
Palladium-catalyzed oxidative annulation of in situ generated enones to pyrroles: a concise route to functionalized indoles†
Tenglong
Guo
a,
Quanbin
Jiang
a and
Zhengkun
Yu
*ab
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, Liaoning 116023, P. R. China. E-mail: zkyu@dicp.ac.cn; Fax: +86–411-8437-9227
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, P. R. China
First published on 12th August 2015
Abstract
Palladium(II)-catalyzed, copper(II)-mediated indole synthesis was achieved from the reactions of N-substituted simple pyrroles with enones generated in situ from 3-chloropropiophenones. A benzene ring was thus constructed onto a pyrrole backbone, affording substituted indole derivatives. A domino dehydrochlorination/C–H olefination /Diels–Alder cycloaddition/dehydrogenative aromatization sequence was established as the reaction pathway. The present methodology provides a concise route to highly functionalized indole derivatives.
Indoles are very important structural motifs in many organic compounds.1,2 Although Fischer indoles were first reported in 1883,3 the recently known indole syntheses are predominantly directed towards construction of a pyrrole ring onto a prefunctionalized benzene precursor.4 However, the alternative method which uses a pyrrole ring as the “template”-like structure to establish a benzene ring onto it is much less common.5 In this aspect, anionic benzannulation of N-methylpyrroles with alkenes or alkynes was employed to form functionalized indoles.6 Intramolecular cyclization of vinylpyrroles7a and alkynylated pyrroles7b occurred to give multi-substituted indoles under various conditions, respectively. Diels–Alder reactions of 2-vinylpyrroles with maleimides,8a and 2- or 3-nitropyrroles with dienes,8b were realized for the same purpose. Lewis acid-catalyzed pyrrolyl-supported enynals with enols or enol ethers,9a and ruthenium(0)-mediated annulation of pyrroles with propargyl alcohols9b yielded indoles under relatively harsh conditions. Palladium-catalyzed oxidative decarboxylative annulation of N-methylpyrrole-2-carboxylic acid with alkynes generated indoles as reaction intermediates.10 Substituted indoles were also obtained from one-pot, two-step reactions of pyrroles with β-nitroacrylates.11 Benzannulation of pyrroles with rhodium enalcarbenoids formed substituted indoles.12 Because establishment of an indole core is often encountered in the synthesis of functionalized molecules,4 concise and direct indole synthesis from relatively simple pyrroles has been strongly desired.
As transition-metal-catalyzed C–H activation has recently become a promising straightforward route to construct carbon–carbon and carbon–heteroatom bonds, oxidative cross-coupling of a pyrrole with two olefin molecules to form a benzene ring seems to be a potential route to access indole derivatives. However, in contrast to other N-heterocycles which can be used as coupling partners for carbon–carbon bond formation,13 direct oxidative cross-coupling of pyrroles with olefins has seldom been documented due to the poor regioselectivity, polyolefination, and significant polymerization of the pyrrole substrates under oxidative conditions. In 2006, Gaunt et al. reported palladium-catalyzed olefination of simple pyrroles by acrylates through N-protecting groups differentiating in steric and electronic properties,14a and later Yao's lab achieved palladium-catalyzed solvent-controlled switchable olefination of 3,4-disubstituted pyrroles with electron-deficient olefins14b (Scheme 1a). Under acid,15 iodine,16 or organo-catalysis17 conditions pyrrole substrates usually underwent Friedel–Crafts alkylation with α,β-unsaturated ketones (enones) (Scheme 1b). Intrigued by the advantages of using in situ generated enones as olefin sources for carbon–carbon bond formation,18–21 we reasonably envisioned the oxidative annulation reactions of β-chloroalkyl ketones to pyrroles. Herein, we report direct synthesis of indoles from pyrroles and 3-chloropropiophenones in a one-shot style (Scheme 1c).
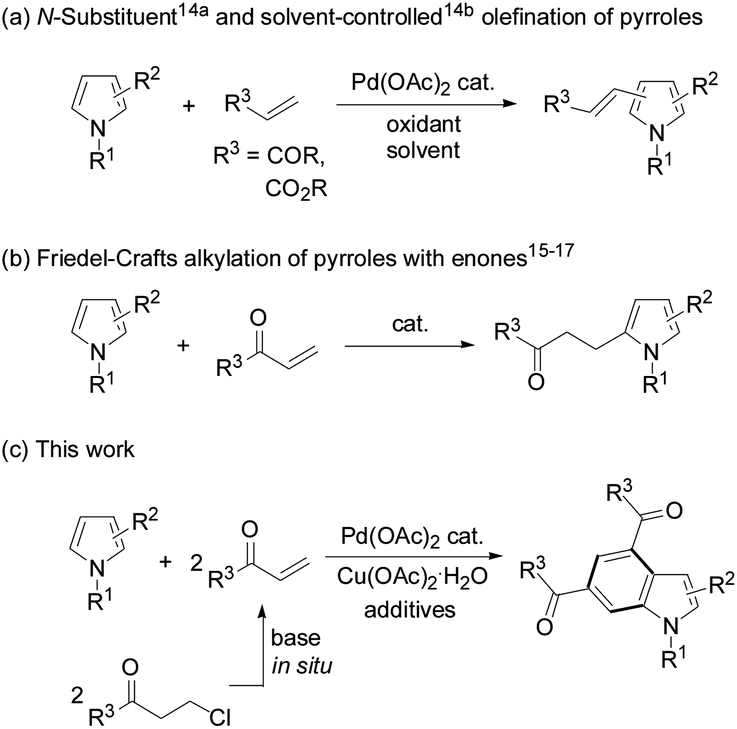 | ||
| Scheme 1 Direct reactions of pyrroles with electron-deficient olefins. | ||
Initially, the reaction of N-methyl-2-phenylpyrrole (1a) with 3-chloropropiophenone (2a) was conducted to screen the reaction conditions (Table 1). In the presence of 10 mol% Pd(OAc)2 as the catalyst, Cu(OAc)2·H2O (6.0 equiv.) as the oxidant, and NaOAc (4 equiv.) as the base, the target product 3a was obtained in 41% yield from the reaction in DMSO at 100 °C (Table 1, entry 1). Among the screened solvents, i.e., DMSO, DMF, toluene, THF, and dioxane, a mixture of DMF/DMSO (v/v, 9/1) effected the reaction best (Table 1, entry 3). Cu(OAc)2, AgOAc, Ag2CO3, benzoquinone, tBuOOtBu, and dioxygen were used to promote the reaction, and only Cu(OAc)2·H2O was found to be the most efficient oxidant (Table 1, entries 1–4). The reaction hardly occurred without an oxidant, and the air atmosphere facilitated the reaction (Table 1, entries 5 and 6). A combination of additives tetra-butyl ammonium bromide (TBAB) and pivalic acid (PivOH) enhanced the yield of 3a to 70% (Table 1, entries 7–9). With a lower loading of the oxidant (4 equiv.) at a higher temperature (130 °C) the target product was also obtained in a decent yield (68%), while varying the temperature around 130 °C was detrimental to the reaction efficiency (Table 1, entries 10–13). Various palladium sources and bases were also investigated, and Pd(OAc)2 and NaOAc were shown to be the most efficient catalyst and base for the desired reaction, respectively (see the ESI†). To ease the work-up procedure by using a less amount of oxidant, the conditions for entry 12 were considered as those suitable for the desired reaction (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant | Solvent | Additive (equiv.) | Temp (°C) | Yieldb (%) |
a Conditions: 1a (0.2 mmol), 2a (0.8 mmol), catalyst (0.02 mmol), base (0.8 mmol), oxidant (1.2 mmol), solvent (2.5 mL), air, 24 h.
b Isolated yields based on 1a.
c DMF/DMSO (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 9:1).
d Under 0.1 MPa N2 atmosphere.
e Cu(OAc)2·H2O (0.8 mmol).
f TBAB (0.1 mmol) and PivOH (0.2 mmol). :v = 9:1).
d Under 0.1 MPa N2 atmosphere.
e Cu(OAc)2·H2O (0.8 mmol).
f TBAB (0.1 mmol) and PivOH (0.2 mmol).
|
|||||
| 1 | Cu(OAc)2·H2O | DMSO | 100 | 41 | |
| 2 | Cu(OAc)2·H2O | DMF | 100 | 42 | |
| 3 | Cu(OAc)2·H2O | DMF/DMSOc | 100 | 64 | |
| 4 | Cu(OAc)2 | DMF/DMSOc | 100 | 61 | |
| 5d | DMF/DMSOc | 100 | <1 | ||
| 6 | Air | DMF/DMSOc | 100 | 13 | |
| 7 | Cu(OAc)2·H2O | DMF/DMSOc | TBAB (0.5) | 100 | 65 |
| 8 | Cu(OAc)2·H2O | DMF/DMSOc | PivOH (1.0) | 100 | 66 |
| 9 | Cu(OAc)2·H2O | DMF/DMSOc | TBAB/PivOHf | 100 | 70 |
| 10e | Cu(OAc)2·H2O | DMF/DMSOc | TBAB/PivOHf | 100 | 60 |
| 11e | Cu(OAc)2·H2O | DMF/DMSOc | TBAB/PivOHf | 80 | 48 |
| 12e | Cu(OAc)2·H2O | DMF/DMSOc | TBAB/PivOHf | 130 | 68 |
| 13e | Cu(OAc)2·H2O | DMF/DMSOc | TBAB/PivOHf | 140 | 52 |
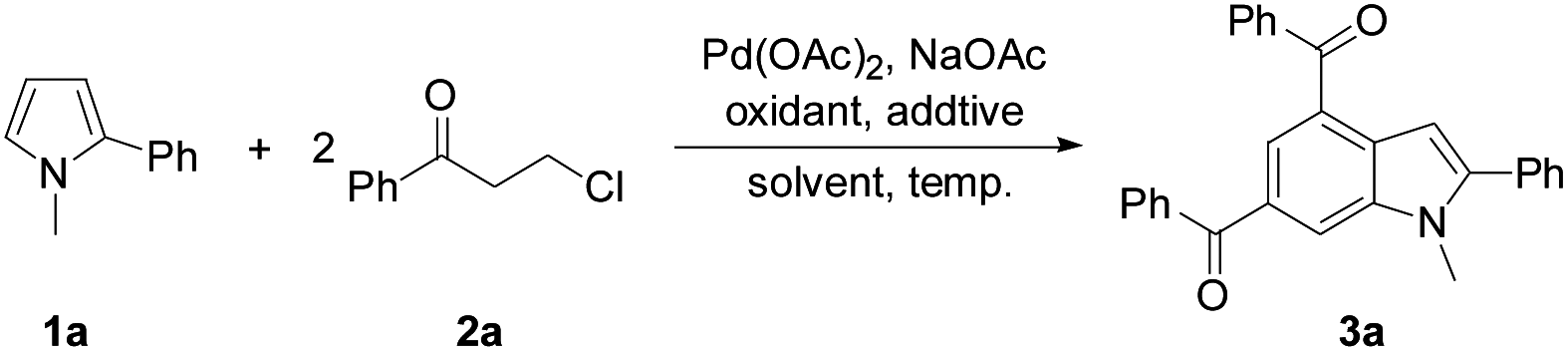
Next, the scope of 3-chloroalkyl ketones 2 was explored to probe into the protocol generality (Table 2). Pyrrole 1a reacted with 3-chloroalkyl aryl ketones bearing a substituent on the aryl moiety proceeded to form the target products 3a–3g in 67–73% yields, revealing no obvious electronic impact from the substituents. Increasing the steric hindrance of the aryl moiety in 2 by introducing two methyls led to the corresponding products 3h–3j (50–65%), which is attributed to the steric effect of the ortho-methyl on the aryl moiety of 2. The presence of bulky 1-naphthyl deteriorated the yield of 3k (52%). Heteroaryl 3-chloropropionones also underwent the reactions to afford indoles 3l (71%) and 3m (63%), respectively. In contrast to 3-chloroalkyl aryl ketones, 5-chloropentan-3-one exhibited a low reactivity to 1a, and its reaction with 1a only gave 3n in 35% yield.
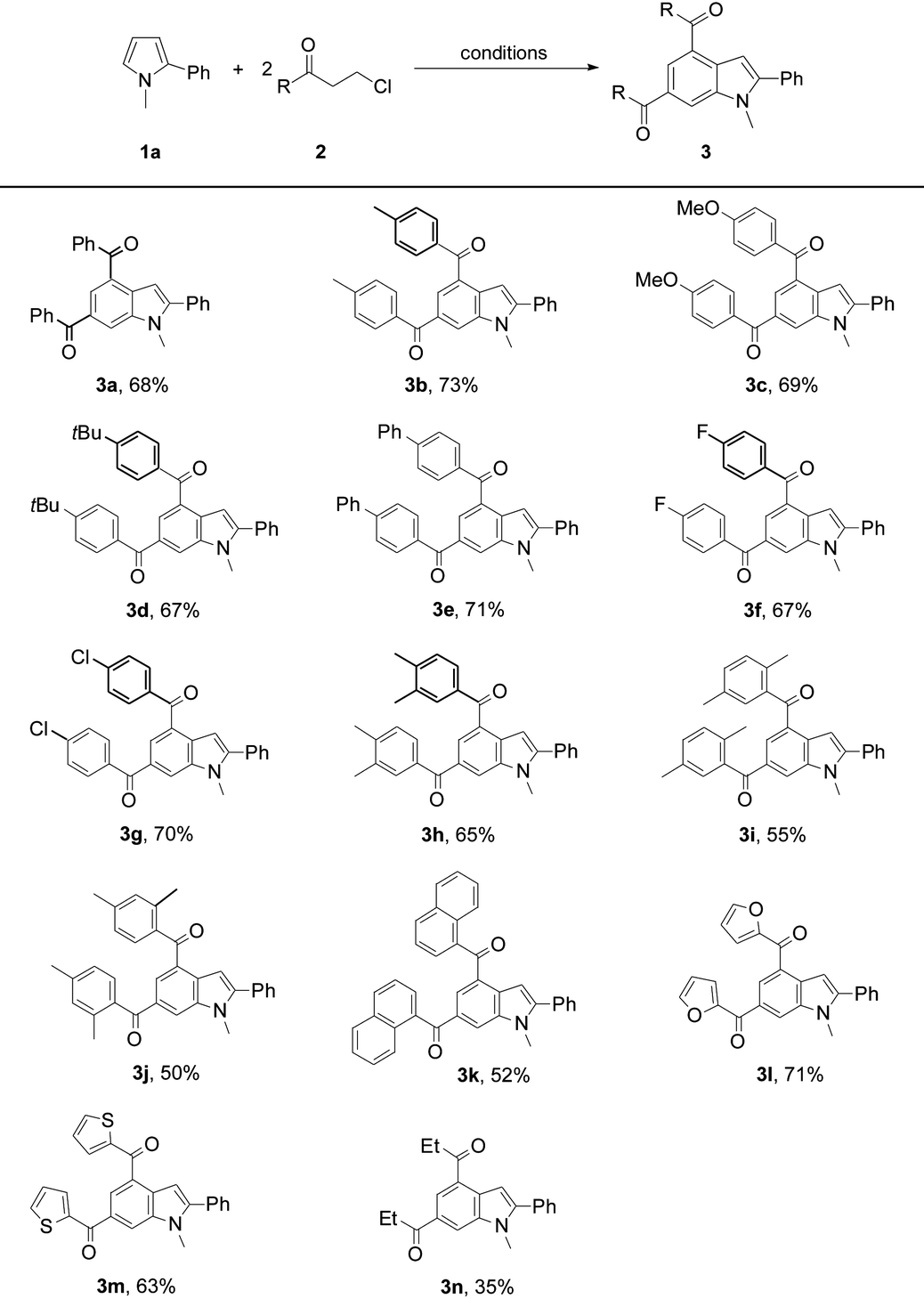
The substrate scope was further investigated by reacting 2a with a variety of pyrrole substrates 1 (Table 3). The steric hindrance from the N-R moieties (R = Et, allyl, Bn, and Me) differentiated the reactivity of these 2-phenylpyrroles, leading to the target indole products 4a–4d (50–68%). A methyl substituent on the aryl group of N-methyl-2-arylpyrroles did not obviously affect the reaction efficiency and 4d–4f were obtained in 66–68% yields. However, electron-donating methoxy, and electron-withdrawing groups such as fluoro, ester, cyano, and chloro, on the aryl group deteriorated the production of 4g (51%), and 4h–4k (52–62%). N-Methyl-2-(1-naphthyl)pyrrole reacted to give 4l (61%). The reaction also tolerated thienyl on the pyrrole ring, yielding 2-thienylindole 4m (42%). 1,3-Dimethyl-2-phenyl-pyrrole exhibited a decent reactivity to form 4n (63%), showing no obvious steric effect from the pyrrole backbone. However, the reaction of 1,2-dimethylpyrrole only formed 4o in 36% yield, exhibiting a negative electronic effect from the 2-substituent of the pyrrole backbone. Owing to the multiple reactive sites in 1-methylpyrrole, the target product 4p was only obtained in a low yield (17%). Unexpectedly, the reaction of N-methyl-2-(4′-nitro-phenyl)pyrrole with 2a afforded indole 4q (38%) and the reaction intermediate 5-alkenylated pyrrole 5a (35%). In the cases of using 2-phenyl-substituted N-H or N-Boc pyrroles, the target indole products could not be obtained. It should be noted that indole 4d was further structurally confirmed by X-ray crystallographic analysis (see the ESI†).
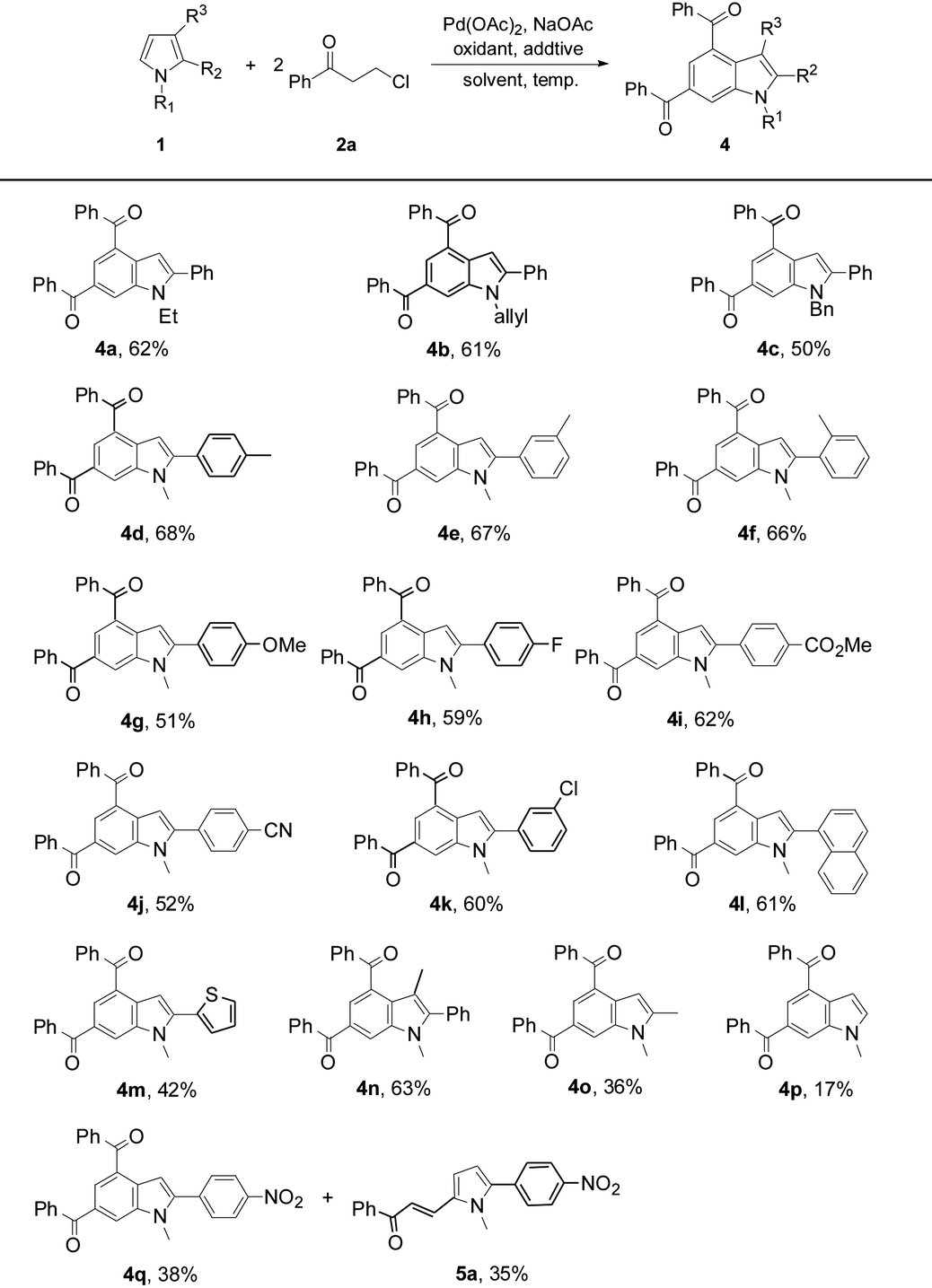
In order to investigate the reaction mechanism, controlled experiments were conducted to identify the possible reaction intermediates. We have recently established that compound 2a can be dehydrochlorinated to form phenyl vinyl ketone (enone) 6 under basic conditions.21b Thus, enone 6 (4 equiv.) was used to react with 1a under the conditions similar to those shown in Tables 2 and 3 (eqn (1)). In this case, a base was not necessary, and 3a was formed in 59% yield, suggesting that in situ generation of enone 6 from 3-chloropropiophenone is superior to direct use of the corresponding enone substrate for the synthesis of indole 3a. To obtain the 5-alkenylated pyrrole intermediate of type 5 pyrrole 1a was treated with 2a in a 1:1 molar ratio by lowering both the base and oxidant loadings and shortening the reaction time to 2 h (eqn (2)). Fortunately, 5-alkenylated pyrrole 5b was obtained in 26% yield. Then, 5b was treated with 2a under the controlled conditions as shown in Scheme 2. With 10 mol% Pd(OAc)2 as the catalyst, 3a was formed in 50% isolated yield. Without Pd(OAc)2 the reaction occurred to form 3a in 65% yield, and using Cu(OAc)2·H2O as the sole mediator under a nitrogen atmosphere also led to 3a in a decent yield (72%). The air atmosphere promoted the formation of 3a (29%). These results have revealed that Pd(OAc)2 only acted as the catalyst for the oxidative alkenylation of pyrrole 1a, Cu(OAc)2·H2O promoted both the alkenylation of 1a and dehydrogenative cyclization of 5b with the in situ generated enone 6 to produce indole 3a. Although the air atmosphere facilitated the formation of the target product 3a, an additional oxidant is required to reach a satisfactory reaction efficiency.
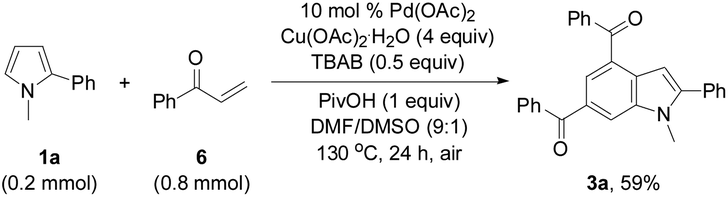 | (1) |
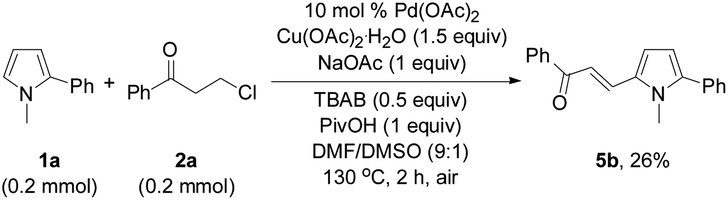 | (2) |
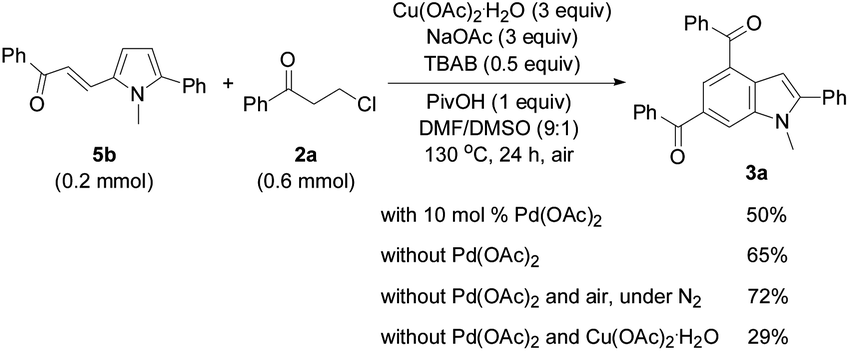 | ||
| Scheme 2 Controlled reactions of 5b with 2a. | ||
Taking the observed regioselectivity into account, we assume that 3a is formed through the Diels–Alder reaction of intermediate 5b with enone 6 generated in situ from 2a followed by dehydrogenative aromatization. Indeed, the Diels–Alder reaction intermediate, that is, tetrahydroindole 7, was isolated in 18% yield from the reaction of 5b with 2a (3 equiv.) in the presence of a NaOAc base (3 equiv.) under an inert atmosphere without the catalyst, oxidant, and additives. The dehydrogenative aromatization reaction of 7 was conducted by means of Cu(OAc)2·H2O as the oxidant, affording the target product 3a in 79% yield (Scheme 3).
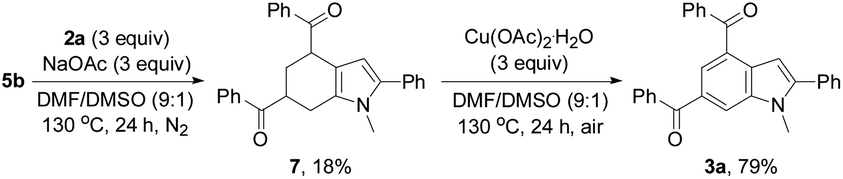 | ||
| Scheme 3 Identification of the Diels–Alder reaction intermediate. | ||
A plausible mechanism is proposed in Scheme 4. Pyrrole 1a initially undergoes palladation at its 5-position to form a palladated species A and HOAc via C–H activation. Species A reacts with the in situ generated enone 6 from 2a to yield an olefin insertion species B. Subsequent reductive elimination produces 5-alkenylated pyrrole 5b and the Pd(0) species. A Diels–Alder cycloaddition of enone 6 to 5b forms tetrahydroindole 7, which is subsequently oxidized to indole 3a. Both the Cu(II) oxidant and air facilitate regeneration of the catalyst.
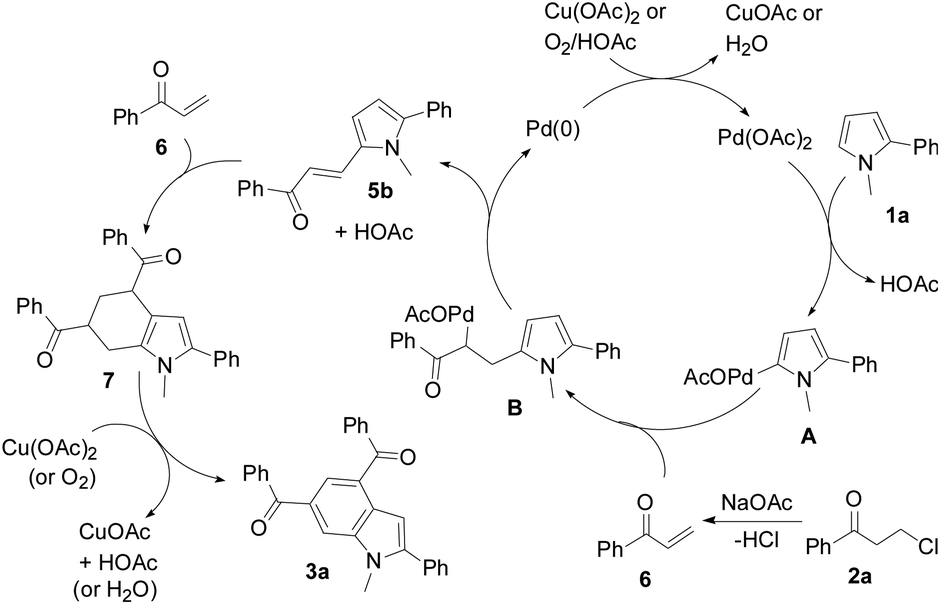 | ||
| Scheme 4 Proposed mechanism. | ||
In conclusion, palladium-catalyzed oxidative annulation of 3-chloropropiophenones and their analogues to N-substituted simple pyrroles has been successfully achieved to synthesize functionalized indoles. The synthetic protocol features construction of a substituted benzene ring onto a pyrrole backbone through the in situ generated enones. A domino dehydro-chlorination/C–H olefination/Diels–Alder cycloaddition/dehydrogenative aromatization sequence was established as the reaction pathway. The present protocol provides a concise route to functionalized indoles.
Experimental section
A typical procedure for the synthesis of indoles from pyrrroles: synthesis of 3a
A mixture of N-methyl-2-phenylpyrrole (1a) (32 mg, 0.2 mmol), 3-chloropropiophenone (2a) (133 mg, 0.8 mmol), Pd(OAc)2 (4.5 mg, 0.02 mmol), Cu(OAc)2·H2O (160 mg, 0.8 mmol), TBAB (32 mg, 0.1 mmol), PivOH (20 mg, 0.2 mmol), and NaOAc (66 mg, 0.8 mmol) in 2.5 mL DMF/DMSO (v/v = 9:1) was stirred at 130 °C under an air atmosphere for 24 h. After being cooled to ambient temperature, 10 mL CH2Cl2 was added and the resultant mixture was filtered through a short pad of celite, followed by rinsing with 10 mL CH2Cl2. The combined filtrate was washed with brine (10 mL) and separated. The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: petroleum ether (60–90 °C)/EtOAc/CH2Cl2 (30:1:3, v/v/v) to afford 3a as a yellow liquid (56 mg, 68%).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21472185) and the National Basic Research Program of China (2015CB856600).Notes and references
- N. K. Kaushik, N. Kaushik, P. Attri, P. N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620 CrossRef CAS PubMed.
- W. Zhu, Y. Wu, S. Wang, W. Li, X. Li, J. Chen, Z.-S. Wang and H. Tian, Adv. Funct. Mater., 2011, 21, 756 CrossRef CAS PubMed.
- E. Fischer and F. B. Jourdan, Ber. Dtsch. Chem. Ges., 1883, 16, 2241 CrossRef PubMed.
- For selected recent reviews, see: (a) T. L. Guo, F. Huang, L. K. Yu and Z. K. Yu, Tetrahedron Lett., 2015, 56, 296 CrossRef CAS PubMed; (b) J. Le Bras and J. Muzart, Synthesis, 2014, 1555 Search PubMed; (c) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466 CrossRef CAS PubMed; (d) M. Platon, R. Amardeil, L. Djakovitch and J.-C. Hierso, Chem. Soc. Rev., 2012, 41, 3929 RSC; (e) S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Biomol. Chem., 2011, 9, 641 RSC.
- J. Moskal and A. M. van Leusen, J. Org. Chem., 1986, 51, 4131 CrossRef CAS.
- B. K. Dinda, S. Basak and D. Mal, Eur. J. Org. Chem., 2014, 5521 CrossRef CAS PubMed.
- (a) J. Zhao, C. O. Hughes and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 7436 CrossRef CAS PubMed; (b) K. Hayashi, K. Yoshida and A. Yanagisawa, J. Org. Chem., 2013, 78, 3464 CrossRef CAS PubMed.
- (a) W. E. Noland, N. P. Lanzatella, L. Venkatraman, N. F. Anderson and G. C. Gullickson, J. Heterocycl. Chem., 2009, 46, 1154 CrossRef CAS PubMed; (b) C. D. Rosa, M. Kneeteman and P. Mancini, Tetrahedron Lett., 2007, 48, 1435 CrossRef PubMed.
- (a) N. Asao and H. Aikawa, J. Org. Chem., 2006, 71, 5249 CrossRef CAS PubMed; (b) N. Thies, C. G. Hrib and E. Haak, Chem. – Eur. J., 2012, 18, 6302 CrossRef CAS PubMed.
- M. Yamashita, H. Horiguchi, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 7481 CrossRef CAS PubMed.
- A. Palmieri, S. Gabrielli, D. Lanari, L. Vaccaro and R. Ballini, Adv. Synth. Catal., 2011, 353, 1425 CrossRef CAS PubMed.
- S. G. Dawande, V. Kanchupalli, J. Kalepu, H. Chennamsetti, B. S. Lad and S. Katukojvala, Angew. Chem., Int. Ed., 2014, 53, 4076 CrossRef CAS PubMed.
- K. Ozaki, H. Zhang, H. Ito, A. W. Lei and K. Itami, Chem. Sci., 2013, 4, 3416 RSC.
- (a) E. M. Beck, N. P. Grimster, R. Hatley and M. J. Gaunt, J. Am. Chem. Soc., 2006, 128, 2528 CrossRef CAS PubMed; (b) Y. L. Su, H. P. Zhou, J. X. Chen, J. Y. Xu, X. M. Wu, A. J. Lin and H. Q. Yao, Org. Lett., 2014, 16, 4884 CrossRef CAS PubMed.
- W. Wang, X. Liu, W. Cao, J. Wang, L. Lin and X. Feng, Chem. – Eur. J., 2010, 16, 1664 CrossRef CAS PubMed.
- B. Das, N. Chowdhury and K. Damodar, Tetrahedron Lett., 2007, 48, 2867 CrossRef CAS PubMed.
- D. Hack and D. Enders, Synthesis, 2013, 2904 CAS.
- T. L. Guo, Q. B. Jiang, F. Huang, J. P. Chen and Z. K. Yu, Org. Chem. Front., 2014, 1, 707 RSC.
- (a) Y. P. Shang, X. M. Jie, J. Zhou, P. Hu, S. J. Huang and W. P. Su, Angew. Chem., Int. Ed., 2013, 52, 1299 CrossRef CAS PubMed; (b) J. Zhou, G. Wu, M. Zhang, X. M. Jie and W. P. Su, Chem. – Eur. J., 2012, 18, 8032 CrossRef CAS PubMed; (c) Y. Moon, D. Kwon and S. Hong, Angew. Chem., Int. Ed., 2012, 51, 11333 CrossRef CAS PubMed.
- (a) M. V. Leskinen, K.-T. Yip, A. Vuorinen, I. Pápai, A. J. Neuvonen and P. M. Pihko, J. Am. Chem. Soc., 2014, 136, 6453 CrossRef CAS PubMed.
- (a) T. L. Guo, Q. B. Jiang and Z. K. Yu, Chin. J. Catal., 2015, 36, 78 CrossRef CAS; (b) Q. B. Jiang, T. L. Guo, Q. F. Wang, P. Wu and Z. K. Yu, Adv. Synth. Catal., 2013, 355, 1874 CrossRef CAS PubMed; (c) Q. B. Jiang, T. L. Guo and Z. K. Yu, ChemCatChem, 2015, 7, 660 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization and NMR spectra. CCDC 1002185 for 4d. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00203f |
| This journal is © the Partner Organisations 2015 |