
Syntheses of sceptrins and nakamuric acid and insights into the biosyntheses of pyrrole–imidazole dimers†
Xiaolei
Wang
a,
Yang
Gao
bc,
Zhiqiang
Ma
a,
Rodrigo A.
Rodriguez
d,
Zhi-Xiang
Yu
b and
Chuo
Chen
*a
aDepartment of Biochemistry, The University of Texas Southwestern Medical Center, Dallas, TX 75390, USA. E-mail: Chuo.Chen@UTSouthwestern.edu
bCollege of Chemistry, Peking University, Beijing 100871, China
cKey Laboratory of Pesticide & Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, Hubei, Wuhan 430079, China
dDepartment of Chemistry, The Scripps Research Institute, La Jolla, CA 92037, USA
First published on 12th June 2015
Abstract
Sceptrins and nakamuric acid are structurally unique antibiotics isolated from marine sponges. Recent studies suggest that the biosynthesis of these dimeric pyrrole–imidazole alkaloids involves a single-electron transfer (SET)-promoted [2 + 2] cycloaddition to form their cyclobutane core skeletons. We describe herein the biomimetic syntheses of racemic sceptrin and nakamuric acid. We also report the asymmetric syntheses of sceptrin, bromosceptrin, and dibromosceptrin in their natural enantiomeric form. We further provide mechanistic insights into the pathway selectivity of the SET-promoted [2 + 2] and [4 + 2] cycloadditions that lead to the divergent formation of the sceptrin and ageliferin core skeletons. Both the [2 + 2] and [4 + 2] cycloadditions are stepwise reactions, with the [2 + 2] pathway kinetically and thermodynamically favored over the [4 + 2] pathway. For the [2 + 2] cycloaddition, the dimerization of pyrrole–imidazole monomers is rate-limiting, whereas for the [4 + 2] cycloaddition, the cyclization is the slowest step.
Introduction
Sceptrin (1a)1 and ageliferin (3a)2 are the founding members of the dimeric pyrrole–imidazole alkaloids that have attracted synthetic chemists’ attention for decades (Fig. 1).3,4 Previously, the Baran and the Birman groups each developed a [2 + 2] photocycloaddition approach to accomplish the total synthesis of 1.5,6 The Baran group further demonstrated that 1 could be converted into 3 in a stereospecific manner.7 We also used a biomimetic radical cyclization approach to complete the asymmetric synthesis of 1a and 3.8 In addition, the Harran group achieved the synthesis of 3a using a ring-expansion strategy,9 and the Ohta group synthesised 12,12′-dimethylageliferin using a thermal Diels–Alder cycloaddition strategy.10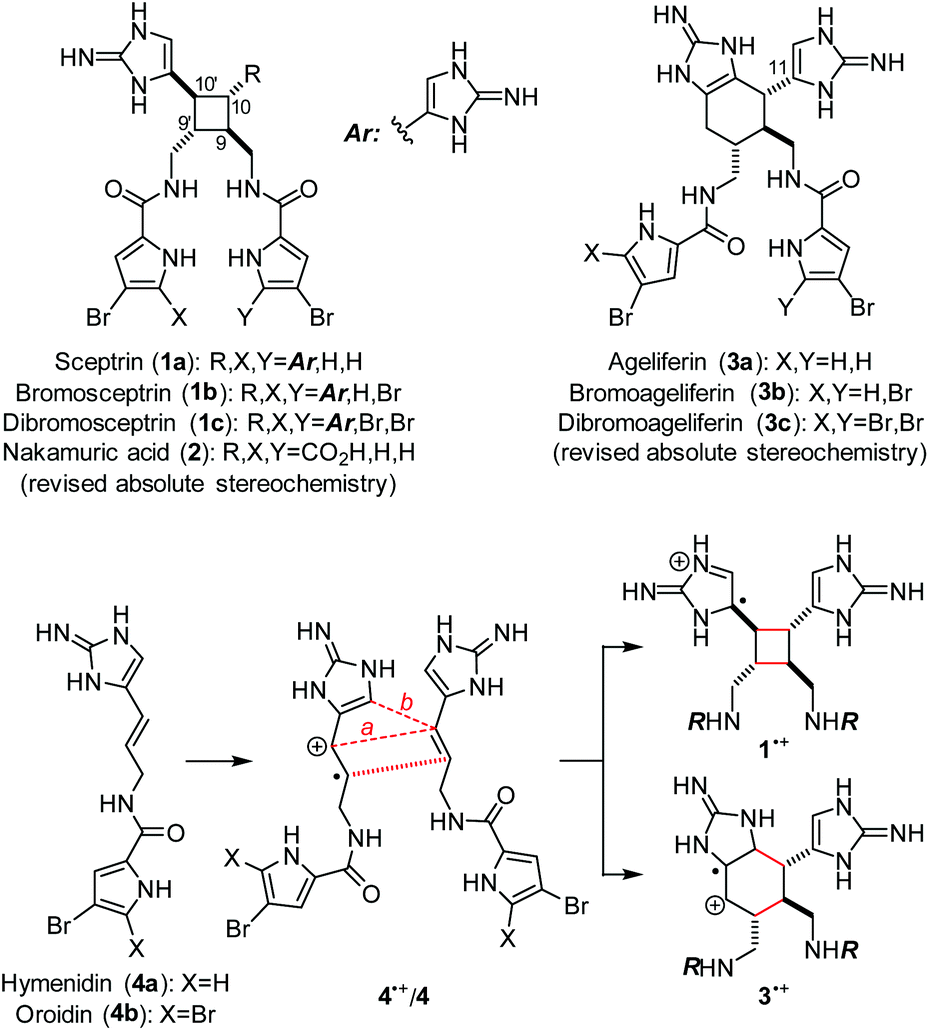 | ||
| Fig. 1 Sceptrins (1) and ageliferins (3) are formally the [2 + 2] and [4 + 2] cycloaddition products of hymenidin/oroidin (4). Nakamuric acid (2) is a pseudo-symmetric [2 + 2]-type pyrrole–imidazole dimer. | ||
Although the biogenesis of these pyrrole–imidazole dimers has been a subject of long debates,11 it is generally agreed that 1a and 3a are derived from hymenidin (4a) through formal [2 + 2] and [4 + 2] cycloadditions, respectively. Recently, Molinski and Romo have provided evidence through metabiosynthetic studies that the biogenic dimerization of 4 is promoted by an enzymatic single-electron transfer (SET) reaction.12 A SET oxidation of 4 would give radical cation 4˙+ that is highly reactive toward [2 + 2] (path a) and [4 + 2] (path b) cycloadditions13 with another molecule of 4 to give 1˙+ and 3˙+, correspondingly. A subsequent SET reduction would provide 1 and 3 after tautomerization. Guided by this biosynthetic hypothesis, we have developed an asymmetric approach to accomplish the syntheses of 1a and 3, and revised their absolute stereochemistries recently.8e We report herein the syntheses of all members of sceptrins (1) and the pseudo-symmetric [2 + 2] dimer nakamuric acid (2).5b,14 Additionally, we provide a mechanistic understanding of the pathway selectivity of the cycloaddition of 4˙+/4.
Results and discussion
We are interested in using total syntheses to examine the biosynthetic hypotheses for the pyrrole–imidazole dimers.8 The prior syntheses of 1/2 by the Baran group and the Birman group involved the use of a [2 + 2] photocycloaddition to build the cyclobutane core with the formation of the C9/C10 and C9′/C10′ linkages.5,6 Recent developments in photoredox chemistry by Yoon and others15,16 have inspired us to explore the possibility of using a SET-promoted [2 + 2] cycloaddition to establish the C9/C9′ and C10/C10′ linkages of 1/2 in a biomimetic fashion.Our study started with the design of a SET-promoted [2 + 2] cycloaddition to assemble the core skeleton of 1 and 2. After exploring various substrate systems, we decided to use radical cation 5 to mimic 4˙+/4. We envisioned that 5 would undergo a formal [2 + 2] and a [4 + 2] cycloaddition to give 6 and 7, correspondingly (Fig. 2). Initial studies using 8 as the model substrate led to the identification of a suitable photoredox catalyst. Irradiation of 8 in the presence of a catalytic amount of Ir(ppy)3 gave the [2 + 2] cycloadduct 9 and the [4 + 2] cycloadduct 10 smoothly.8f
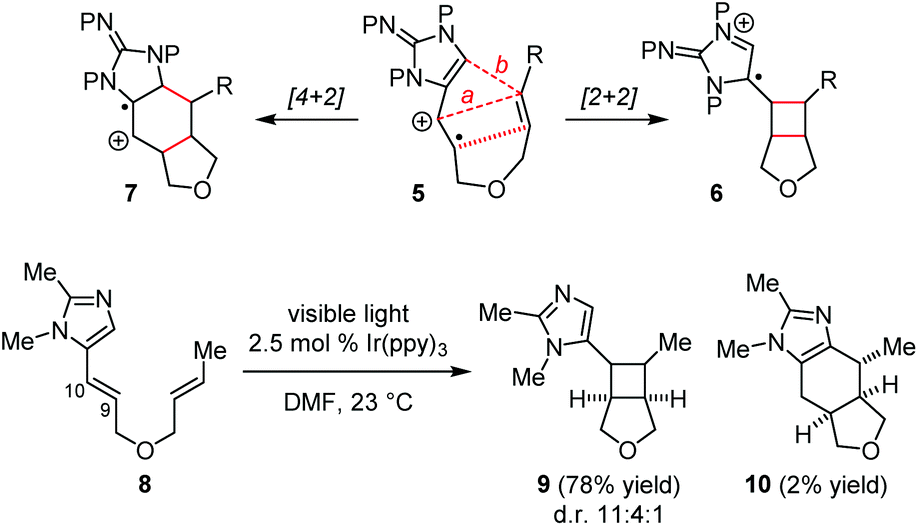 | ||
| Fig. 2 Design and proof-of-concept of our biomimetic synthetic strategy. | ||
With this success, we went on to study the synthesis of nakamuric acid (2). Starting from aldehyde 11,8c,d a Julia–Kocienski olefination gave allylic ether 12 with a modest E/Z selectivity (Fig. 3). Subsequent deprotection yielded allylic alcohol 13, which was coupled with 2,3-dihydrofuran to provide 14 through alkoxy selenylation. Next, oxidation of the phenylselenide, elimination of the resulting phenylselenoxide, and reduction of the azido group afforded 15.
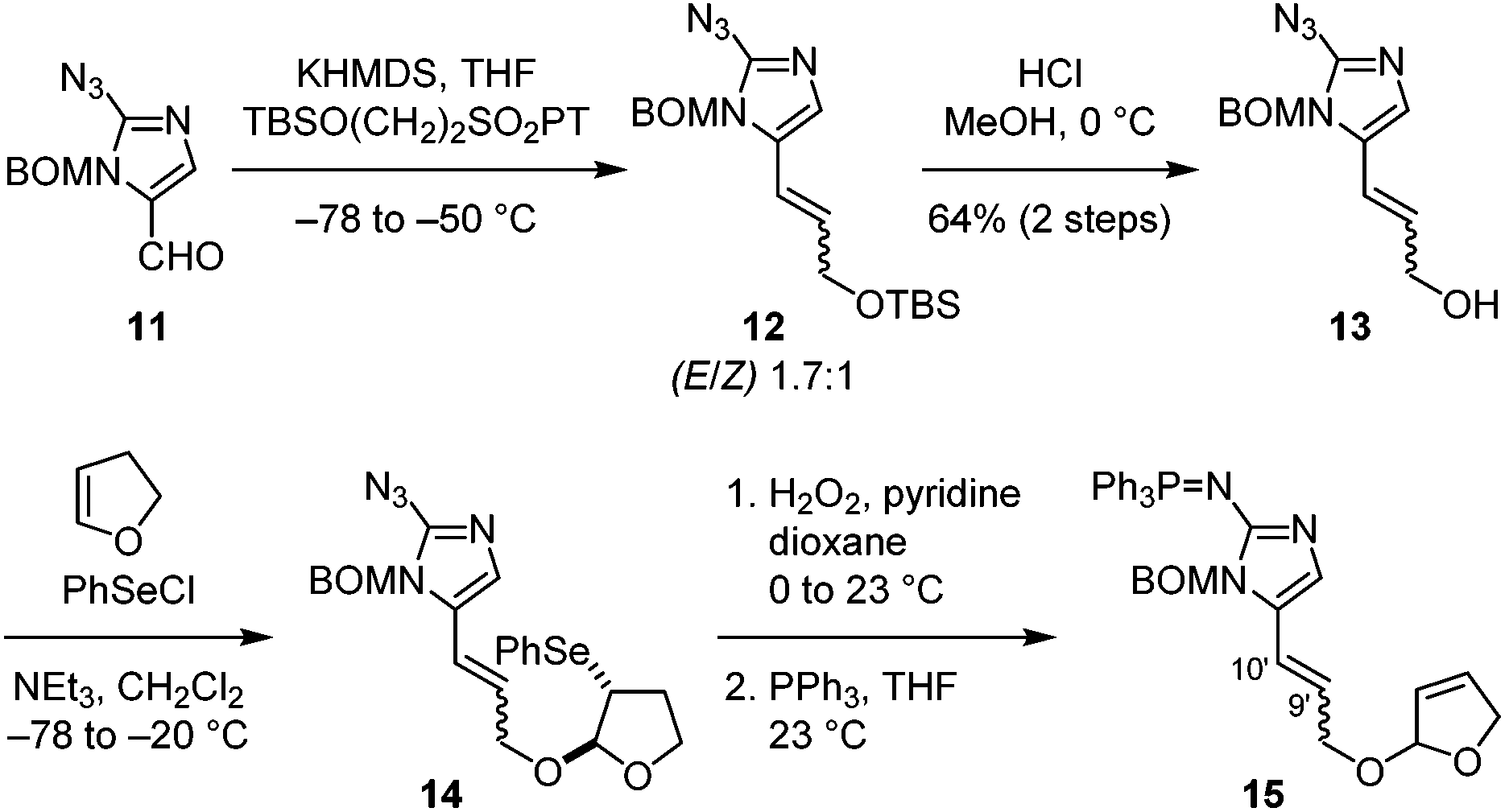 | ||
| Fig. 3 Preparation of the [2 + 2] cycloaddition precursor. | ||
Recently, the Yoon group has developed a remarkably mild method to promote SET-mediated [2 + 2] cycloadditions.16 Following their protocols, we irradiated 15 with visible light in the presence of 3 mol% Ir(ppy)3 and obtained the desired [2 + 2] cycloadduct meso-16 together with its C10′ epimer (d.r. 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Fig. 4). Attempts to induce the [2 + 2] cycloaddition prior to the Staudinger reduction failed under various conditions presumably due to the high oxidative potential of vinylazidoimidazole. Reduction of the azido group gave a phosphine imide-protected vinylaminoimidazole that is more electron-rich and can be readily oxidized by a photoredox catalyst. Because Yoon has found that iridium(III)-complexes could also promote [2 + 2] cycloadditions via an energy-transfer mechanism,16g we irradiated 15 with a catalytic amount of 9-fluorenone that has a triplet energy (ET = 55 kcal mol−1)17 the same as that of Ir(ppy)3 in order to probe the mechanism of the Ir(ppy)3-catalyzed [2 + 2] cycloaddition of 15. As no reaction occurred with 9-fluorenone, we believe that 16 was produced via a SET mechanism. Notably, isomerization of the C9′–10′ olefin of 15 was observed during the reaction, indicating that the cycloaddition of 15˙+ instead of the SET oxidation of 15 was the rate-limiting step.
:1) (Fig. 4). Attempts to induce the [2 + 2] cycloaddition prior to the Staudinger reduction failed under various conditions presumably due to the high oxidative potential of vinylazidoimidazole. Reduction of the azido group gave a phosphine imide-protected vinylaminoimidazole that is more electron-rich and can be readily oxidized by a photoredox catalyst. Because Yoon has found that iridium(III)-complexes could also promote [2 + 2] cycloadditions via an energy-transfer mechanism,16g we irradiated 15 with a catalytic amount of 9-fluorenone that has a triplet energy (ET = 55 kcal mol−1)17 the same as that of Ir(ppy)3 in order to probe the mechanism of the Ir(ppy)3-catalyzed [2 + 2] cycloaddition of 15. As no reaction occurred with 9-fluorenone, we believe that 16 was produced via a SET mechanism. Notably, isomerization of the C9′–10′ olefin of 15 was observed during the reaction, indicating that the cycloaddition of 15˙+ instead of the SET oxidation of 15 was the rate-limiting step.
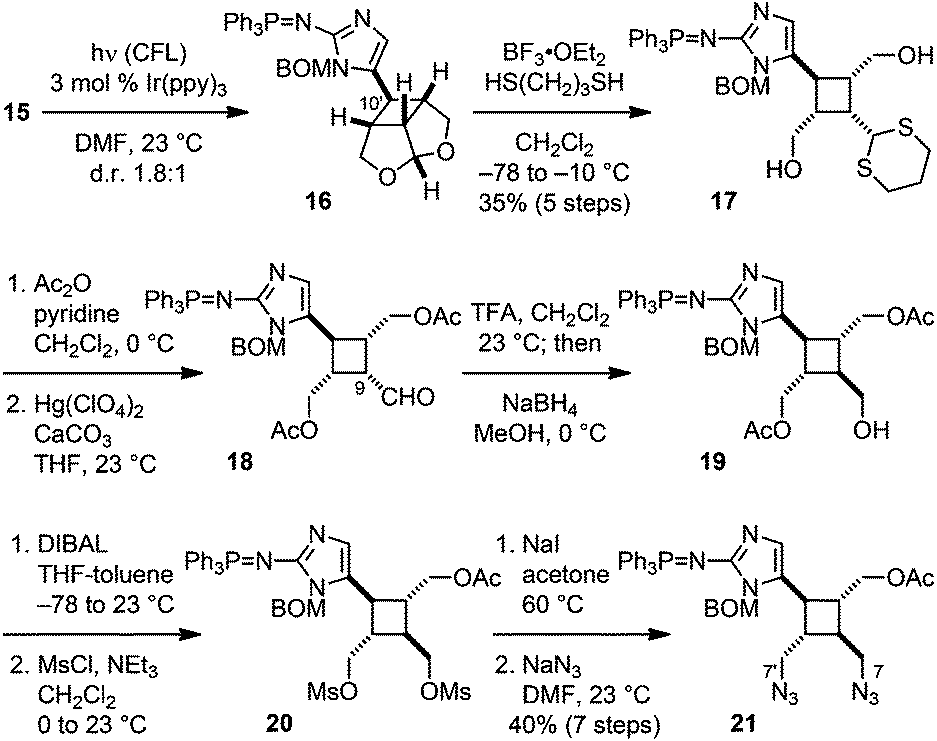 | ||
| Fig. 4 Construction of the cyclobutane core skeleton. | ||
In the following stage of the synthesis, the core skeleton of 2 was revealed by subjecting 16 to transthioketalization to provide 17. Next, protection of the hydroxyl groups and removal of the dithiane protecting group gave 18. Epimerization of the C9 stereogenic center followed by reduction of the C9 aldehyde yielded 19. Consistent with Baran's prior observations,5b introduction of the N7 and N7′ groups was challenging due to steric hindrance. Eventually, we realized this demanding transformation by applying the three-step procedure that we had previously developed for the synthesis of ageliferin (3a).8c,d After removing one of the acetyl protecting groups of 19, the resulting diol was mesylated to afford rac-20. Subsequent iodination and azidation gave 21 as a trifluoroacetic acid salt upon HPLC purification. Using pyridine instead of triethylamine8c,d as the base for the mesylation step effectively suppressed the side-reaction that led to the formation of the mesylaminoimidazolium salt of 20.
To install the pyrrole groups, the azido groups of 21 were reduced to yield 22 (Fig. 5). Although this hydrogenolysis reaction proceeded more efficiently in methanol than in ethyl acetate, employing methanol as the solvent also led to the undesired formation of the methylamines of 22. Diamine 22 was unstable and decomposed under various amidation conditions. We found that the carbodiimide-mediated amide coupling was the only effective method to introduce the pyrrole groups to give 23. Subsequent removal of the acetyl group yielded alcohol 24. Oxidation of the hydroxyl group to carboxylic acid was achieved by Dess–Martin oxidation and Lindgren oxidation in one-pot to provide 25. To remove the benzyloxymethyl protecting group, 25 was first treated with boron trichloride to cleave the benzyl group. The resulting hydroxymethyl group was then hydrolyzed by treating with ammonium hydroxide in situ to give 26. Finally, the triphenylphosphine imide group was hydrolyzed under acidic conditions to afford rac-nakamuric acid (2). Because most aminoimidazole-containing intermediates in this synthesis were not stable and readily decomposed upon exposure to acid or air, this synthetic route was optimized to minimize the number of purification processes.
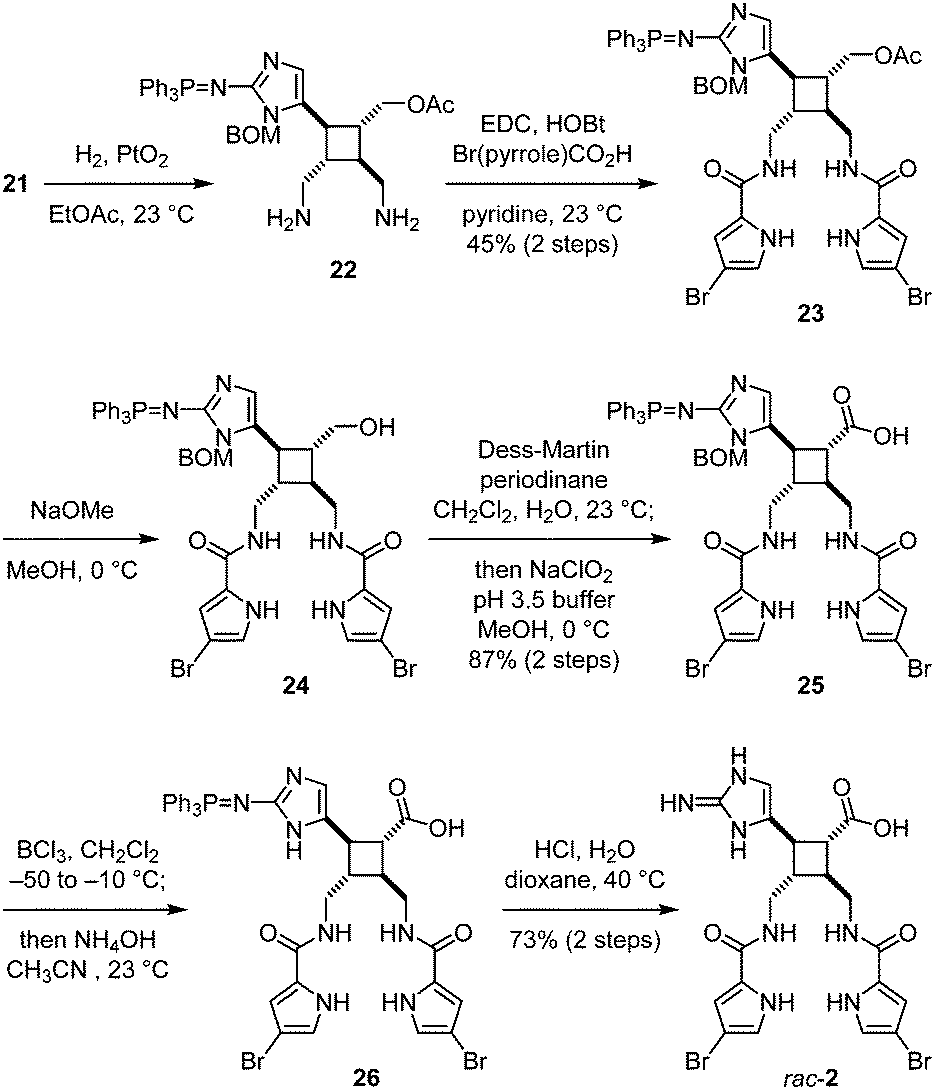 | ||
| Fig. 5 Synthesis of rac-nakamuric acid (2). | ||
After completing the synthesis of nakamuric acid (2), we turned our attention to the synthesis of sceptrin (1a). Starting with alcohol 24, we adapted the method developed by Stoltz for the dragmacidin synthesis18 to construct the second aminoimidazole group. A Dess–Martin oxidation of 24 followed by a Henry reaction with nitromethane gave 27 after the reduction of the nitro group using the Carreira's conditions,19 and protection of the resulting amino group as a tert-butylcarbamate (Fig. 6). Notably, no debromination of the 4-bromopyrrole occurred when the reduction of the nitro group by zinc was performed at 4 °C. Subsequent oxidation of the hydroxyl group yielded ketone 28. The benzyloxymethyl and tert-butylcarbamate protecting groups were then removed to provide aminoketone 29. The aminoimidazole group was constructed by condensing 29 with cyanamide20 to furnish rac-sceptrin (1a). The triphenylphosphine protecting group was also hydrolyzed under these reaction conditions.
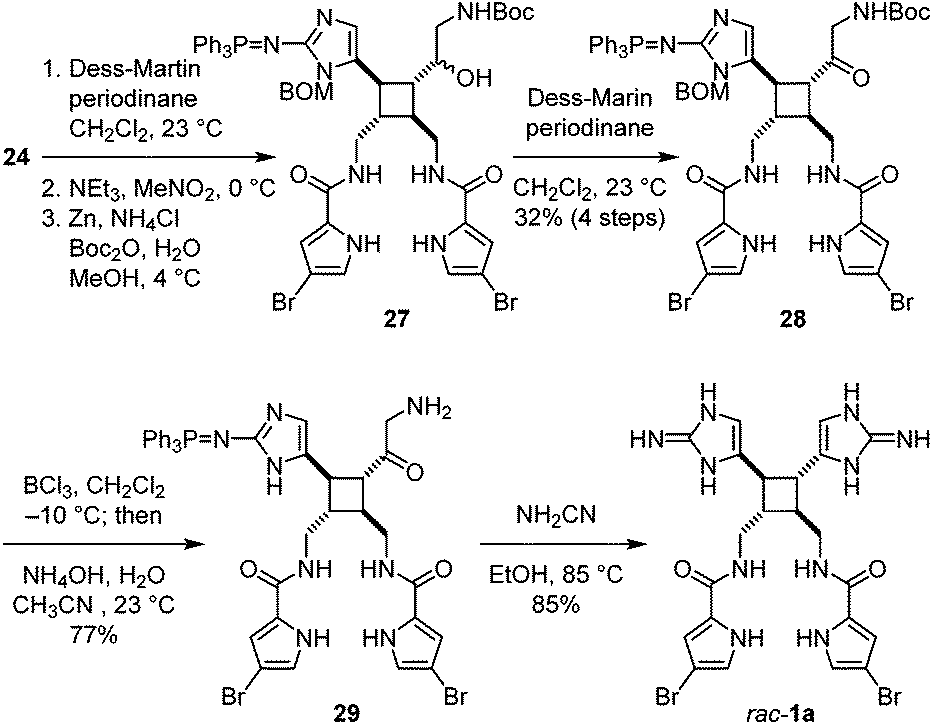 | ||
| Fig. 6 Synthesis of rac-sceptrin (1a). | ||
To achieve the asymmetric synthesis of 1, we first attempted to desymmetrize 17 or its derivatives. However, we could only obtain products with low optical purities. Equally frustrating was the resolution of 21 or its derivatives. The formation of imidazolium salts with L-menthol chloroformate proceeded with low efficiency. Separation of the two diastereomeric salts on HPLC was also difficult. Despite some success on a small scale, this method is far from being practical. Therefore, we turned our attention to the chiral-pool approach that we developed for the asymmetric syntheses of 3.8a–e
Previously, we took advantage of the stereocenter at the C11 position to control the stereoselective formation of the core skeleton of 3. We thus decided to use this strategy for the stereoselective establishment of the sceptrin core skeleton. In contrast to using D-serine as the source of chirality for 3, we started our synthesis of 1 from D-glutamic acid (30). Diazotization of 30 led to the formation of 31 (Fig. 7).21 Reduction of the carboxylic acid group of 31 provided alcohol 32.22 Subsequent protection of the hydroxyl group and the reduction of the lactone yielded hemiacetal 33, which was dehydrated to afford dihydrofuran 34.23 Coupling of fragments 13 and 34 through alkoxy selenylation24 proceeded with excellent stereoselectivity. Episelenium formation with 34 occurred on its less hindered face, allowing alcohol 13 to approach from the more hindered face to give 35 as the only diastereomeric product. Subsequent oxidative elimination of the phenylselenide group provided 36. Staudinger reduction of the azido group gave 8,11-cis-37 that underwent a smooth intramolecular [2 + 2] cycloaddition under photoredox conditions. Irradiation of 37 with visible light in the presence of 3 mol% Ir(ppy)3 yielded 38 as a 1.8:1 mixture of C10′ diastereomers.
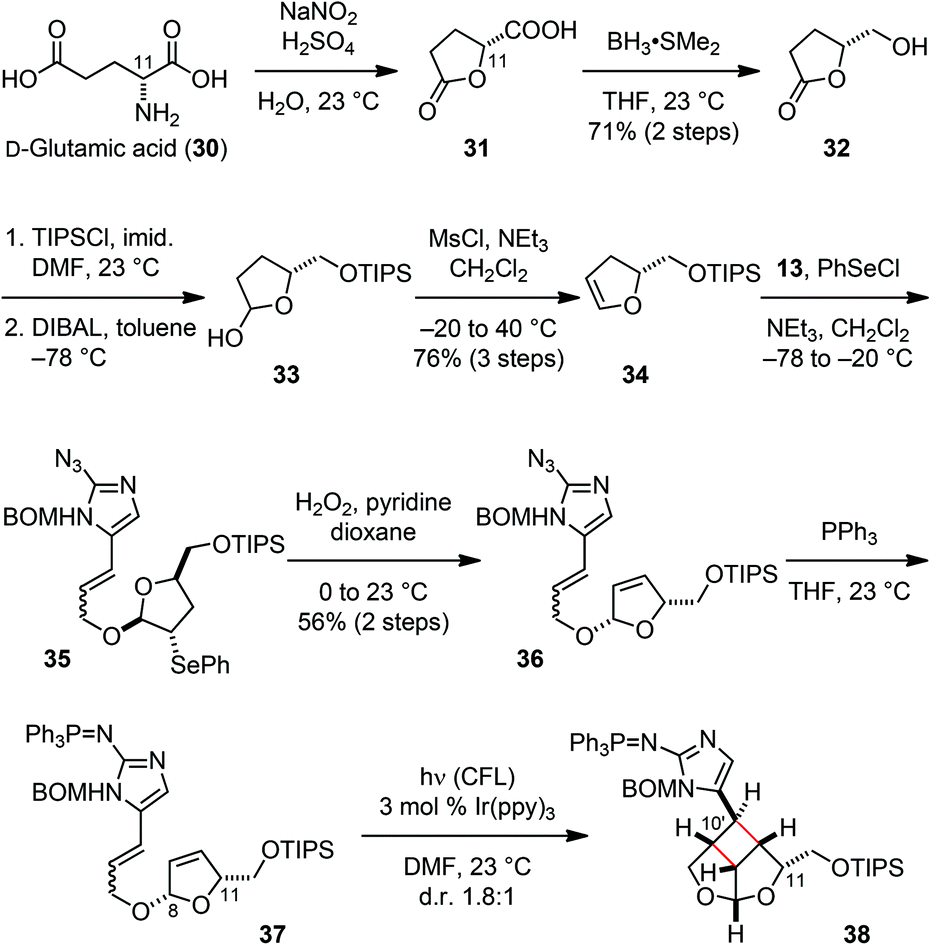 | ||
| Fig. 7 Construction of the core skeleton of nat-sceptrins (1). | ||
To complete the synthesis of 1, the sceptrin core of pseudosymmetric 38 was revealed by transthioketalization to give 39 (Fig. 8). The hydroxyl groups were then protected and the dithiane group was hydrolyzed. Epimerization at the C9 stereocenter, reduction of the aldehyde group, and removal of the acetate protecting groups yielded 40. Subsequently, mesylation, iodination, azidation, and desilylation afforded 41. To construct the second aminoimidazole group, the hydroxyl group of 41 was oxidized and the resulting mesyl aldehyde was condensed with Boc-guanidine to give 42. Hydrolysis of the Boc protecting group occurred during HPLC purification. Next, reduction of the azido groups of 42 yielded diamine 43. Carrying out the reduction in tert-butanol provided improved efficiency. Coupling of 43 with 4-bromopyrrole-2-carboxylic acid or 4,5-dibromopyrrole-2-carboxylic acid gave 44a or 44c, correspondingly. Sequential coupling of 43 with these two acids gave 44b. Finally, deprotection of 44 afforded sceptrins in its natural enantiomeric form (nat-1) as indicated by CD analyses (Fig. 9).25 This result is consistent with our previous work that using L-glutamic acid as the source of chirality for this synthetic sequence gave ent-1a, affirming the notion8e that the absolute stereochemistry of sceptrins and ageliferins should be revised to those of 1 and 3 as shown in Fig. 1.
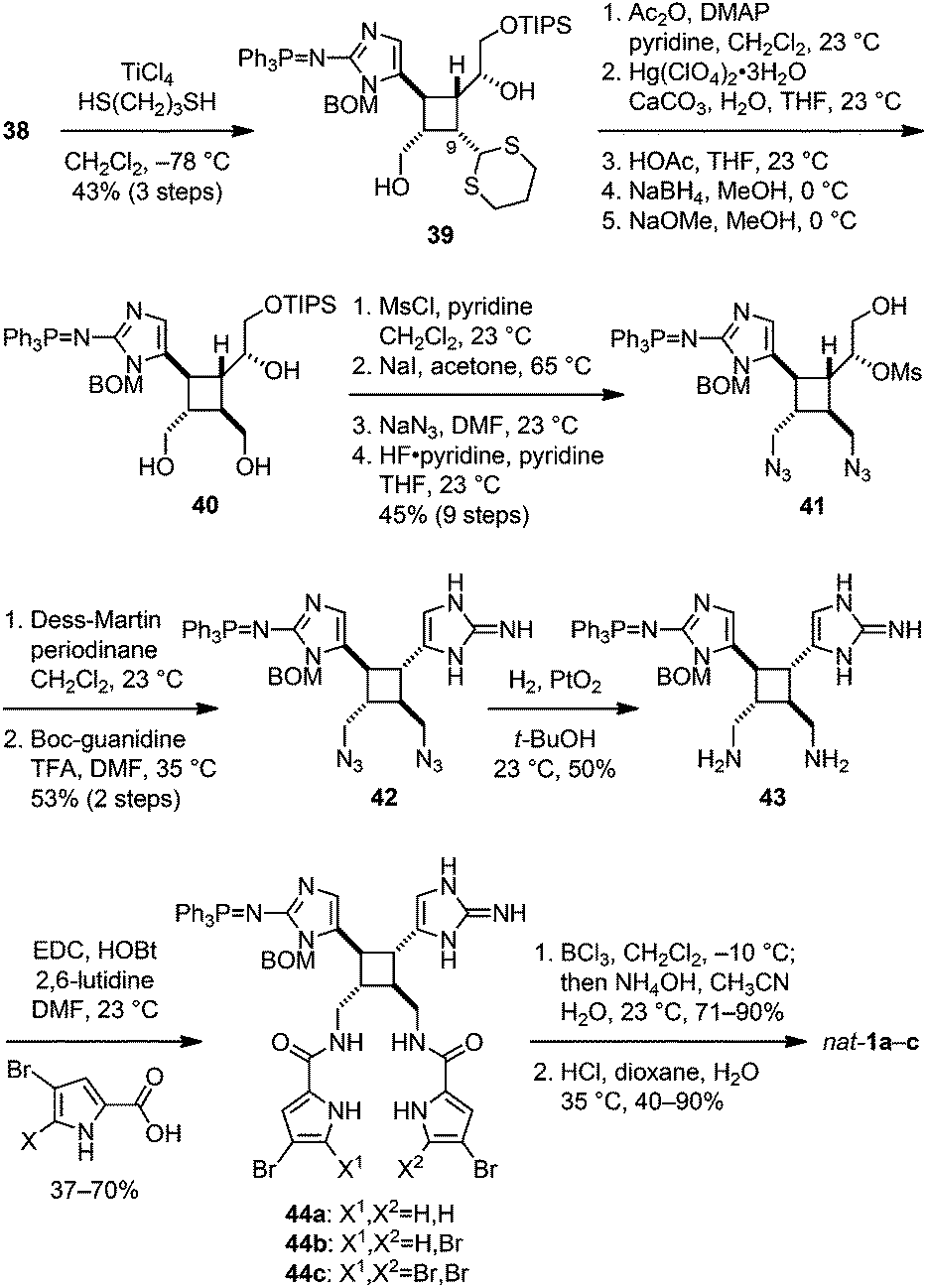 | ||
| Fig. 8 Synthesis of nat-sceptrin (1a). | ||
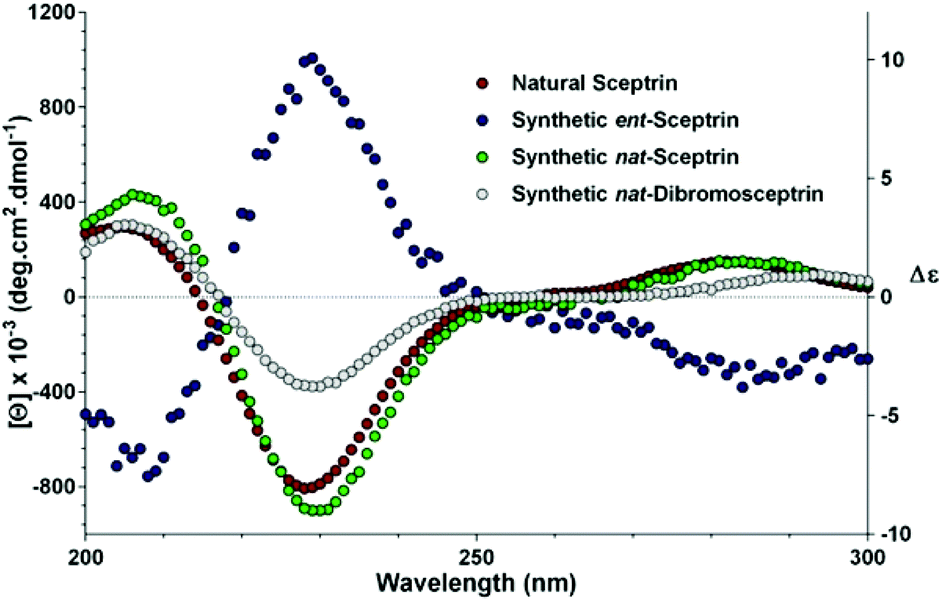 | ||
| Fig. 9 Comparison of the CD spectra of sceptrins in water. Red, natural sceptrin TFA in methanol;26b blue, synthetic sceptrin TFA from L-glutamic acid in water; green, synthetic sceptrin TFA from D-glutamic acid in water; grey, synthetic dibromosceptrin TFA from D-glutamic acid in water. | ||
To gain insights into the biogenic dimerization of 4, we analyzed the energy profiles of the SET-promoted [2 + 2] and [4 + 2] cycloadditions of clathrodin (4c) by DFT calculations at the (U)B3LYP-6-311G(d,p) level (Fig. 10). Both the Gibbs free energies and the enthalpies of the reaction intermediates and transition states were evaluated. Consistent with our experimental results on the model systems,8d,f these cycloadditions are most likely stepwise radical addition reactions, as we were not able to locate the transition states corresponding to the concerted cycloadditions. These reactions start from the formation of INT1 from 4c˙+ and 4c, with an activation enthalpy of 1.1 kcal mol−1. Formation of INT1 is exothermic by 11.6 kcal mol−1. Then, bifurcation from INT1 to give either [2 + 2] or [4 + 2] cycloadduct takes place. For the formation of 1d˙+, the first step viaTS1 with an activation enthalpy of 1.1 kcal mol−1 is a bimolecular process, whereas the second step of converting INT1 to 1d˙+ with an activation enthalpy of 1.9 kcal mol−1 is a unimolecular process. Therefore, the first step of the [2 + 2] pathway is the rate-determining step considering the entropy penalty in TS1 with respect to the reactants 4d˙+ and 4d.
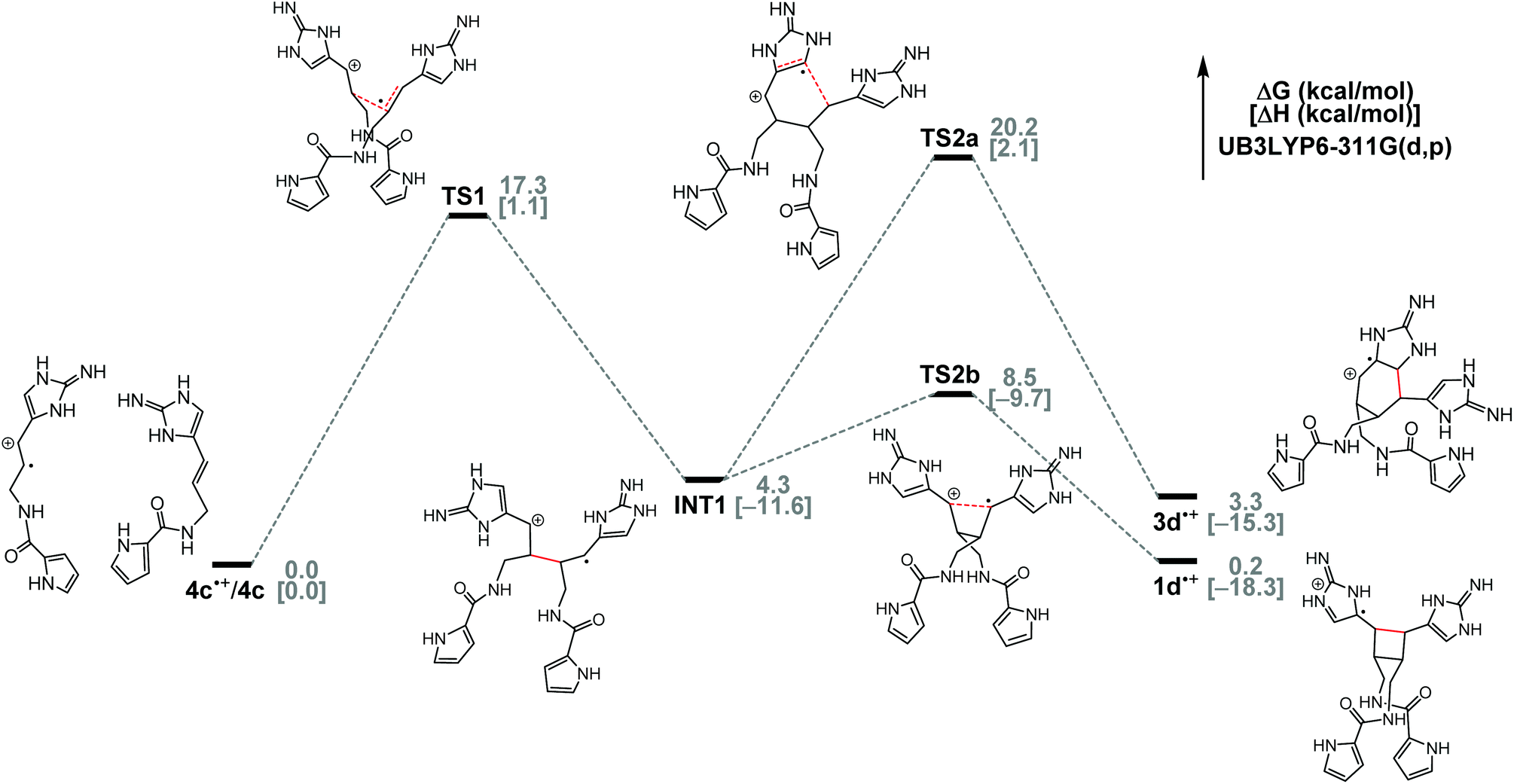 | ||
| Fig. 10 Energy diagram for the [2 + 2] and [4 + 2] cycloadditions of 4c˙+/4c. | ||
DFT calculations also found that the [2 + 2] pathway is highly favored because TS2b to 1d˙+ is lower in free energy than TS2a to 3d˙+ by 11.7 kcal mol−1 (ΔΔH‡, 11.8 kcal mol−1). Consistently, our model studies showed the same pathway selectivity. SET-promoted cycloaddition of 8 gave the [2 + 2] cycloaddition product predominantly, with a pathway selectivity of nearly 40:1 (Fig. 2). It is likely that, in ageliferin-producing sponges, the oxidoreductase promoting the SET-dimerization reaction stabilizes TS2a to facilitate the formation of 3. In nature, 1a is one of the most abundant pyrrole–imidazole alkaloids.26 When 1a and 3a were isolated from the same sponges, 1a is always by far the major constituent.1b,2b,14
Conclusions
We demonstrate herein that SET can promote [2 + 2] and [4 + 2] cycloadditions of oroidin/hymenidin/clathrodin (4) to give sceptrins (1) and ageliferins (3) as proposed by Molinski, Romo, and us. Following this hypothesis, we completed the biomimetic syntheses of sceptrins (1) and nakamuric acid (2), using photoredox chemistry to promote SET in assembling the core skeleton of these pyrrole–imidazole dimers. We also confirmed that the original absolute stereochemistry of 1 determined by crystallographic studies was incorrect. Whereas X-ray analysis has been regarded as an “unambiguous” way to establish the structure of small molecules in the field of organic chemistry, cautions are still needed to avoid misinterpretation of the diffraction data.27 Finally, our computational studies provided direct support that SET can promote cycloadditions of 4 to give 1 and 3. Importantly, the relative natural abundance of 1 and 3 is consistent with the pathway selectivity of the SET-promoted but not the thermal cycloadditions.Acknowledgements
We thank Prof. Phil Baran (The Scripps Research Institute), Dr Vincent Lynch (University of Texas at Austin), Prof. Jon Clardy (Harvard University), and Prof. Tadeusz Molinski (University of California, San Diego) for helpful discussions. We also thank Dr Matthias Köck (Alfred Wegener Institute) for providing the natural sample of sceptrin, Dr Julie Muñoz (Alfred Wegener Institute) for a copy of the CD spectrum of natural sceptrin TFA, and Dr Heping Shi (UT Southwestern) for assistance in CD experiments. Financial support was provided by NIH (NIGMS R01-GM079554), the Welch Foundation (I-1868), and UT Southwestern.Notes and references
- (a) R. P. Walker, D. J. Faulkner, D. V. Engen and J. Clardy, J. Am. Chem. Soc., 1981, 103, 6772–6773 CrossRef CAS; (b) M. Assmann and M. Köck, Z. Naturforsch., C: Biosci., 2002, 57, 157–160 CAS.
- (a) J. Kobayashi, M. Tsuda, T. Murayama, H. Nakamura, Y. Ohizumi, M. Ishibashi, M. Iwamura, T. Ohta and S. Nozoe, Tetrahedron, 1990, 46, 5579–5586 CrossRef CAS; (b) P. A. Keifer, R. E. Schwartz, M. E. S. Koker, R. G. Hughes, Jr., D. Rittschof and K. L. Rinehart, J. Org. Chem., 1991, 56, 2965–2975 CrossRef CAS.
- (a) H. Hoffmann and T. Lindel, Synthesis, 2003, 1753–1783 CAS; (b) D. E. N. Jacquot and T. Lindel, Curr. Org. Chem., 2005, 9, 1551–1565 CrossRef CAS; (c) H. Du, Y. He, R. Sivappa and C. J. Lovely, Synlett, 2006, 965–992 CAS; (d) M. Köck, A. Grube, I. B. Seiple and P. S. Baran, Angew. Chem., Int. Ed., 2007, 46, 6586–6594 CrossRef PubMed; (e) S. M. Weinreb, Nat. Prod. Rep., 2007, 24, 931–948 RSC; (f) H.-D. Arndt and M. Riedrich, Angew. Chem., Int. Ed., 2008, 47, 4785–4788 CrossRef CAS PubMed; (g) B. Heasley, Eur. J. Org. Chem., 2009, 1477–1489 CrossRef CAS PubMed; (h) B. Forte, B. Malgesini, C. Piutti, F. Quartieri, A. Scolaro and G. Papeo, Mar. Drugs, 2009, 7, 705–753 CrossRef CAS PubMed; (i) A. Al-Mourabit, M. A. Zancanella, S. Tilvi and D. Romo, Nat. Prod. Rep., 2011, 28, 1229–1260 RSC.
- (a) X. Wang, Z. Ma, X. Wang, S. De, Y. Ma and C. Chen, Chem. Commun., 2014, 50, 8628–8639 RSC; (b) Y. Ma, S. De and C. Chen, Tetrahedron, 2014, 71, 1145–1173 CrossRef PubMed.
- (a) P. S. Baran, A. L. Zografos and D. P. O'Malley, J. Am. Chem. Soc., 2004, 126, 3726–3727 CrossRef CAS PubMed; (b) D. P. O'Malley, K. Li, M. Maue, A. L. Zografos and P. S. Baran, J. Am. Chem. Soc., 2007, 129, 4762–4775 CrossRef PubMed.
- V. B. Birman and X.-T. Jiang, Org. Lett., 2004, 6, 2369–2371 CrossRef CAS PubMed.
- (a) P. S. Baran, D. P. O'Malley and A. L. Zografos, Angew. Chem., Int. Ed., 2004, 43, 2674–2677 CrossRef CAS PubMed; (b) P. S. Baran, K. Li, D. P. O'Malley and C. Mitsos, Angew. Chem., Int. Ed., 2006, 45, 249–252 CrossRef CAS PubMed.
- (a) X. Tan and C. Chen, Angew. Chem., Int. Ed., 2006, 45, 4345–4348 CrossRef CAS PubMed; (b) Z. Ma, J. Lu, X. Wang and C. Chen, Chem. Commun., 2011, 47, 427–429 RSC; (c) X. Wang, Z. Ma, J. Lu, X. Tan and C. Chen, J. Am. Chem. Soc., 2011, 133, 15350–15353 CrossRef CAS PubMed; (d) X. Wang, X.-L. Wang, X. Tan, J. Lu, K. W. Cormier, Z. Ma and C. Chen, J. Am. Chem. Soc., 2012, 134, 18834–18842 CrossRef CAS PubMed; (e) Z. Ma, X. Wang, X. Wang, R. A. Rodriguez, C. E. Moore, S. Gao, X. Tan, Y. Ma, A. L. Rheingold, P. S. Baran and C. Chen, Science, 2014, 346, 219–224 CrossRef CAS PubMed; (f) X. Wang and C. Chen, Tetrahedron, 2015, 71, 3690–3693 CrossRef CAS PubMed.
- H. Ding, A. G. Roberts and P. G. Harran, Chem. Sci., 2012, 4, 303–306 RSC.
- (a) I. Kawasaki, N. Sakaguchi, N. Fukushima, N. Fujioka, F. Nikaido, M. Yamashita and S. Ohta, Tetrahedron Lett., 2002, 43, 4377–4380 CrossRef CAS; (b) I. Kawasaki, N. Sakaguchi, A. Khadeer, M. Yamashita and S. Ohta, Tetrahedron, 2006, 62, 10182–10192 CrossRef CAS PubMed.
- (a) Ref. 1a;; (b) Ref. 2a;; (c) Ref. 2b;; (d) Ref. 7a;; (e) A. Al Mourabit and P. Potier, Eur. J. Org. Chem., 2001, 237–243 CrossRef; (f) B. H. Northrop, D. P. O'Malley, A. L. Zografos, P. S. Baran and K. N. Houk, Angew. Chem., Int. Ed., 2006, 45, 4126–4130 CrossRef CAS PubMed.
- (a) E. P. Stout, Y.-G. Wang, D. Romo and T. F. Molinski, Angew. Chem., Int. Ed., 2012, 51, 4877–4881 CrossRef CAS PubMed; (b) E. P. Stout, B. I. Morinaka, Y.-G. Wang, D. Romo and T. F. Molinski, J. Nat. Prod., 2012, 75, 527–530 CrossRef CAS PubMed.
- (a) D. J. Bellville, D. D. Wirth and N. L. Bauld, J. Am. Chem. Soc., 1981, 103, 718–720 CrossRef CAS; (b) N. L. Bauld and R. Pabon, J. Am. Chem. Soc., 1983, 105, 633–634 CrossRef CAS; (c) D. W. Reynolds and N. L. Bauld, Tetrahedron, 1986, 42, 6189–6194 CrossRef CAS; (d) N. L. Bauld, Tetrahedron, 1989, 45, 5307–5363 CrossRef CAS; (e) W. Yueh and N. L. Bauld, J. Am. Chem. Soc., 1995, 117, 5671–5676 CrossRef CAS.
- C. Eder, P. Proksch, V. Wray, R. W. M. v. Soest, E. Ferdinandus, L. A. Pattisina and Sudarsono, J. Nat. Prod., 1999, 62, 1295–1297 CrossRef CAS PubMed.
- (a) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (b) T. P. Yoon, ACS Catal., 2013, 3, 895–902 CrossRef CAS PubMed; (c) M. A. Ischay and T. P. Yoon, Eur. J. Org. Chem., 2012, 3359–3372 CrossRef CAS PubMed; (d) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (e) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (f) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed; (g) J. J. Douglas, J. D. Nguyen, K. P. Cole and C. R. J. Stephenson, Aldrichimica Acta, 2014, 47, 15–25 CAS; (h) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (i) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (j) F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859–917 CrossRef; (k) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed.
- (a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed; (b) J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604–14605 CrossRef CAS PubMed; (c) M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572–8574 CrossRef CAS PubMed; (d) S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed; (e) E. L. Tyson, E. P. Farney and T. P. Yoon, Org. Lett., 2012, 14, 1110–1113 CrossRef CAS PubMed; (f) M. A. Ischay, M. S. Ament and T. P. Yoon, Chem. Sci., 2012, 3, 2807–2811 RSC; (g) Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed; (h) J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed; (i) A. E. Hurtley, Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 8991–8994 CrossRef CAS PubMed.
- (a) D. Valentine, Jr. and G. S. Hammond, J. Am. Chem. Soc., 1972, 94, 3449–3454 CrossRef; (b) F.-C. Chen, S.-C. Chang, G. He, S. Pyo, Y. Yang, M. Kurotaki and J. Kido, J. Polym. Sci., Part B: Polym. Phys., 2003, 21, 2681–2690 CrossRef PubMed.
- N. K. Garg, R. Sarpong and B. M. Stoltz, J. Am. Chem. Soc., 2002, 124, 13179–13184 CrossRef CAS PubMed.
- A. Breder, G. M. Chinigo, A. W. Waltman and E. M. Carreira, Chem. – Eur. J., 2011, 17, 12405–12416 CrossRef CAS PubMed.
- (a) Ref. 5b;; (b) L. H. Foley and G. Büchi, J. Am. Chem. Soc., 1982, 104, 1776–1777 CrossRef CAS.
- (a) O. H. Gringore and F. P. Rouessac, Org. Synth., 1985, 63, 121–126 CrossRef CAS; (b) A. T. Austin and J. Howard, J. Chem. Soc., 1961, 3278–3284 RSC; (c) A. T. Austin and J. Howard, J. Chem. Soc., 1961, 3284–3289 RSC; (d) A. T. Austin and J. Howard, J. Chem. Soc., 1961, 3593–3603 RSC; (e) M. Taniguchi, K. Koga and S. Yamada, Tetrahedron, 1974, 30, 3547–3552 CrossRef CAS.
- U. Ravid, R. M. Silverstein and L. R. Smith, Tetrahedron, 1978, 34, 1449–1452 CrossRef CAS.
- A. Takle and P. Kocieński, Tetrahedron, 1990, 46, 4503–4516 CrossRef CAS.
- A. P. Kozikowski, K. L. Sorgi and R. J. Schmiesing, J. Chem. Soc., Chem. Commun., 1980, 477–479 RSC.
- (a) Ref. 7b;; (b) J. Muñoz, C. Moriou, L. Ermolenko, J.-F. Gallard, P. D. Marie and A. Al-Mourabit, Tetrahedron Lett., 2012, 53, 5828–5832 CrossRef PubMed.
- (a) Ref. 1a;; (b) F. Cafieri, R. Carnuccio, E. Fattorusso, O. Taglialatela-Scafati and T. Vallefuoco, Bioorg. Med. Chem. Lett., 1997, 7, 2283–2288 CrossRef CAS; (c) M. Assmann, E. Lichte, J. R. Pawlik and M. Köck, Mar. Ecol.: Prog. Ser., 2000, 207, 255–262 CrossRef CAS; (d) E. Richelle-Maurer, M. J. De Kluijver, S. Feio, S. Gaudêncio, H. Gaspar, R. Gomez, R. Tavares, G. Van de Vyver and R. W. M. Van Soest, Biochem. Syst. Ecol., 2003, 31, 1073–1091 CrossRef CAS; (e) M. Assmann, E. Lichte and M. Köck, Boll. Mus. Ist. Biol. Univ. Genova, 2004, 68, 187–193 Search PubMed.
- (a) P. G. Jones, Chem. Soc. Rev., 1984, 13, 157–172 RSC; (b) D. Rogers, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1981, 37, 734–741 CrossRef; (c) H. D. Flack and G. Bernardinelli, J. Appl. Crystallogr., 2000, 33, 1143–1148 CAS; (d) H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed; (e) H. D. Flack, Acta Chim. Slov., 2008, 55, 689–691 CAS; (f) R. W. W. Hooft, L. H. Stravera and A. L. Spek, J. Appl. Crystallogr., 2008, 41, 96–103 CrossRef CAS PubMed; (g) A. L. Thompson and D. J. Watkin, Tetrahedron: Asymmetry, 2009, 20, 712–717 CrossRef CAS PubMed; (h) H. D. Flack, Chimia, 2014, 68, 26–30 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, experimental procedures, and characterization data. See DOI: 10.1039/c5qo00165j |
| This journal is © the Partner Organisations 2015 |