
An efficient robust fluorite CeZrO4−δ oxide catalyst for the eco-benign synthesis of styrene
Abstract
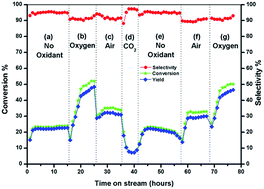
In this work, we have reported CeO2, ZrO2, physically mixed (PH)-CeO2/ZrO2 and fluorite CeZrO4−δ oxides and their catalytic activities for the oxidative dehydrogenation (ODH) of ethyl benzene (EB) to styrene (ST) using molecular oxygen, air and carbon dioxide as oxidants. The catalysts were prepared by a gel-combustion method followed by calcination at 600 °C for 6 h and subjected to catalytic activity measurements. All the catalysts were characterized and studied by various physicochemical methods. The reaction parameters were varied systematically such as different catalysts, oxidants, temperatures, EB flow and oxidant flow. CeZrO4−δ accounted for a 47% styrene yield for 72 h without any significant deactivation under optimized reaction conditions. A thorough analysis of the spent catalysts demonstrated the robustness of the catalyst for this reaction under different oxidants and reaction conditions. Pristine CeO2 deactivated easily and the activity decreased with time on stream of the reaction.
 Please wait while we load your content...
Please wait while we load your content...

