DOI:
10.1039/C5QO00205B
(Research Article)
Org. Chem. Front., 2015,
2, 1366-1373
Successive Cu/Pd transmetalation relay catalysis in stereoselective synthesis of tetraarylethenes†
Received
30th June 2015
, Accepted 5th August 2015
First published on 6th August 2015
Abstract
A new and efficient strategy for the synthesis of tetraaryl-substituted olefins has been developed. With a Cu/Pd-catalyzed isomerization/insertion/oxidative coupling cascade reaction of cyclopropene with internal alkynes, a wide variety of cis-tetrasubstituted olefins were synthesized in good yields as single stereoisomers. The photophysical properties of these novel tetraarylethenes were fully characterized and proved to be good AIE (aggregation-induced emission) luminogens. Experimental studies and theoretical calculation indicated that Cu(I) and Pd(II) were the actual catalysts. A novel deprotonative Cu-catalyzed cyclopropene cycloisomerization and subsequent successive Cu/Pd transmetalation relay mechanism was proposed for the discovered reaction.
Introduction
Multimetallic cooperative catalysis has recently been developed to achieve exquisite one-pot cascade reactions, which require multiple reaction steps with the traditional one-catalyst one-reaction approach.1 This multi-catalyst cascade concept, represents one of the most efficient synthetic methodologies in terms of environmental benignness, atom-economy and also step-economy. More importantly, with the synergistic cooperation of different catalytic species, potentially unprecedented transformations with a single metal catalyst could be achieved. However, it is still a challenge for the rational design of multimetallic catalysts to achieve programmed activation of the reactants toward the expected products, rather than the undesired side reactions.2 Recently we utilized transmetalation as a key strategy to bridge two catalytic cycles and developed an efficient copper–palladium transmetalation relay catalysis for the synthesis of tetrasubstituted furan derivatives from cyclopropenes.3 Cyclopropene went through copper catalyzed cycloisomerization forming furanyl copper intermediate M1, and subsequent transmetalation to palladium generating the key furanyl palladium intermediate M2, which underwent Heck, carbonylation, or dimerization reaction generating the tetrasubstituted furan and bifuran derivatives. In this Tandem Metal Relay Catalysis (TMRC) strategy, two different metals, i.e. copper and palladium, allow the activation and further functionalization of cyclopropenes in one pot. Herein, we present our recent efforts on the stereoselective synthesis of tetraarylethenes with an unprecedented successive double Cu/Pd relay catalysis.
Tetrasubstituted olefins are privileged scaffolds presented in biologically important drugs such as Tamoxifen or Vioxx, and natural products such as Nileprost analogues.4 The congested nature of the tetrasubstituted double bond makes tetrasubstituted olefins very useful molecules for applications in materials research such as molecular motors and liquid crystals.5 More importantly, tetraarylethene showed an unusual photophysical phenomenon of aggregation-induced emission (AIE) that is non-emissive or low-emissive in solution, but induced to emit efficiently by aggregate formation in the solid state.6,7 However most organic chromophores are highly emissive in solution but weakly luminescent in the solid state due to the ubiquitous aggregation-caused quenching (ACQ) effect.8 Because organic light-emitting materials are often used in the solid state, for example, as thin films in the fabrication of organic light-emitting diodes (OLEDs), the development of AIE active functional materials is very important. Recently the tetraphenylethene (TPE) type molecules have been widely applied in fluorescence sensors for ions and pH, fluorescent light-up bioprobes for analytical sensing and optical imaging, and multistimuli-responsive nanomaterials.9
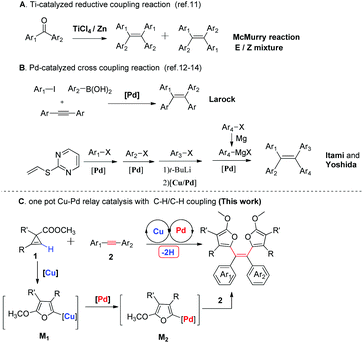
The regio- and stereoselective synthesis of tetraarylethenes is still one of the most challenging subjects in organic synthesis (Scheme 1).10 For the construction of double bonds, Wittig olefination and olefin metathesis remain the most important and general approaches. However, it is very difficult to apply these methods in the construction of sterically hindered tetraarylethene. McMurry reaction,11 in which two aromatic ketones are coupled to alkenes in the presence of titanium chloride and a reducing reagent, was utilized by Tang and others as a major approach for the synthesis of tetraarylethenes as the AIE luminogens. Under these conditions, mixtures of E/Z isomers are generally obtained. The regio- and stereoselectivity problem became the most challenging problems for the synthesis of tetraarylethene because of the similarity of different aromatic substituents. In 2003, Larock reported a palladium-catalyzed three-component cross-coupling reaction of an aryl iodide, an internal alkyne, and an arylboronic acid for the efficient synthesis of tetraarylethene.12 Itami and Yoshida made use of vinyl 2-pyrimidyl sulfide as a key template, sequentially installed four different aromatics onto the double bond through two Heck reactions and another two palladium-catalyzed cross-coupling reactions.13 These two methods demonstrated the brilliant applications of transition-metal catalyzed cross-coupling reactions between aromatic iodides and organometallic reagents.14 In this article, we report the synthesis of tetraarylethenes with an unprecedented Cu/Pd-catalyzed isomerization/insertion/oxidative coupling cascade reaction of cyclopropene and internal alkyne. The obtained tetraarylethenes embedded with two cis-furan rings represent a new type of AIE luminogens. The X-ray crystal structures, optical properties and structure–function relationships of these tetraarylethenes were also discussed. After detailed theoretical calculations and experimental mechanism study, an unprecedented successive double Cu/Pd transmetalation relay reaction mechanism was uncovered.
 |
| | Scheme 1 Synthesis of tetraarylethene. | |
Results and discussion
Initial studies and reaction optimization
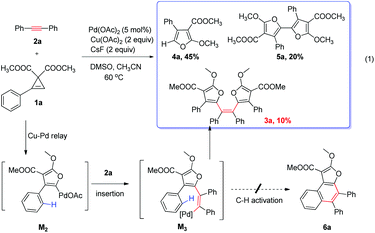
We began our study with the reaction of cyclopropene151a with diphenylacetylene 2a in the presence of copper and palladium catalysts (Scheme 2). It was anticipated that the furanyl palladium M2 was formed through a Cu/Pd relay process, followed by the insertion to the triple bond and intramolecular C–H activation to form the tricyclic aromatics 6a. However, in the complicated reaction mixture, two major byproducts, trisubstituted furan 4a and bifuran 5a were isolated in 45% and 20% yield respectively. Moreover, another new compound was isolated in less than 10% yield. To our great surprise, it is not the expected tricyclic product 6a, but the tetraarylethene 3a with two cis tetrasubstituted furan rings. This unexpected result prompted us to develop a general synthetic method for the challenging tetraarylethene. The alkyl substituted cyclopropene 1b was also amenable to this reaction (Table 1), and the isolated yellow solid tetraarylethene 3b showed a very bright color under irradiation of 365 nm.
 |
| | Scheme 2 Initial observation of the formation of tetraarylethene. | |
Table 1 Optimization of reaction conditionsa
|
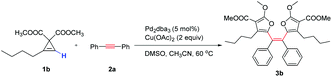
|
| Entry |
Catalyst |
Ligand |
Base |
Yieldb/% |
|
Reaction conditions: cyclopropene 1b (0.2 mmol), diphenylacetylene 2a (0.1 mmol), catalyst (5% mol), oxidant (0.2 mmol), base (0.1 mmol), DMSO (0.6 mmol), CH3CN (1 mL), 60 °C.
Isolated yield.
Without DMSO.
DMSO (1 mL) as the solvent.
Cyclopropene 1b (0.25 mmol).
Without Cu(OAc)2. DPEphos = bis(2-diphenylphosphinophenyl)ether; Boc-Pro = N-(tert-butoxycarbonyl)-L-proline.
|
| 1 |
Pd(OAc)2 |
|
— |
<5 |
| 2 |
Pd(OAc)2 |
|
CsF |
20 |
| 3 |
Pd(OAc)2 |
|
KF |
8 |
| 4 |
Pd(OAc)2 |
|
Cs2CO3 |
55 |
| 5 |
Pd(OAc)2 |
|
Et3N |
10 |
| 6c |
Pd(OAc)2 |
|
Cs2CO3 |
8 |
| 7d |
Pd(OAc)2 |
|
Cs2CO3 |
20 |
| 8 |
Pd(OAc)2 |
DPEphos |
Cs2CO3 |
28 |
| 9 |
Pd(OAc)2 |
PCy3 |
Cs2CO3 |
20 |
| 10 |
Pd(OAc)2 |
Boc-Pro |
Cs2CO3 |
22 |
| 11 |
PdCl2 |
|
Cs2CO3 |
12 |
| 12 |
PdCl2(PPh3)2 |
|
Cs2CO3 |
37 |
| 13 |
Pd2(dba)3 |
|
Cs2CO3 |
42 |
| 14e |
Pd2(dba)3 |
|
Cs2CO3 |
72 |
| 15 |
— |
|
Cs2CO3 |
0 |
| 16f |
Pd2(dba)3 |
|
Cs2CO3 |
0 |
Then we started to optimize the reaction conditions (Table 1, for details, see the ESI†). We noticed that the base is crucial for this transformation. No desired product was observed without a base (entry 1). A detailed screening of different bases found that Cs2CO3 was the best choice (entries 2–5). Removal of the DMSO additive or running the reaction in DMSO solvent both decreased the yield (entries 6 and 7). Then different palladium catalysts and ligands were examined. When different phosphine or amino acids were introduced as the ligand, the yield of 3b decreased (entries 8–10). The best conditions found consisted of performing the reaction in the presence of 5 mol% Pd2(dba)3 and one equivalent of Cs2CO3 at 60 °C. Under these conditions the target product 3b was isolated in 72% yield when a slightly excess amount of cyclopropene 1b was used (entry 14). Very importantly, only the cis-isomer was observed in this reaction. Only trisubstituted furan was formed in the presence of copper without the palladium catalyst (entry 15). No product was formed with the palladium catalyst alone (entry 16).
Substrate scope
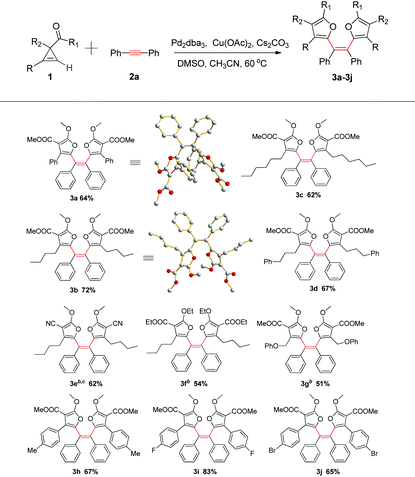
With the optimized reaction conditions established, the scope of various cyclopropenes was firstly investigated (Table 2). Both aliphatic cyclopropenes and aromatic cyclopropenes generated the corresponding cis products in good yields (3a–3j). The structures of 3a and 3b were confirmed by X-ray crystallography.16 Different ester groups, other electron-withdrawing groups such as CN, and the phenol group on the alkyl chain, were well tolerated in this reaction (3e–3g). Cyclopropenes with different substituents on the phenyl ring such as Br, F and methyl were also amenable to this transformation, giving corresponding products in 68–83% yields (3h–3j).
Table 2 Scope of cyclopropenesa
|
Reaction conditions: cyclopropene 1a–1j (0.25 mmol), diphenylacetylene 2a (0.1 mmol), Pd2(dba)3 (5% mol), Cu(OAc)2 (0.2 mmol), Cs2CO3 (0.1 mmol), DMSO (0.6 mmol), CH3CN (1 mL), 60 °C.
Pd2(dba)3 (10% mol).
|
|
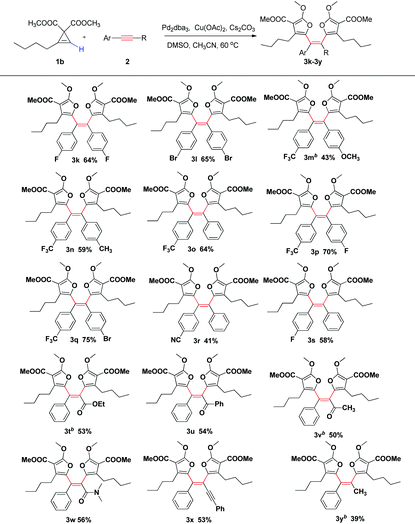
Then we investigated the scope of various disubstituted acetylenes (Table 3). Not only symmetric diaryl alkynes, but also asymmetric alkynes are all suitable for this transformation, and a large variety of different tetraarylethenes were prepared in moderate to good yields. A series of functional groups such as F, Br, CH3, OCH3, CF3, and CN, were all tolerated under these reaction conditions, thus allowing further functionalization if need be (3k–3s). Electro-deficient internal alkyne with an ester, ketone, or amide group also reacted efficiently to furnish the cis tetrasubstituted alkenes as the single isomer in moderate yields (3t–3w). The structure of 3w was also confirmed by single crystal X-ray analysis. To our delight, 1,4-diphenylbuta-1,3-diyne also reacted smoothly to give the corresponding cis tetrasubstituted olefin with an alkyne substituent in a respectable 53% yield (3x). The internal alkyne with one alkyl group is more challenging for this reaction, and the produced cis tetrasubstituted olefin was isolated in 39% with 10 mol% catalyst loading (3y). Thus this new reaction displayed high functional group tolerance and proved to be a general method for the synthesis of tetraarylethene derivatives.
Table 3 Scope of internal alkynesa
|
Reaction conditions: cyclopropene 1b (0.25 mmol), alkyne (0.1 mmol), Pd2(dba)3 (5% mol), Cu(OAc)2 (0.2 mmol), Cs2CO3 (0.1 mmol), DMSO (0.6 mmol), CH3CN (1 mL), 60 °C.
Pd2(dba)3 (10% mol).
|
|
Photophysical property study
Organic molecules bearing heteroaryls are making significant contributions in organic materials. The unprecedented cis-difuryl tetrasubstituted alkenes prompted us to investigate their photophysical properties. As indicated in Table 4, they all display an absorption at 369–390 nm in THF solution, which are greatly red shifted compared with TPE at 299 nm17 (For absorption and fluorescence spectra, see the ESI†). In the fluorescence spectra, they exhibited cyan to green emission at 492–510 nm with very low quantum yields (0.47–0.77%). The most interesting compounds are the alkyl substituted products (3b, 3o, 3w). The fluorescence quantum yields (ΦF) of these compounds in the aggregation state are 9.69%, 3.00%, and 5.07%, respectively, which are much higher than those in solution. In contrast, compound 3a synthesized from the phenyl substituted cyclopropene 1a, showed a very weak emission in solution, and almost no emission in the solid state. These differences could be explained by their crystal structures and packing mode of compounds 3a, 3b, and 3w. For compounds 3b and 3w, the butyl group serves an important hand to inhibit the possible intramolecular π–π interactions. At the same time, the intermolecular CH⋯O interaction makes the whole molecular pack into a relative rigid structure. For example, compound 3b takes a very interesting head-to-tail packing mode (Fig. 2b). For compound 3a, there is an obvious intramolecular π–π stacking between the phenyl group on one furan and the other furan ring. The distance between them is only 3.74 Å. This intramolecular interaction may contribute to their non-emission in the solid state.
Table 4 Optophysics data of the tetraarylethenes
| Compound |
λ
abs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/nm a/nm |
λ
emb/nm (in THF) |
λ
emb/nm (solid) |
Φ
F, (in THF)
|
Φ
F, solidd |
|
Only the longest absorption maxima are shown.
Excited at the longest absorption maxima.
Calculated using fluorescein as the standard.
Absolute quantum yield determined by a calibrated integrating sphere system.
|
|
3a
|
390 |
492 |
|
— |
— |
|
3b
|
379 |
500 |
501 |
0.47 |
9.69 |
|
3o
|
369 |
510 |
521 |
0.53 |
3.00 |
|
3w
|
390 |
495 |
514 |
0.77 |
5.07 |
To further study if these compounds are really AIE active, we chose a THF/H2O system to examine their photophysical properties, since these compounds are very soluble in THF, but completely insoluble in water. With increased fraction of water, the absorption of 3b in THF was almost the same (Fig. 1), but the fluorescence of 3b strengthened gradually. When the water fraction increased to 90%, the quantum yield increased by 45 times relative to its pure THF solution (Fig. 2a). These compounds represent a new type of AIE luminogens and could be potentially utilized as optoelectronic materials.
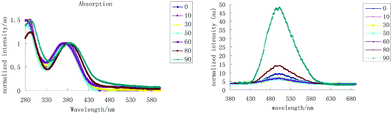 |
| | Fig. 1 Absorption (left) and fluorescence (right) spectra of 3b in THF/water with different fractions of water (fw%). | |
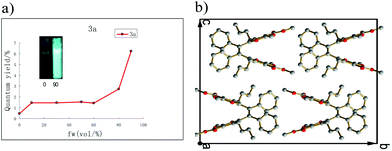 |
| | Fig. 2 (a) Variations of fluorescence quantum yields of 3b with water fractions in THF/water mixtures. (b) Head to tail packing of 3b in the solid state. | |
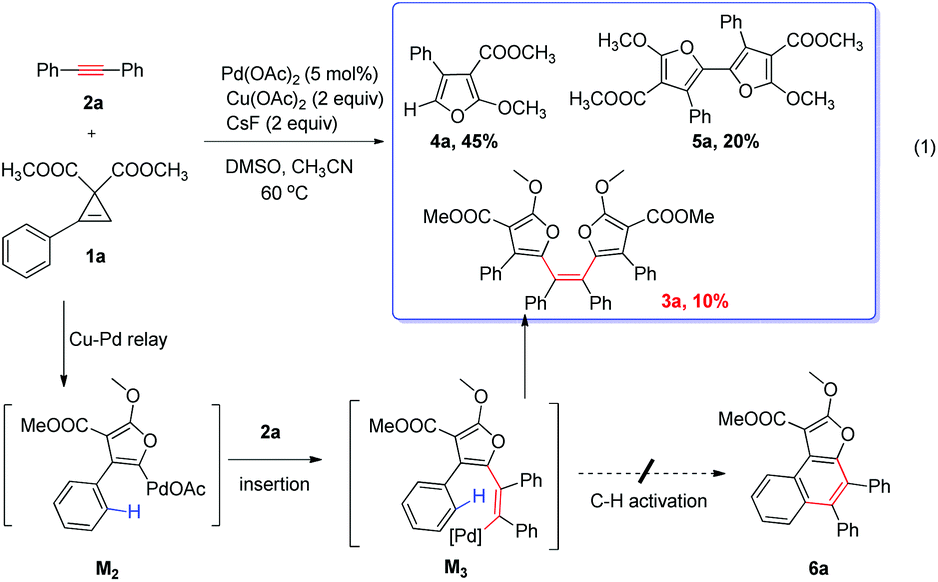
Mechanism discussion
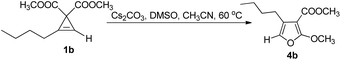
Then we started to explore the reaction mechanism. Trisubstituted furan 4b could not react with 2a to form the product under standard conditions, indicating that furan is not the reaction intermediate (eqn (1)). Thus a stepwise reaction is not likely and furanyl copper to furanyl palladium transmetalation (M1 to M2) mechanism is quite possible.| |  | (1) |
The first step of this transformation is the copper catalyzed cycloisomerization to form furanyl copper intermediate M1. Importantly, we noticed that the reaction rate of Cu(I) catalyzed cyclopropene isomerization was much faster than that of Cu(II).18 Then we carried out the kinetic study of the first step reaction (Fig. 3). The in situ formed CuOAc catalyzed the isomerization reaction efficiently and completed in less than 1 hour, but the reaction catalyzed by Cu(OAc)2 was much slower and there is an obvious induction period of about 30 minutes. These data indicated that Cu(I) might be the actual catalyst and Cu(II) could be reduced to Cu(I) to catalyze this reaction by a base, Pd(0), or even solvents.19,2c
| |  | (2) |
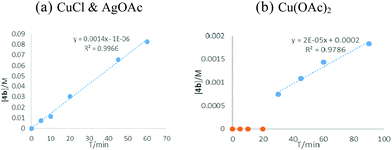 |
| | Fig. 3 Kinetic study of Cu(I) and Cu(II) catalyzed cycloisomerization of cyclopropene. | |
Since only Pd(0) and Cu(II) were used in this reaction and Pd(II) is required in the transmetalation step, the redox reaction between Pd(0) and Cu(II) possibly led to Pd(II) and Cu(I) simultaneously. Then we investigated the redox reaction of Cu(II) and Pd(0) in the presence or absence of Cs2CO3 by X-ray photoelectron spectroscopy (for details, see the ESI†). Pd2dba3 and Cu(OAc)2 were reacted under a N2 atmosphere at 60 °C for 30 min, after removal of the solvent, the obtained black solid was subjected to XPS analysis. From the copper spectra, binding energy of Cu 2p3/2 was 932.0 eV and Cu 2p1/2 was 954.1 eV, which were the typical binding energies for Cu(I) species.20 At the same time, Pd(II) species were observed with the binding energy of 338.4 eV (Pd 3d5/2) and 343.6 eV (Pd 3d3/2). In addition, in the reaction without a base, palladium black was also observed (336.4 eV for Pd 3d5/2 and 341.8 eV for Pd 3d3/2),20b but no palladium black formation was observed in the reaction with Cs2CO3. These results also highlighted the importance of the base in the standard reactions.
The above mechanistic experiments imply that Cu(II) is not likely to be the actual catalyst in cyclopropene cycloisomerization, and it needs to be transformed to the actual catalyst, most likely Cu(I), before the reaction can occur. To investigate the origin of this advantage of Cu(I) over Cu(II) in cyclopropene cycloisomerization at the molecular level, we further directly compared the reactivity of Cu(I) and Cu(II) employing computational density functional theory (DFT) modeling. The first step of cyclopropene cycloisomerization is the cyclopropene ring opening by breaking the corresponding C–C bond. As shown in Fig. 4, we discovered that the Cu(I) catalyst CuOAc is kinetically more reactive in cyclopropene ring opening than the Cu(II) catalyst Cu(OAc)2, by having a substantially lower ring opening barrier by 5.3 kcal mol−1 from Cu(I)-cyclopropene reactant complex A to ring-opened Cu(I) enolate B through transition state TSA–B. Thus the experimental implication of an actual Cu(I) catalyst is supported by DFT modeling. Inspecting the transition states and products of ring opening processes as shown in Fig. 5, which constitute the lowest energy reaction profiles for both Cu(I) and Cu(II) systems resulting from our extensive reaction pathway search,21 it is notable that during the C–C cleavage, the acetate ligand of the Cu catalyst migrates from the Cu to the C2 position. This mechanistic feature of cyclopropene ring opening is consistent with a very recent computational study by Xia et al. on the mechanism of cyclopropene cycloisomerization to produce trisubstituted furan catalyzed by a CuI catalyst.22 However, as shown below, the subsequent mechanism after ring opening to generate furanyl copper intermediate M1 in this work is completely different from that of producing trisubstituted furan found earlier.22
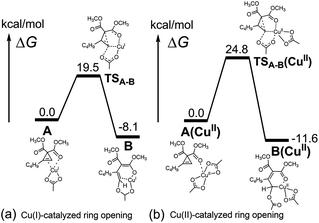 |
| | Fig. 4 Comparison between DFT calculations of (a) Cu(I)-catalyzed cyclopropene ring opening and (b) Cu(II)-catalyzed cyclopropene ring opening. | |
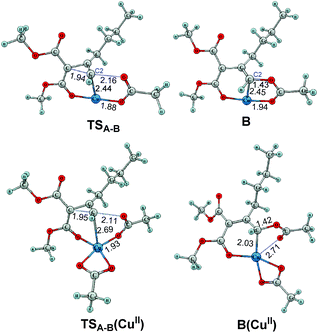 |
| | Fig. 5 DFT optimized structures of transition state and intermediate in cyclopropene ring opening mechanism shown in Fig. 4 for Cu(I) and Cu(II) catalytic systems. | |
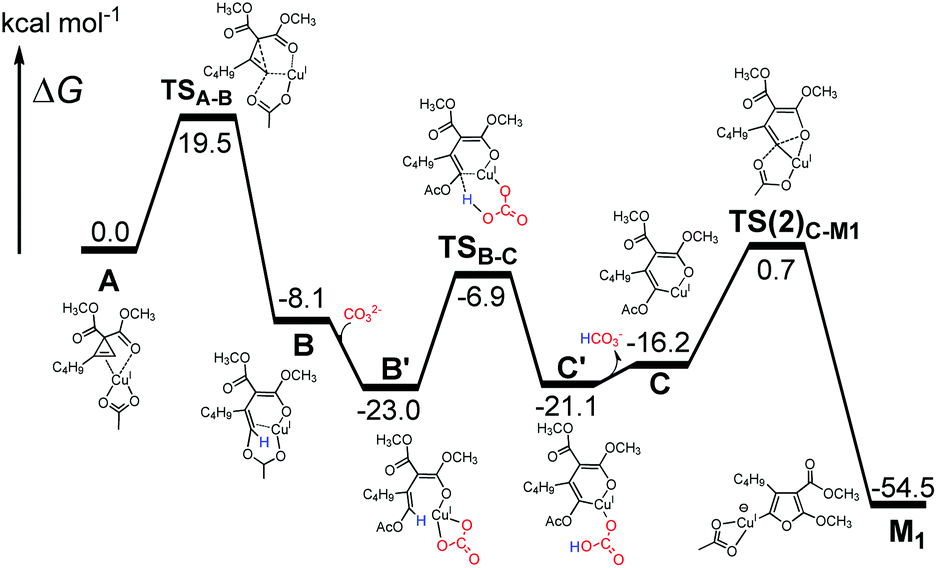
From DFT theoretical modeling, a detailed reaction pathway to generate furanyl copper intermediate M1 from cyclopropene–Cu(I) complex A is depicted in Fig. 6. After cyclopropene ring opening occurs to form intermediate B, we discovered that the added carbonate base took part in the reaction by abstracting the proton from the C2 position of Bvia transition state TSB–C merely with a reaction barrier of 16.1 kcal mol−1. Thus, one important deprotonative role played by the carbonate base (Cs2CO3), which is the key additive in the reaction, is discovered from our DFT calculations. Without this pivotal deprotonation process, as in the case of the CuI catalyzed cyclopropene cycloisomerization found by Xia et al.,22 furan rather than furanyl copper intermediate M1 is found to be generated directly from intermediate B, through a very high-lying transition state (23.6 kcal mol−1 higher than TSB–C) of O-attack at the C2 position. Recalling that furan has been ruled out experimentally to act as an intermediate in the reactions (eqn (1)), deprotonation of B is crucial for the formation of furanyl copper intermediate M1. After this deprotonation by an added base, alkenyl copper intermediate C is generated and subsequent cyclization by O-attack of C2 position to form M1 can be accomplished through a second-order saddle point TS(2)C–M123 on the potential energy surface by overcoming a barrier of 16.9 kcal mol−1. Generally, the relatively low calculated reaction barriers below 20 kcal mol−1 in this reaction mechanism of M1 formation shown in Fig. 6 is consistent with the experimentally observed fast reaction rate under mild reaction condition (60 °C).
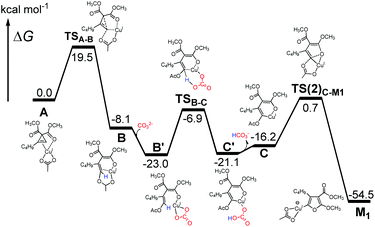 |
| | Fig. 6 DFT calculated reaction profile from cyclopropene–Cu(I) complex A to key furanyl copper intermediate M1. | |
After furanyl copper intermediate M1 is produced, transmetalations are possible. To explore whether transmetalations are thermodynamically feasible, we calculated thermodynamic free energy driving forces of first and second transmetalations the results of which are depicted in Fig. 7. We can see that the first and second transmetalations from Cu site of M1 to Pd catalytic center are all thermodynamically feasible processes, with the former transmetalation bearing a relatively larger thermodynamic driving force. Importantly, as shown in Fig. 7b, compared with alternative aromatic C–H activation, the second transmetalation is more favorable thermodynamically. This explains well why C–H activation was not observed in our experiments (Scheme 2).
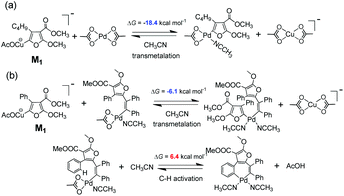 |
| | Fig. 7 DFT calculated free energy driving forces of (a) first transmetalation, and (b) second transmetalation in comparison with C–H activation. | |
Based on these results and our previous reactions, a plausible Cu/Pd relay catalyzed cross-coupling reaction was proposed in Scheme 3. Cu(OAc)2 reacted with Pd2dba3 to form Pd(II), and was itself reduced to CuOAc. Cyclopropene reacted with CuOAc through the ring opening reaction to obtain the ring-opened Cu(I) enolate intermediate B. With the help of a base (stronger and more beneficial CO32−, or weaker AcO−) in the reaction system, deprotonation process occurs to generate alkenyl copper intermediate C. Subsequent cyclization by O-attack generates the furanyl copper intermediate M1. It was transmetalated to a Pd(II) catalyst, generating key furanyl palladium M2, which went through cis alkyne insertion to form the alkenyl palladium M3, followed by second Cu/Pd transmetalation with M1 and the subsequent reductive elimination to afford the final products 3 and Pd(0). Finally a redox reaction between Pd(0) and Cu(II) would lead to both Pd(II) catalyst regeneration and more Cu(I) catalysts, all participating in the next catalytic cycle. Importantly, the cyclopropene cycloisomerization mechanism revealed in this work is characterized by deprotonation after cyclopropene ring opening, and was found optimal for the generation of furanyl copper intermediate M1, while the mechanism without deprotonation as recently proposed by Xia et al.22 is suitable for producing trisubstituted furan, but cannot cope with the generation of furanyl copper intermediate M1 necessitated in this study as well as in our previous work.3
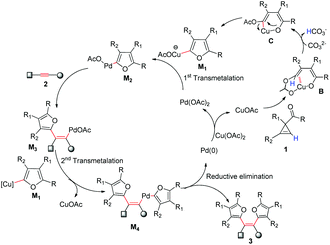 |
| | Scheme 3 Proposed mechanism of tetraarylethenes production involving Cu/Pd transmetalation relay catalysis based on experiments and theoretical modeling results. | |
Conclusions
In summary, a new Cu/Pd-catalyzed reaction between cyclopropene and internal alkyne has been successfully developed. Different from many other transition-metal catalyzed C–H activation reactions, this oxidative insertion coupling reaction proceeded under very mild conditions. Based on the experimental and computational studies, Cu(I) and Pd(II) were identified as the actual catalysts, and a novel deprotonative Cu-catalyzed cyclopropene cycloisomerization mechanism was proposed for the discovered reaction. The subsequent thermodynamically favored Cu/Pd transmetalations afford the carbon–palladium bond, which is the major reason for the mildness of the reaction. The successive double transmetalation relay is the most important feature of this reaction. A wide variety of tetrasubstituted alkenes, especially tetraarylethenes with two cis tetrasubstituted furans, were prepared conveniently, efficiently, and stereoselectively by this one-pot cascade reaction. The photophysical properties of these novel tetraarylethenes were fully characterized and these tetraarylethenes proved to be good AIE luminogens. This new type of tetraarylethenes will potentially have more applications as the AIE active materials, and we believe that this synthetic method should serve as a new powerful tool for the synthesis of highly functionalized tetrasubstituted alkenes.
Acknowledgements
We are grateful for the fundamental research and subject construction funds of Shandong University (no. 2014JC008, 104.205.2.5) and National Natural Science Foundation of China (no. 21290194, 21221002 and 21473215). We thank Prof. Dr Di Sun for the analysis of X-ray structure.
Notes and references
- For reviews, see:
(a) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187 CrossRef CAS PubMed;
(b) J. M. Lee, Y. Na, H. Han and S. Chang, Chem. Soc. Rev., 2004, 33, 302 RSC.
-
(a) K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 7567 CrossRef CAS PubMed;
(b) A. S. K. Hashmi, M. Ghanbari, M. Rudolph and F. Rominger, Chem. – Eur. J., 2012, 18, 8113 CrossRef CAS PubMed;
(c) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Ackermann, J. D. B. Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133 CrossRef CAS PubMed;
(d) P. Hu, M. Zhang, X. Jie and W. Su, Angew. Chem., Int. Ed., 2012, 51, 227 CrossRef CAS PubMed;
(e) Y.-F. Wang, K. K. Toh, J.-Y. Lee and S. Chiba, Angew. Chem., Int. Ed., 2011, 50, 5927 CrossRef CAS PubMed;
(f) B. Chen and S. Ma, Chem. – Eur. J., 2011, 17, 754 CrossRef CAS PubMed;
(g) L. Zhou, F. Ye, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2010, 132, 13590 CrossRef CAS PubMed;
(h) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Rudolph, T. D. Ramamurthi and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8243 CrossRef CAS PubMed;
(i) L. J. Gooβen, G. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef PubMed.
-
(a) C. Song, L. Ju, M. Wang, P. Liu, Y. Zhang, J. Wang and Z. Xu, Chem. – Eur. J., 2013, 19, 3584 CrossRef CAS PubMed;
(b) C. Song, S. Dong, L. Feng, X. Peng, M. Wang, J. Wang and Z. Xu, Org. Biomol. Chem., 2013, 11, 6258 RSC;
(c) C. Song, D. Sun, X. Peng, J. Bai, R. Zhang, S. Hou, J. Wang and Z. Xu, Chem. Commun., 2013, 49, 9167 RSC;
(d) C. Song, J. Wang and Z. Xu, Org. Biomol. Chem., 2014, 12, 5802 RSC;
(e) F. Wei, H. Li, C. Song, Y. Ma, L. Zhou, C.-H. Tung and Z. Xu, Org. Lett., 2015, 17, 2860 CrossRef CAS PubMed;
(f) S. Zhang, Y. Chen, J. Wang, Y. Pan, Z. Xu and C.-H. Tung, Org. Chem. Front., 2015, 2, 578 RSC.
-
(a) D. W. Robertson, J. A. Katzenellenbogen, J. R. Hayes and B. S. Katzenellenbogen, J. Med. Chem., 1982, 25, 167 CrossRef CAS;
(b) F. Caturla, M. Amat, R. F. Reinoso, M. Córdoba and G. Warrellow, Bioorg. Med. Chem. Lett., 2006, 16, 3209 CrossRef CAS PubMed;
(c) A. Takahashi, Y. Kirio, M. Sodeoka, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1989, 111, 643 CrossRef CAS , and references therein.
-
(a) R. Eelkema, M. M. Pollard, N. Katsonis, J. Vicario, D. J. Broer and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 14397 CrossRef CAS PubMed;
(b) N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- For reviews on AIE effect, see:
(a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC;
(b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC;
(c) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441 CrossRef CAS PubMed;
(d) J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483 RSC.
-
(a) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed;
(b) S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef PubMed.
-
(a) M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858 RSC;
(b) J. Liang, R. T. K. Kwok, H. Shi, B. Z. Tang and B. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 8784 CrossRef CAS PubMed;
(c) H. Wang, J. Liu, A. Han, N. Xiao, Z. Xue, G. Wang, J. Long, D. Kong, B. Liu, Z. Yang and D. Ding, ACS Nano, 2014, 8, 1475 CrossRef CAS PubMed;
(d) M. Gao, C. K. Sim, C. W. T. Leung, Q. Hu, G. Feng, F. Xu, B. Z. Tang and B. Liu, Chem. Commun., 2014, 50, 8312 RSC;
(e) Y. Liu, C. Deng, L. Tang, A. Qin, R. Hu, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2011, 133, 660 CrossRef CAS PubMed;
(f) Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed.
-
(a) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed;
(b) Y. Nishihara, Y. Okada, J. Jiao, M. Suetsugu, M.-T. Lan, M. Kinoshita, M. Iwasaki and K. Takagi, Angew. Chem., Int. Ed., 2011, 50, 8660 CrossRef CAS PubMed;
(c) Y. Nishihara, M. Miyasaka, M. Okamoto, H. Takahashi, E. Inoue, K. Tanemura and K. Takagi, J. Am. Chem. Soc., 2007, 129, 12634 CrossRef CAS PubMed.
-
(a) S. Gauthier, J. Mailhot and F. Labrie, J. Org. Chem., 1996, 61, 3890 CrossRef CAS;
(b) A. Detsi, M. Koufaki and T. Calogeropoulou, J. Org. Chem., 2002, 67, 4608 CrossRef CAS PubMed.
-
(a) C. Zhou, D. E. Emrich and R. C. Larock, Org. Lett., 2003, 5, 1579 CrossRef CAS PubMed;
(b) C. Zhou and R. C. Larock, J. Org. Chem., 2005, 70, 3765 CrossRef CAS PubMed;
(c) C. Zhou and R. C. Larock, Org. Lett., 2005, 7, 259 CrossRef CAS PubMed.
-
(a) K. Itami, M. Mineno, N. Muraoka and J. Yoshida, J. Am. Chem. Soc., 2004, 126, 11778 CrossRef CAS PubMed;
(b) K. Itami, T. Kamei and J. Yoshida, J. Am. Chem. Soc., 2003, 125, 14670 CrossRef CAS PubMed.
-
(a) V. Kotek, H. Dvorakova and T. Tobrman, Org. Lett., 2015, 17, 608 CrossRef CAS PubMed;
(b) B. M. Monks and S. P. Cook, J. Am. Chem. Soc., 2012, 134, 15297 CrossRef CAS PubMed;
(c) M. Jiang, T. Jiang and J.-E. Bäckwall, Org. Lett., 2012, 14, 3538 CrossRef CAS PubMed;
(d) N. Sakai, R. Komatsu, N. Uchida, R. Ikeda and T. Konakahara, Org. Lett., 2010, 12, 1300 CrossRef CAS PubMed;
(e) Y. Cheng, Z. Duan, L. Yu, Z. Li, Y. Zhu and Y. Wu, Org. Lett., 2008, 10, 901 CrossRef CAS PubMed;
(f) J. Song, Q. Shen, F. Xu and X. Lu, Org. Lett., 2007, 9, 2947 CrossRef CAS PubMed;
(g) A. Pinto, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2007, 46, 3291 CrossRef CAS PubMed;
(h) A. Pinto, L. Neuville, P. Retailleau and J. Zhu, Org. Lett., 2006, 8, 4927 CrossRef CAS PubMed;
(i) G. Zhu, X. Tong, J. Cheng, Y. Sun, D. Li and Z. Zhang, J. Org. Chem., 2005, 70, 1712 CrossRef CAS PubMed.
- For recent reviews on cyclopropenes, see:
(a) J. M. Fox and N. Yan, Curr. Org. Chem., 2005, 9, 719 CrossRef CAS;
(b) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed;
(c) I. Marek, S. Simaan and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364 CrossRef CAS PubMed;
(d) Z.-B. Zhu, Y. Wei and M. Shi, Chem. Soc. Rev., 2011, 40, 5534 RSC;
(e) F. Miege, C. Meyer and J. Cossy, Beilstein J. Org. Chem., 2011, 7, 717 CrossRef CAS PubMed. For recent advances, see:
(f) V. Tarwade, X. Liu, N. Yan and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 5382 CrossRef CAS PubMed;
(g) D. H. T. Phan, K. G. M. Kou and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16354 CrossRef CAS PubMed;
(h) C. Li, H. Zhang, J. Feng, Y. Zhang and J. Wang, Org. Lett., 2010, 12, 3082 CrossRef CAS PubMed;
(i) C. Li, Y. Zeng, H. Zhang, J. Feng, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 6413 CrossRef CAS PubMed;
(j) Y. Liu and S. Ma, Chem. Sci., 2011, 2, 811 RSC;
(k) F. Miege, C. Meyer and J. Cossy, Angew. Chem., Int. Ed., 2011, 50, 5932 CrossRef CAS PubMed;
(l) J. Li, C. Sun, S. Demerzhan and D. Lee, J. Am. Chem. Soc., 2011, 133, 12964 CrossRef CAS PubMed;
(m) K. Krämer, P. Leong and M. Lautens, Org. Lett., 2011, 13, 819 CrossRef PubMed;
(n) F. Liu, X. Bugaut, M. Schedler, R. Fröhlich and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 12626 CrossRef CAS PubMed;
(o) X. Bugaut, F. Liu and F. Glorius, J. Am. Chem. Soc., 2011, 133, 8130 CrossRef CAS PubMed;
(p) L. H. Phun, J. Aponte-Guzman and S. France, Angew. Chem., Int. Ed., 2012, 51, 3198 CrossRef CAS PubMed;
(q) Y. Liu and S. Ma, Org. Lett., 2012, 14, 720 CrossRef CAS PubMed;
(r) X. Xie, Y. Li and J. M. Fox, Org. Lett., 2013, 15, 1500 CrossRef CAS PubMed;
(s) S. Ni, J. Chen and S. Ma, Org. Lett., 2013, 15, 3290 CrossRef CAS PubMed;
(t) Y. Yang, Z. Zhang, X. Zhang, D. Wang, Y. Wei and M. Shi, Chem. Commun., 2014, 50, 115 RSC;
(u) A. Parra, L. Amenós, M. Guisán-Ceinos, A. López, J. L. G. Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833 CrossRef CAS PubMed;
(v) B. Tian, Q. Liu, X. Tong, P. Tian and G.-Q. Lin, Org. Chem. Front., 2014, 1, 1116 RSC;
(w) H. Zhang, K. Wang, B. Wang, H. Yi, F. Hu, C. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 13234 CrossRef CAS PubMed;
(x) A. S. K. Hashmi, M. A. Grundl, D. Riedel, M. Rudolph and J. W. Bats, Organometallics, 2012, 31, 523 CrossRef CAS;
(y) A. S. K. Hashmi, F. Naumann, A. R. Nass, A. Degen, M. Bolte and J. W. Bats, Chem. – Eur. J., 1999, 5, 2836 CrossRef CAS;
(z) H. Zhang, B. Wang, H. Yi, Y. Zhang and J. Wang, Org. Lett., 2015, 17, 3322 CrossRef CAS PubMed.
- CCDC 1021785–1021787 (3a, 3b, 3w) contains the supplementary crystallographic data for this paper.
- N.-W. Tseng, J. Liu, J. C. Y. Ng, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2012, 3, 493 RSC.
- Copper(I) catalyzed cycloisomerization of cyclopropene into furan, see:
(a) J. Chen and S. Ma, Chem. – Asian J., 2010, 5, 2415 CrossRef CAS PubMed;
(b) S. Ma and J. Zhang, J. Am. Chem. Soc., 2003, 125, 12386 CrossRef CAS PubMed;
(c) S. Chuprakov and V. Gevorgyan, Org. Lett., 2007, 9, 4463 CrossRef CAS PubMed.
- Copper(II) was reduced in situ to Cu(I), see:
(a) P. C. Too, S. H. Chua, S. H. Wong and S. Chiba, J. Org. Chem., 2011, 76, 6159 CrossRef CAS PubMed;
(b) G. Zhang, H. Yi, G. Zhang, Y. Deng, R. Bai, H. Zhang, J. T. Miller, A. J. Kropf, E. E. Bunel and A. Lei, J. Am. Chem. Soc., 2014, 136, 924 CrossRef CAS PubMed.
-
(a) C. Qi, X. Sun, C. Lu, J. Yang, Y. Du, H. Wu and X.-M. Zhang, J. Organomet. Chem., 2009, 694, 2912 CrossRef CAS PubMed;
(b) H. Liu, J. P. Wei, Z. Qiao, Y. Fu and X. Jiang, Chem. – Eur. J., 2014, 20, 8308 CrossRef CAS PubMed;
(c) M.
L. Kantam, R. Chakravarti, B. Neelima, R. Arundhati and B. Sreedhar, Appl. Catal., A, 2007, 333, 136 CrossRef CAS PubMed.
- For Cu(I) system, we also found alternative ring opening pathway by locating the ring opening transition state without migrating acetate ligand to C2 position, but this transition state is 11.5 kcal mol−1 higher than TSA–B in energy, thus this pathway is unfavorable.
- G. P. Huang and Y. Z. Xia, ACS Catal., 2015, 5, 859 CrossRef CAS.
- We have tried to use various initial geometric structures to optimize the transition state (first-order saddle point), but after intensive search for transition state, we failed to locate any proper TS but found that second-order saddle point is the viable transition structure for O-attack to form M1.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and analytical data. CCDC 1021785–1021787. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00205b |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.