
Synthesis and reactivity of bis(2,2,2-trifluoroethyl)cyclopropane-1,1-dicarboxylates†
Erin L.
Armstrong
and
Michael A.
Kerr
*
University of Western Ontario, Department of Chemistry, London, Ontario, Canada N6A 5B7. E-mail: makerr@uwo.ca
First published on 14th July 2015
Abstract
Diazo-bis-222-trifluoroethylmalonate was treated with a variety of alkenes under the influence of rhodium catalysis to yield the corresponding bis(2,2,2-trifluoroethyl)cyclopropane-1,1-dicarboxylates in good yields. These donor–acceptor cyclopropanes were found to have greatly enhanced electrophilicity in reactions with indole.
The use of donor–acceptor cyclopropanes as synthetic starting materials has seen a surge in popularity in recent years.1 Aside from interesting and useful methodologies, they have played integral roles in the total synthesis of complex natural products.2 Research into expanding the possibilities of donor–acceptor reactivity has typically been focused on (1) altering the donor group on the cyclopropane, (2) altering the catalysts (usually but not always Lewis acids) or (3) using unusual (i.e. hyperbaric) reaction conditions3 (Fig. 1). Less work has been done which investigates changes to the acceptor group, which is normally a single carbonyl or geminal dicarbonyl moiety.
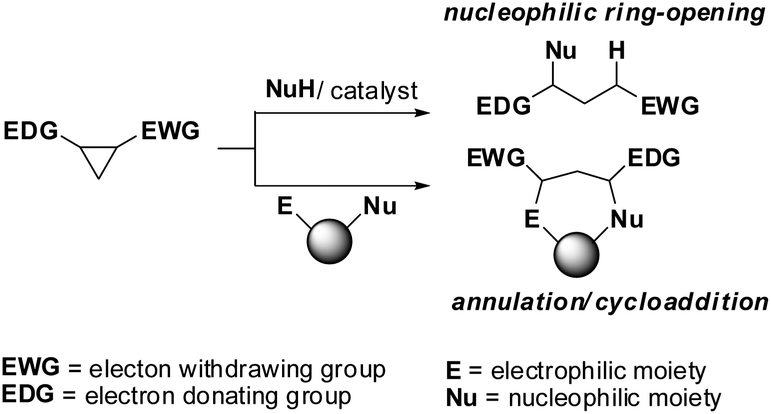 | ||
| Fig. 1 The reactivity of donor–acceptor cyclopropanes. | ||
During efforts to promote a stubbornly sluggish annulation reaction we became aware of the reports by Waser4 and Trost5 in which a cyclopropane equipped with a geminal bis(2,2,2-trifluoroethyl) dicarboxylate moiety was used in place of the more common bisdialkyldicarboxylate groups, in order to improve reactivity and yields (Scheme 1). The Waser work used Lewis acid activation to promote nucleophilic additions by indoles to an imido substituted cyclopropane 1 to give adducts 2 whereas the Trost chemistry involved π-allyl chemistry of a vinyl cyclopropane 3 and subsequent formal cycloadditions of compounds such as 4 to yield adducts 5. To our knowledge these are the only examples of cyclopropanes used in synthesis which are equipped with such electron withdrawing groups. The cyclopropanes in the Waser and Trost work were very specific in nature (phthalimido or vinyl substituted); however for our purposes we required cyclopropanes with a more general substitution. To our knowledge, a general preparation of this class of cyclopropanes has not been reported and so we have investigated the synthesis and reactivity of a variety of cyclopropanes such as 8 using a rhodium carbenoid insertion of diazomalonates 7 to alkenes. We report the results of this research herein.
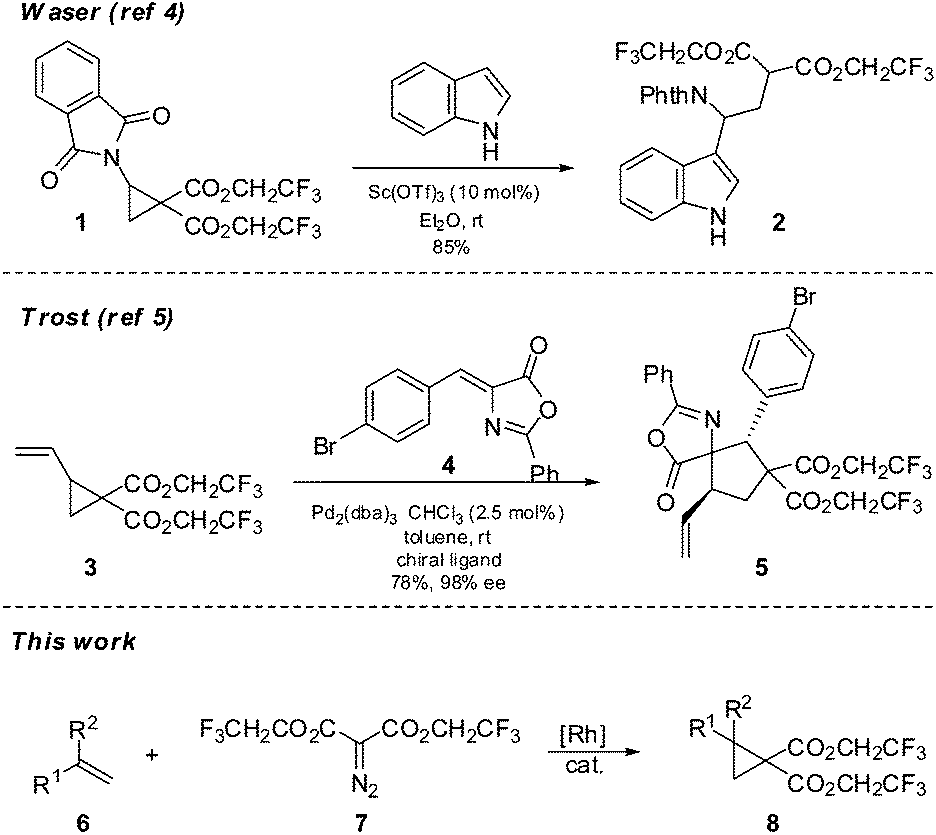 | ||
| Scheme 1 The prior use of fluoroester-substituted donor–acceptor cyclopropanes. | ||
Initially we felt that the most direct synthesis of the target cyclopropanes 10 would be a saponification of the methyl ester derivatives 9 followed by a simple reesterification with trifluoroethanol. While this certainly worked, the reaction sequence was plagued by poor recovery of the diacid in the saponification step and purification issues during the reesterification step when using coupling reagents. At this time we turned our attention to the use of carbenoid insertion using fluorinated diazomalonate precursors. It should be noted that Waser also employed this strategy for the synthesis of 1 with the more reactive phthalimidoalkenes.
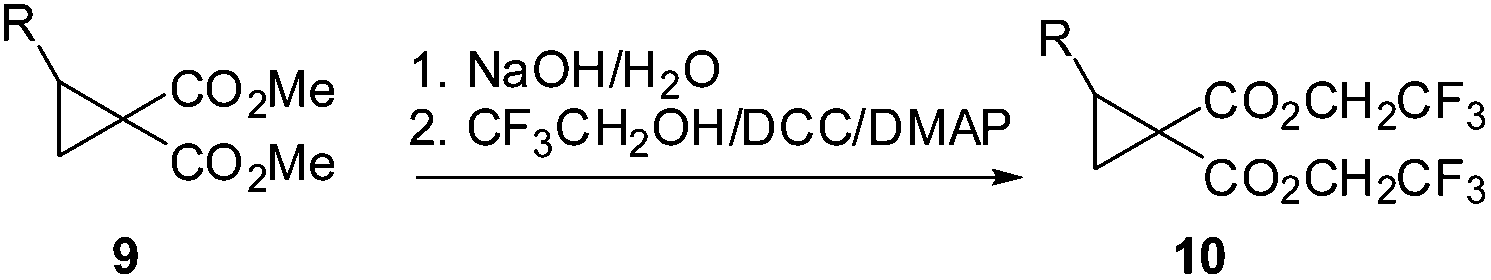
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 produced a trace amount of product 10a while Rh2(TFA)47 as the catalyst resulted in decomposition of the starting materials. It was quickly determined that Rh2(esp)28 was an excellent catalyst for the desired transformation, producing the target compound in 94% yield. Davies Rh2(S-DOSP)49 catalyst was used with an eye toward enantioselection, however the chemical yields were so low that the enantioselectivity was a moot point (Table 1).
6 produced a trace amount of product 10a while Rh2(TFA)47 as the catalyst resulted in decomposition of the starting materials. It was quickly determined that Rh2(esp)28 was an excellent catalyst for the desired transformation, producing the target compound in 94% yield. Davies Rh2(S-DOSP)49 catalyst was used with an eye toward enantioselection, however the chemical yields were so low that the enantioselectivity was a moot point (Table 1).
|
|
|||
|---|---|---|---|
| Entry | Rh cata | Time (h) | Yield (%) |
| a Reactions were performed by adding the diazomalonate to a solution of styrene and the catalyst in CH2Cl2 at 0 °C. The ice bath was removed and the mixture was allowed to stir for the indicated time. | |||
| 1 | Rh2(OAc)4 | 48 h | Trace |
| 2 | Rh2(TFA)4 | 24 h | Decomposition |
| 3 | Rh2(S-DOSP)4 | 48 h | 33% |
| 4 | Rh2(esp)2 | 5 h | 94% |
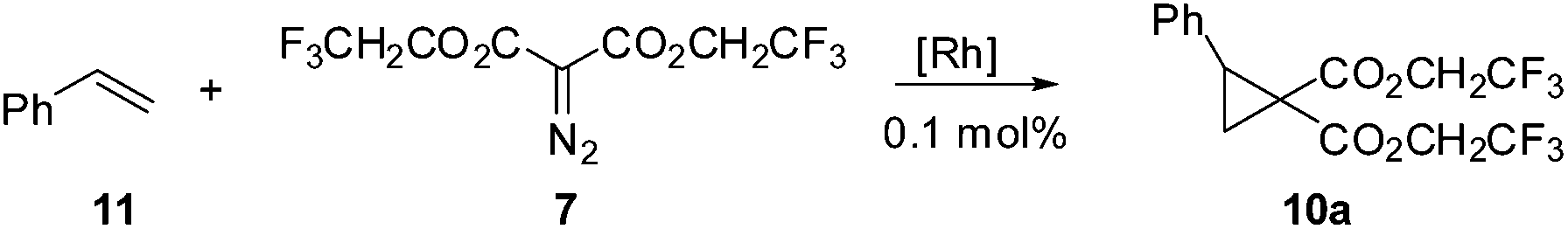
With suitable conditions in hand, we turned our attention to an evaluation of the substrate scope (Scheme 2). All of the styrenes surveyed produced the expected cyclopropanes 10 in good to excellent yields. Although a rigorous study was not done, it appears that the more electron poor styrenes (giving adducts 10c–g) give reduced yields (perhaps not unexpectedly due to the electrophilic nature of the rhodium carbenoid). Simple terminal alkenes produced adducts 10i and 10j in good to acceptable yields.
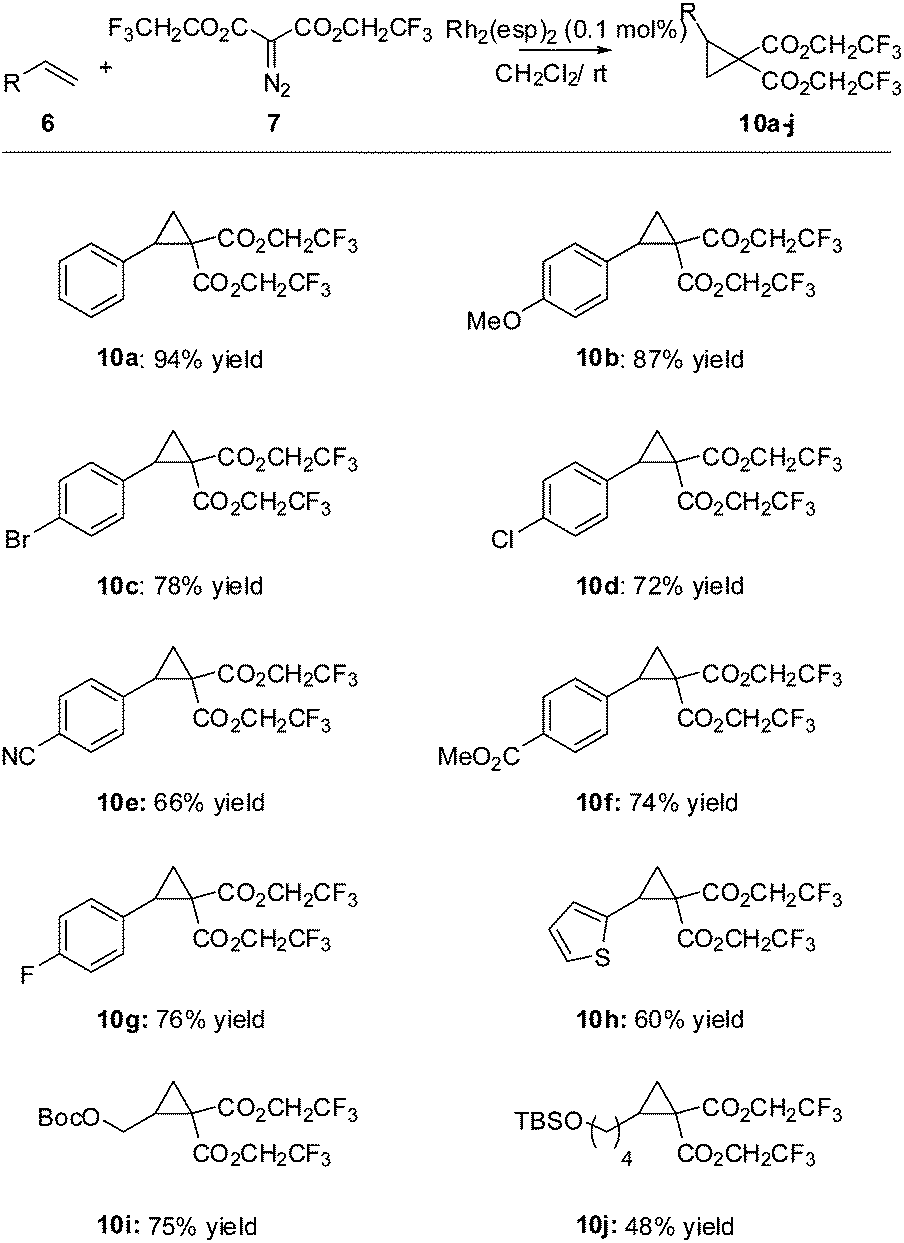 | ||
| Scheme 2 Substrate scope. | ||
Some years ago we reported the first examples of the nucleophilic ring-opening of cyclopropanes by indoles.10 This has proven to be a reliable reaction11 and we have found this to be a good way to evaluate the electrophilicity of donor–acceptor cyclopropanes. To this end, we subjected several of the geminal fluoroester substituted cyclopropanes from Scheme 2 as well as the corresponding methyl ester counterparts to the reaction with N-methylindole under our previously reported conditions of 10% Yb(OTf)3 in acetonitrile (Table 2). All of the cyclopropanes produced the expected adducts in good to excellent yields however the reaction times indicate a greatly enhanced reactivity of the fluorinated substrates. In the case of the parent phenyl cyclopropane 10a, the reaction time at room temperature was reduced from 48 hours to 90 minutes. The substrate bearing the activating methoxy group gave similar yields in both cases with the fluorinated esters resulting in a threefold reduction in reaction time. The yields were the same within experimental error. In the case of the nitrophenyl analogs 10k and 12c,12 expected to be significantly deactivated, the reaction time under refluxing conditions was reduced from 4 hours to 90 minutes. While not an extensive study, it is clear that the electrophilic nature of this class of donor–acceptor cyclopropanes is greatly enhanced by turning to a trifluoroethyl ester in place of a simple alkyl ester.
|
|
||||
|---|---|---|---|---|
| Entry | Cyclopropane | Temp. | Time | Yield (%) |
| 1 | 10a | rt | 1.5 h | 94 |
| 2 | 10b | rt | 50 min | 74 |
| 3 | 10k | Reflux | 1.5 h | 72 |
| 4 | 12a | rt | 48 h | 96(borsm) |
| 5 | 12b | rt | 3 h | 70 |
| 6 | 12c | Reflux | 4 h | 71 |
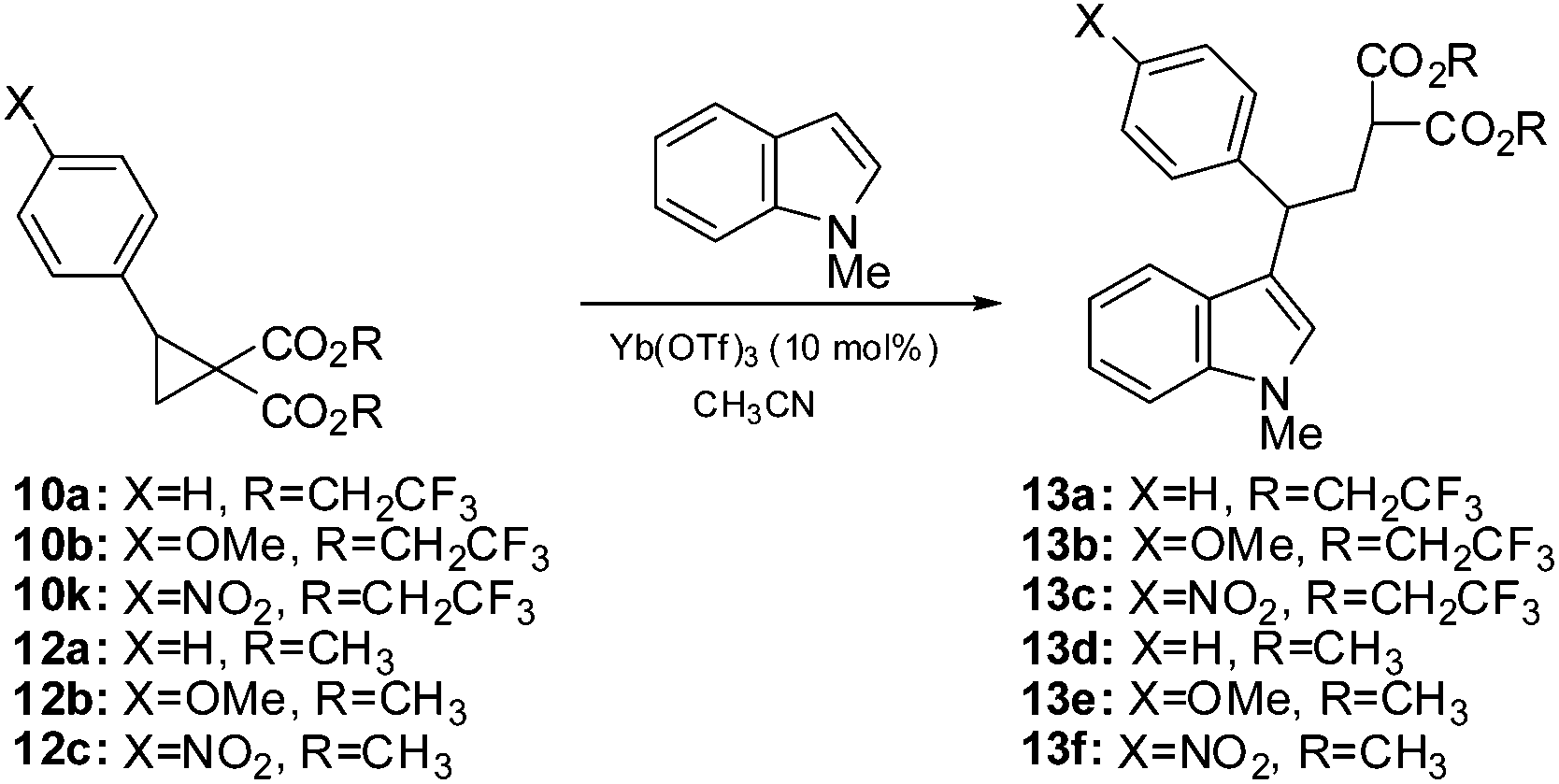
Conclusions
In conclusion, we have reported a simple synthesis of a useful addition to the donor–acceptor cyclopropane family of compounds. Rhodium catalyzed carbenoid insertion of fluorinated diazomalonates to simple alkenes provides a simple preparation of these products. Their enhanced reactivity as electrophiles has been demonstrated by their reaction with indoles under Lewis acid conditions. An exploration of further reactivity into heretofore unsuccessful cycloadditions is currently underway.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada for generous funding of this research. We are grateful to Mr Doug Hairsine of the Western University Mass Spectrometry facility for performing MS analyses.Notes and references
- (a) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (b) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC; (c) T. P. Lebold and M. A. Kerr, Pure Appl. Chem., 2010, 82, 1797 CrossRef CAS; (d) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (e) M. Yu and B. L. Pagenkopf, Tetrahedron, 2005, 61, 321 CrossRef CAS PubMed; (f) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (g) H. N. C. Wong, M. Y. Hon, C. W. Tse, Y. C. Yip, J. Tanko and T. Hudlicky, Chem. Rev., 1989, 89, 165 CrossRef CAS; (h) S. Danishefsky, Acc. Chem. Res., 1979, 12, 66 CrossRef CAS.
- (a) P. Tang and Y. Qin, Synthesis, 2012, 2969 CAS; (b) A. Karadeolian and M. A. Kerr, J. Org. Chem., 2010, 75, 1256 CrossRef PubMed; (c) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC.
- For recent reviews in hyperbaric chemistry see: (a) F. Benito-Lopez, R. J. M. Egberink, D. N. Reinhoudt and W. Verboom, Tetrahedron, 2008, 64, 10023 CrossRef CAS PubMed; (b) K. Matsumoto, H. Hamana and H. Iida, Helv. Chim. Acta, 2005, 2033 CrossRef CAS PubMed. For cycloadditions using hyperbaric chemistry see: (c) J. Zhu, M. D. Ganton, M. A. Kerr and M. S. Workentin, J. Am. Chem. Soc., 2007, 179, 4904 CrossRef PubMed; (d) A. C. Kinsman and M. A. Kerr, Org. Lett., 2000, 2, 3517 CrossRef CAS PubMed; (e) I. C. Barrett and M. A. Kerr, Tetrahedron Lett., 1999, 40, 2439 CrossRef CAS.
- F. de Nanteuil, J. Loup and J. Waser, Org. Lett., 2013, 15, 3738 CrossRef CAS PubMed.
- B. M. Trost and P. J. Morris, Angew. Chem., Int. Ed., 2011, 50, 6167 CrossRef CAS PubMed.
- M. P. Doyle, Chem. Rev., 1986, 86, 919 CrossRef CAS.
- M. P. Doyle, M. R. Colsman and M. S. Chinn, Inorg. Chem., 1984, 23, 3684 CrossRef CAS.
- C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS PubMed.
- P. R. Bruzinshi, D. H. Lake, N. Kong, M. J. Fall and H. M. L. Davies, J. Am. Chem. Soc., 1996, 118, 6897 CrossRef.
- (a) M. R. Emmett and M. A. Kerr, Org. Lett., 2011, 13, 4180 CrossRef CAS PubMed; (b) D. England, T. Kuss, R. Keddy and M. Kerr, J. Org. Chem., 2001, 66, 4704 CrossRef CAS PubMed; (c) M. Kerr and R. Keddy, Tetrahedron Lett., 1999, 40, 5671 CrossRef CAS; (d) P. E. Harrington and M. A. Kerr, Tetrahedron Lett., 1997, 38, 5949 CrossRef CAS.
- (a) R. Talukdar, D. P. Tiwari, A. Saha and M. K. Ghorai, Org. Lett., 2014, 16, 3954 CrossRef CAS PubMed; (b) C. Dulin, K. Murphy and K. Nolin, Tetrahedron Lett., 2014, 55, 5280 CrossRef CAS PubMed; (c) J. Zhu, Y. Liang, L. Wang, Z. Zheng, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2014, 136, 6900 CrossRef CAS PubMed; (d) S. Wales, M. Walker and J. Johnson, Org. Lett., 2013, 15, 2558 CrossRef CAS PubMed; (e) H. Xiong, H. Xu, S. Liao, Z. Xie and Y. Tang, J. Am. Chem. Soc., 2013, 135, 7851 CrossRef CAS PubMed; (f) F. De Simone and J. Waser, Chimia, 2012, 66, 233 CrossRef CAS PubMed; (g) F. De Simone, T. Saget, F. Benfatti, S. Almeida and J. Waser, J. Chem. – Eur. J., 2011, 17, 14527 CrossRef CAS PubMed; (h) B. Bajtos, M. Yu, H. Zhao and B. Pagenkopf, J. Am. Chem. Soc., 2007, 129, 9631 CrossRef CAS PubMed; (i) C. Venkatesh, P. Singh, H. Ila and H. Junjappa, Eur. J. Org. Chem., 2006, 5378 CrossRef CAS PubMed.
- These cyclopropanes were prepared by an alternative method as described in the ESI.† The carbernoid insertion protocol was ineffective in this instance.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectra for characterization. See DOI: 10.1039/c5qo00176e |
| This journal is © the Partner Organisations 2015 |