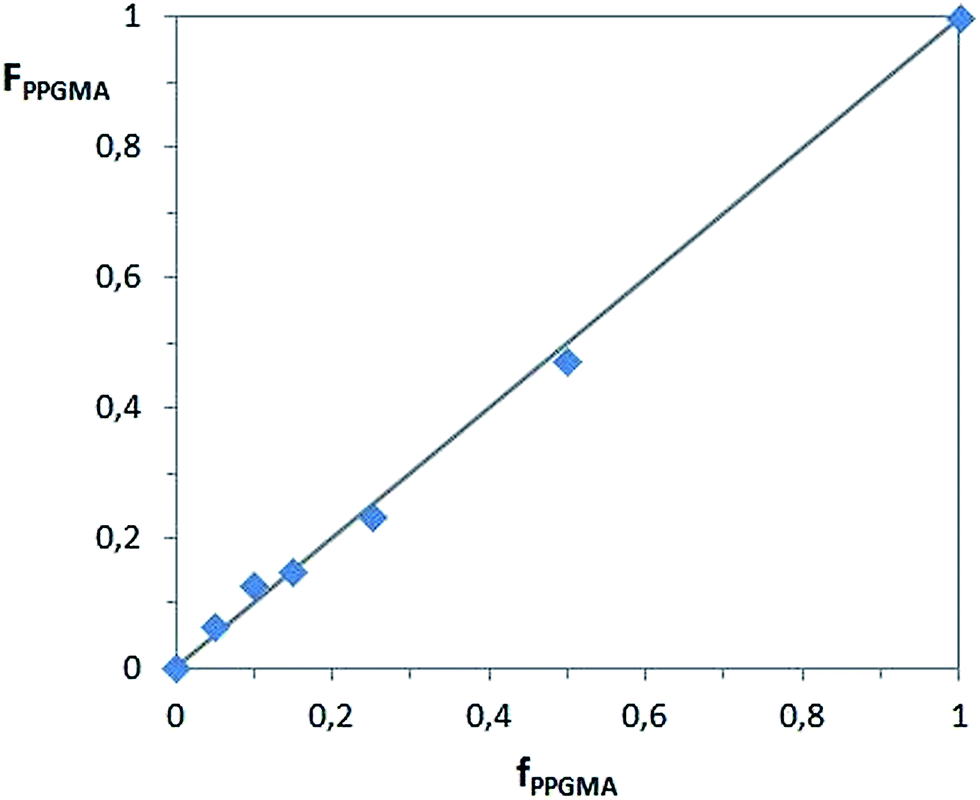 | ||
| Fig. 5 Instantaneous monomer–copolymer composition diagram. | ||
The “side chain extension” polymerizations (Scheme 1c) were conducted under ATRP conditions using a CuCl/PMDETA catalyst system and multifunctional macroinitiators based on PPGMA copolymers with active bromoester groups, obtained by the esterification of hydroxyl groups (Scheme 1b). The content of initiating sites was close to that of hydroxyl groups because the functionalization reaction proceeded with a high efficiency of 91–92% (MI I–MI VII). To avoid irreversible coupling reactions and gel formation, all polymerizations were performed in a highly diluted system (anisole/monomer = 1/1 v/v). Table 2 summarizes the molecular characteristics of the PPGMA based copolymers with side chains extended by PtBMA segments. P[MMA-co-(PPGMA-graft-PtBMA)] copolymers were obtained in monomer conversions of 5–45%, determined by GC. Various compositions were correlated with the polymerization degree of PtBMA and the grafting density of block copolymers (DPtBMA = 18–59 per graft and DGB = 6–14 mol%), which yielded varying content of tBMA in the copolymer (FtBMA = 60–92 wt%). The graft copolymers XII–XIV, based on MI I containing 9 initiating groups in the chain (nBr), which correlated with DGB = 6 mol%, had varying lengths of graft (19 vs. 29 vs. 59 units per graft, respectively) without any influence on the molecular weight distribution (Mw/Mn ≤ 1.25). However, the dispersity index increased for copolymers XV–XVII (DPtBMA = 18–56, Mw/Mn = 1.4), with a larger grafting density of the PPG-bl-PtBMA side chains (12–14 mol%). In the case of XVIII, containing the largest amount of side chains, the reaction was continued up to the highest value of monomer conversion, yielding a drastically high dispersity (Mw/Mn > 2), which led us to assume the occurrence of side reactions accompanied by steric hindrance effects.
| No. | MI (nBr) | [tBMA]0/[MI]0 | Time [h] | GC | NMR | GPCa | |||
|---|---|---|---|---|---|---|---|---|---|
| xtBMA [%] | DPtBMA | Mn,calc × 10−3 [g mol−1] | FtBMA | Mn,GPC × 10−3 [g mol−1] | Mw/Mn | ||||
| a RI detector, CH2Cl2, PS standards; MI are bromoester-functionalized P(MMA-co-PPGMA) copolymers, based on PPGMA copolymers I–III and VII; nBr = DPn × FPPGMA × EfE, where nBr is the number of bromine functional groups obtained by esterification of the hydroxyl groups of PPGMA units in the copolymer, DPn is the polymerization degree of the backbone, FPPGMA is the PPGMA content in the copolymer, EfE is the efficiency of esterification, equal to 91–92%; DPtBMA = xtBMA[tBMA]0/[MI]0; Mn,calc = Mn,NMR + 150 nBr + 142 DPtBMA, where Mn,NMR is the molecular weight of the hydroxyl-functionalized copolymer and 150 is the molecular weight of the bromoester group (–COC(CH3)2Br–1H).b [MI]0/[CuCl]0/[PMDETA]0 = 1/1/1; anisole = 100 vol%; T = 60 °C. | |||||||||
| XII | MI I (9) | 450 | 1 | 5 | 19 | 44.7 | 0.60 | 26.2 | 1.23 |
| XIII | MI I (9) | 400 | 2 | 8 | 29 | 58.4 | 0.72 | 29.0 | 1.25 |
| XIV | MI I (9) | 275 | 3 | 22 | 59 | 87.5 | 0.77 | 35.3 | 1.24 |
| XV | MI II (14) | 175 | 5 | 23 | 40 | 96.2 | 0.89 | 27.2 | 1.37 |
| XVI | MI III (19) | 150 | 2 | 39 | 56 | 179.3 | 0.93 | 36.1 | 1.44 |
| XVII | MI VII (25) | 50 | 2 | 34 | 18 | 91.6 | 0.74 | 44.9 | 1.38 |
| XVIII | MI VII (25) | 100 | 4 | 45 | 47 | 195.2 | 0.92 | 59.0 | 2.06 |
The values of Mn,GPC obtained from conventional GPC were lower than those from the 1H NMR analysis. This discrepancy considerably increased with the length (Fig. 4b) and the number of PtBMA segments in the side chains (Fig. 4c). Both structural parameters of branching could be responsible for a gradual decrease in the hydrodynamic volume of the macromolecules, causing a difference in relation to the linear polymer standards used for calibration in the GPC method.
The tert-butyl groups of the PtBMA segments in the side chains were cleaved by treatment with TFA in dichloromethane, to yield amphiphilic P[MMA-co-(PPGMA-graft-PMAA)] copolymers with acidic groups (FMAA = 28–54 wt%). The modification of hydrophobic polymers to give carboxyl-functionalized ones (Scheme 1d) was observed by significant changes in solubility. Both hydroxyl-functionalized PPGMA copolymers and the copolymers with PtBMA segments in the side chains were completely insoluble in polar solvents, but after acidolysis, the copolymers did not dissolve in methylene chloride, while retaining good solubility in THF, but they could also be dissolved in methanol and in water.
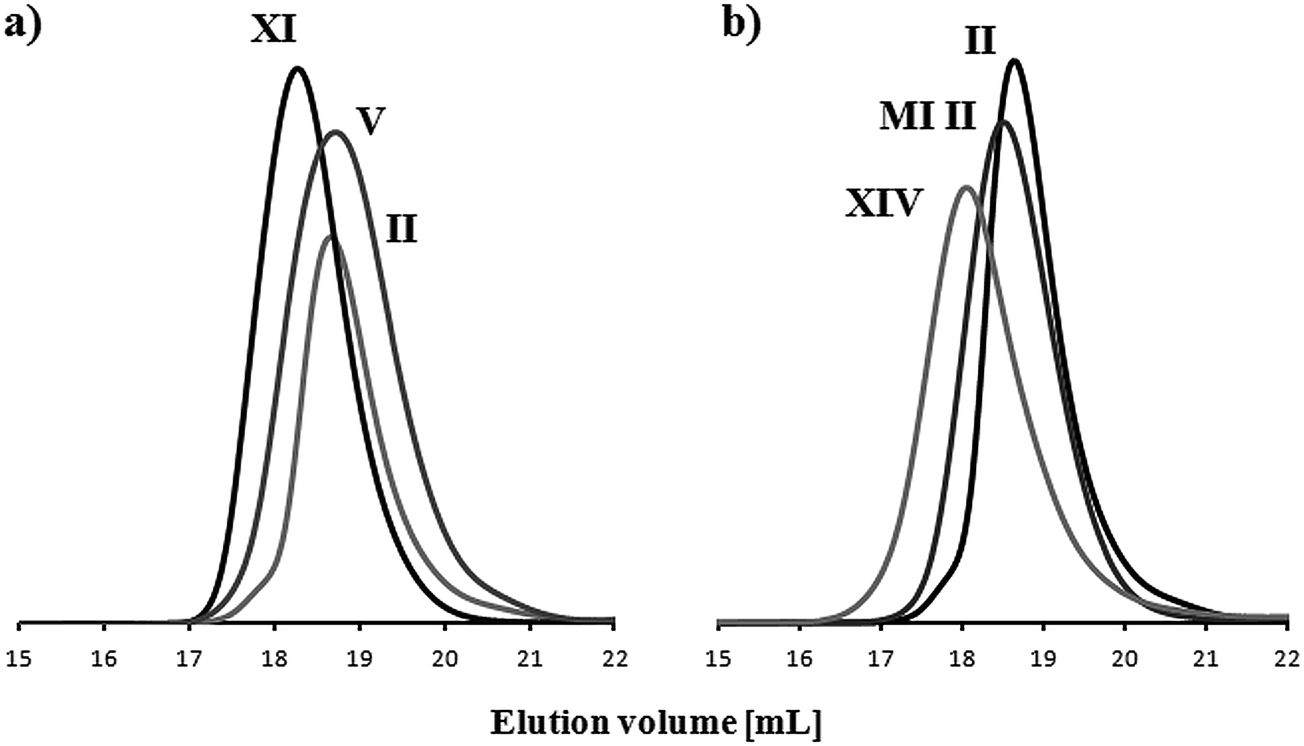
The modification steps starting from hydroxyl-functionalized PPGMA copolymers, including preparation of the multifunctional macroinitiator, “side chain extension” polymerization and acidolysis, were confirmed by spectroscopic analyses. In the 1H NMR spectra (Fig. 6a), the peak assigned to the methyl protons in the bromoester group at 1.94 ppm (G) and the peaks characteristic of the protons of the tert-butyl group at 1.25–1.31 ppm (H), which dramatically decreased after the cleavage of tert-butyl groups during acidolysis, demonstrate the formation of the multifunctional MI, then the grafted copolymer, and the modified copolymer with a fraction of MAA units. In the FT-IR spectra (Fig. 6b), the asymmetric double peak characteristic of the tert-butyl group at 1360–1390 cm−1 for the graft copolymer with PtBMA segments in the side chains significantly diminished after modification. Additionally, the GPC traces of P(MMA-co-PPGMA), P(MMA-co-PPGMA)-Br and P[MMA-co-(PPGMA-graft-PtBMA)] in Fig. 3b show the progressive increase in molecular weight due to the introduction of bromoester groups and the extension of the side chains by PtBMA segments, which is observed by the peak shifting towards a lower elution volume, while narrow dispersities are maintained.
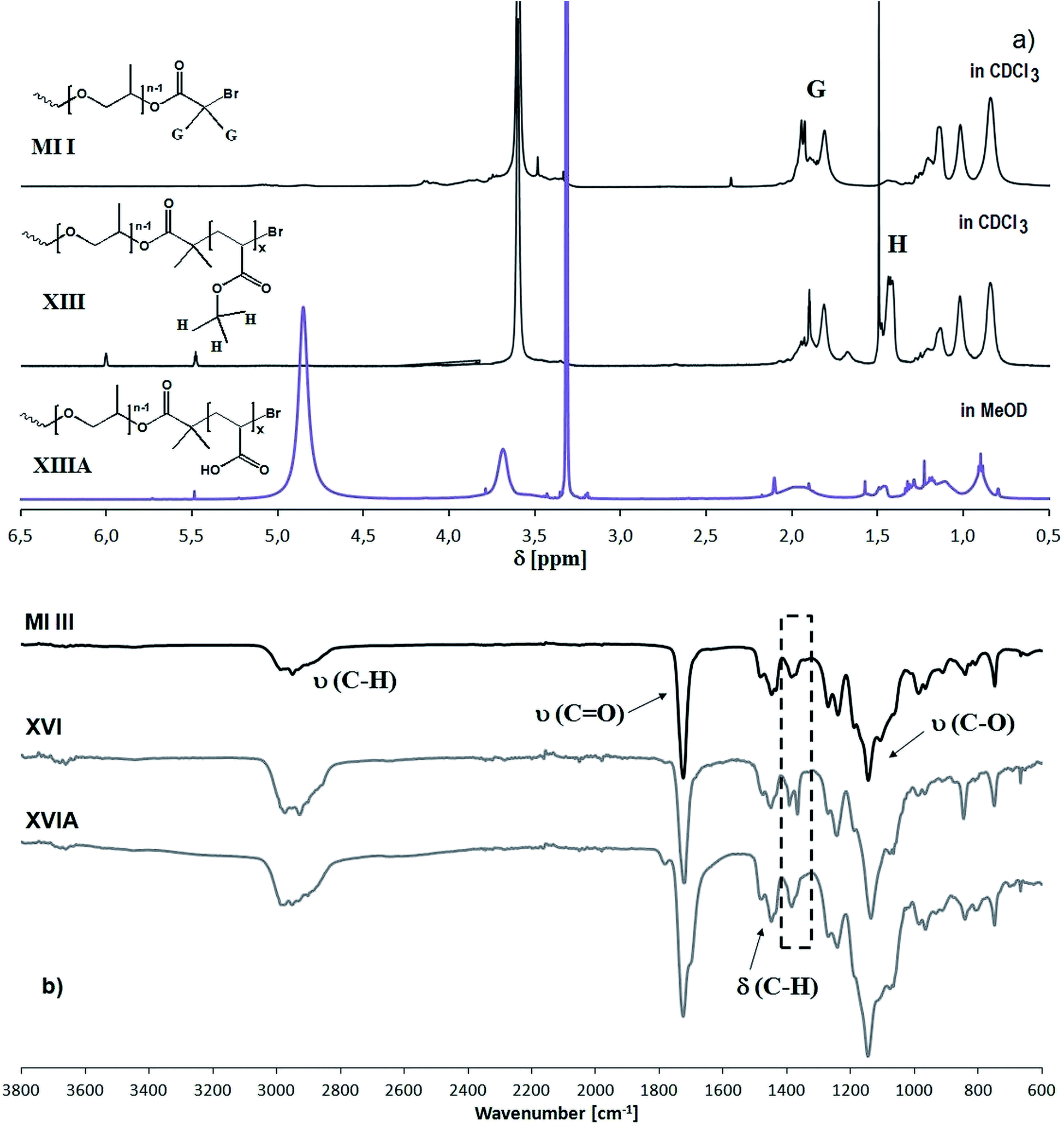 | ||
| Fig. 6 1H NMR (a) and FT-IR (b) spectra of bromoester-functionalized PPGMA-based multifunctional macroinitiator P(MMA-co-PPGMA)-Br, the graft copolymer with PPGMA side chains extended by PtBMA segments, and the amphiphilic copolymer with PMAA segments. | ||
The results of hydrodynamic diameter (dh) measurements, which were performed at 25 °C in deionized water, are summarized in Table 3. The smallest particles, reaching sizes around 165 nm, were formed by XIIIA, with a low grafting degree and medium length of side chains. They became larger for longer grafts at the same graft number (XIIIA vs. XIVA and XVIIA vs. XVIIIA), whereas the latter pair of copolymers exhibited larger diameters due to a higher grafting degree. A representative histogram is presented in Fig. 7a. Another correlation of increasing particle diameter was observed with respect to the content of the hydrophilic fraction in the copolymers, in the following order: XIIIA > XVIA > XVIIIA. According to this relation, the sample XVIIA should give larger aggregates than actually resulted, but this can be explained by short grafts in combination with another important parameter, the efficiency of acidolysis, which in this case yielded almost full transformation of tert-butoxyl groups into carboxyl ones (89%), whereas for the other copolymers the efficiency was significantly lower (52–64%). The amount of MAA units in XVIIA was lower than for XVIA and XVIIIA, which correlated with the result of the zeta potential (ζ-potential, Table 3, Fig. 7b) showing a negative value close to zero, and almost neutral nanoparticle surface. The measurements were performed in an environment of deionized water at 25 °C. The negative values are typical for carboxyl-functionalized copolymers, which are generally well known for pH-sensitivity.
| No. | Ngraft | DGB [%] | NMR | DLS | Zeta potential [mV] | |||
|---|---|---|---|---|---|---|---|---|
| EfA | nMAA/ntBMA | FMAA | dh [nm] | PDI | ||||
| a Ngraft: number of side chains equal to nBr; DGB: percentage degree of grafting based on the content of PPGMA grafts; EfA: efficiency of acidolysis; nMAA: number of MAA units after acidolysis; ntBMA: number of tBMA units before acidolysis; FMAA: content of MAA units in the graft copolymer; dh and PDI: hydrodynamic diameter of particles and polydispersity of sizes; nd – not determined. | ||||||||
| XIIA | 9 | 6 | 0.64 | 12/19 | 0.28 | nd | nd | nd |
| XIIIA | 9 | 6 | 0.55 | 16/29 | 0.29 | 166 | 0.445 | −41 |
| XIVA | 9 | 6 | nd | nd/59 | nd | 203 | 0.336 | nd |
| XVA | 14 | 12 | nd | nd/40 | nd | 231 | 0.549 | nd |
| XVIA | 19 | 12 | 0.52 | 29/56 | 0.37 | 221 | 0.358 | −8 |
| XVIIA | 25 | 14 | 0.89 | 16/18 | 0.54 | 183 | 0.309 | −3 |
| XVIIIA | 25 | 14 | 0.61 | 29/47 | 0.44 | 224 | 0.140 | −26 |
 | ||
| Fig. 7 Size distribution by intensity for XVIIIA (left), and zeta potential distribution (right) in deionized water at 25 °C. | ||
Conclusions
A versatile four-step approach was used to synthesize well-defined amphiphilic graft PPGMA-based copolymers containing acidic MAA units introduced via acidolysis to remove the larger part of the tBMA units in the side chains. The PtBMA segments were prepared by a “side chain extension” polymerization procedure using bromoester-functionalized PPGMA macroinitiators (7–14 mol% of initiating sites, which regulate the grafting degree of PtBMA). These macroinitiators were obtained by the esterification of short-grafted hydroxyl-functionalized copolymers of PPGMA. Hydrophobic P[MMA-co-(PPGMA-graft-PtBMA)] copolymers with varying degrees of grafting and length of PtBMA segments in the side chains were transformed into amphiphilic copolymers with enhanced solubility in polar solvents, including water. The presence of carboxyl groups makes them weak polyelectrolytes with near pH-responsive properties. Self-assembly behavior in aqueous solution lead to the creation of particles with hydrodynamic diameters in the range of 165–230 nm, and negative ions on the surface. Thus, the resulting polymers, due to their precise structures and specific physicochemical properties, are great candidates for novel biocompatible carriers in drug delivery.Acknowledgements
P. M.-B. is a scholar on the project “DoktoRIS” co-financed by the European Union under the European Social Fund.References
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337 CrossRef CAS.
- H. Mori, O. Wakisaka, A. Hirao and S. Nakahama, Macromol. Chem. Phys., 1994, 195, 3213 CrossRef CAS.
- T. Ishizone, S. Han, S. Okuyama and S. Nakahama, Macromolecules, 2003, 36, 42 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015 CrossRef CAS.
- K. L. Robinson, M. A. Khan, M. V. de Paz Báñez, X. S. Wang and S. P. Armes, Macromolecules, 2001, 34, 3155 CrossRef CAS.
- S. Coca, C. B. Jasieczek, K. L. Beers and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1417 CrossRef CAS.
- A. Narumi, Y. Chen, M. Sone, K. Fuchise, R. Sakai, T. Satoh, Q. Duan, S. Kawaguchi and T. Kakuchi, Macromol. Chem. Phys., 2009, 210, 349 CrossRef CAS.
- M. Teodorescu and K. Matyjaszewski, Macromolecules, 1999, 32, 4826 CrossRef CAS.
- J.-F. Lutz, N. Jaheed and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1939 CrossRef CAS.
- M. Ali and H. D. H. Stöver, Macromolecules, 2004, 37, 5219 CrossRef CAS.
- P. Maksym-Bębenek, T. Biela and D. Neugebauer, React. Funct. Polym., 2014, 82, 33 CrossRef PubMed.
- B. S. Shemper, A. E. Acar and L. J. Mathias, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 334 CrossRef CAS.
- B. S. Shemper and L. J. Mathias, Eur. Polym. J., 2004, 40, 651 CrossRef CAS PubMed.
- X. J. Loh, J. Appl. Polym. Sci., 2013, 127, 992 CrossRef CAS.
- W. Wang, H. Liang, R. C. Al Ghanami, L. Hamilton, M. Fraylich, K. M. Shakesheff, B. Saunders and C. Alexander, Adv. Mater., 2009, 21, 1809 CrossRef CAS.
- S. R. Abulateefeh, A. O. Saeed, J. W. Aylott, W. C. Chan, M. C. Garnett, B. R. Saunders and C. Alexander, Chem. Commun., 2009, 6068 RSC.
- H. Tai, W. Wang, T. Vermonden, F. Heath, W. E. Hennink, C. Alexander, K. M. Shakesheff and S. M. Howdle, Biomacromolecules, 2009, 10, 822 CrossRef CAS PubMed.
- H. Tai, D. Howard, S. Takae, W. Wang, T. Vermonden, W. E. Hennink, P. S. Stayton, A. S. Hoffman, A. Endruweit, C. Alexander, S. M. Howdle and K. M. Shakesheff, Biomacromolecules, 2009, 10, 2895 CrossRef CAS PubMed.
- H. Lee, W. Jakubowski, K. Matyjaszewski, S. Yu and S. S. Sheiko, Macromolecules, 2006, 39, 4983 CrossRef CAS.
- S. Y. Yu-Su, S. S. Sheiko, H. Lee, W. Jakubowski, A. Nese, K. Matyjaszewski, D. Anokhin and D. A. Ivanov, Macromolecules, 2009, 42, 9008 CrossRef CAS.
- G. Cheng, A. Boeker, M. Zhang, G. Krausch and A. H. E. Mueller, Macromolecules, 2001, 34, 6883 CrossRef CAS.
- A. Bhattacharyaa and B. N. Misra, Prog. Polym. Sci., 2004, 29, 767 CrossRef PubMed.
- F. Ch, Y. Li, D. Yang, J. Hu, X. Zhang and X. Huang, Chem. Soc. Rev., 2011, 40, 1282 RSC.
- C. M. F. Oliveira and A. S. Gomes, Polym. Bull., 1989, 22, 401 CrossRef CAS.
- C. M. F. Oliveira, C. T. Andrade and M. C. Delpech, Polym. Bull., 1991, 26, 657 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |