
Metal free access to amide compounds via peroxide-mediated N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N double bond cleavage of azobenzenes†
N double bond cleavage of azobenzenes†
Gang
Hong
,
Dan
Mao
,
Xiaoyan
Zhu
,
Shengying
Wu
* and
Limin
Wang
*
Key Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, P. R. China. E-mail: wanglimin@ecust.edu.cn; wsy1986wsy@126.com; Fax: +86-21-64253881; Tel: +86-21-64253881
First published on 16th June 2015
Abstract
A direct amidation of aldehydes or benzylamines with azobenzenes through TBHP-mediated N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond cleavage of azobenzenes has been developed. A series of amide compounds with a wide range of functionalities were obtained with moderate to good yields. To the best of our knowledge, this is the first example of the NN double bond cleavage of azobenzenes used in synthesizing amide compounds.
N double bond cleavage of azobenzenes has been developed. A series of amide compounds with a wide range of functionalities were obtained with moderate to good yields. To the best of our knowledge, this is the first example of the NN double bond cleavage of azobenzenes used in synthesizing amide compounds.
Introduction
Recently the synthesis of amide compounds has attracted wide attention for their extensive applications in pharmaceuticals, agrochemicals and biomolecules.1 Some pharmaceutical compounds containing the amide moiety are depicted in Fig. 1. One of the most traditional methods for the synthesis of amides is the condensation of activated carboxylic acids with amines. Many classical name reactions such as Beckmann,2 Staudinger,3 Ritter reactions4 have been developed to form amide compounds. Furthermore, research during the past decade led to significant progress in the field of direct amidation of aldehydes,5 yet for most of which a transition metal was a must. As a result, it is highly necessary to further develop a new method to synthesize amide compounds under mild conditions from other starting materials.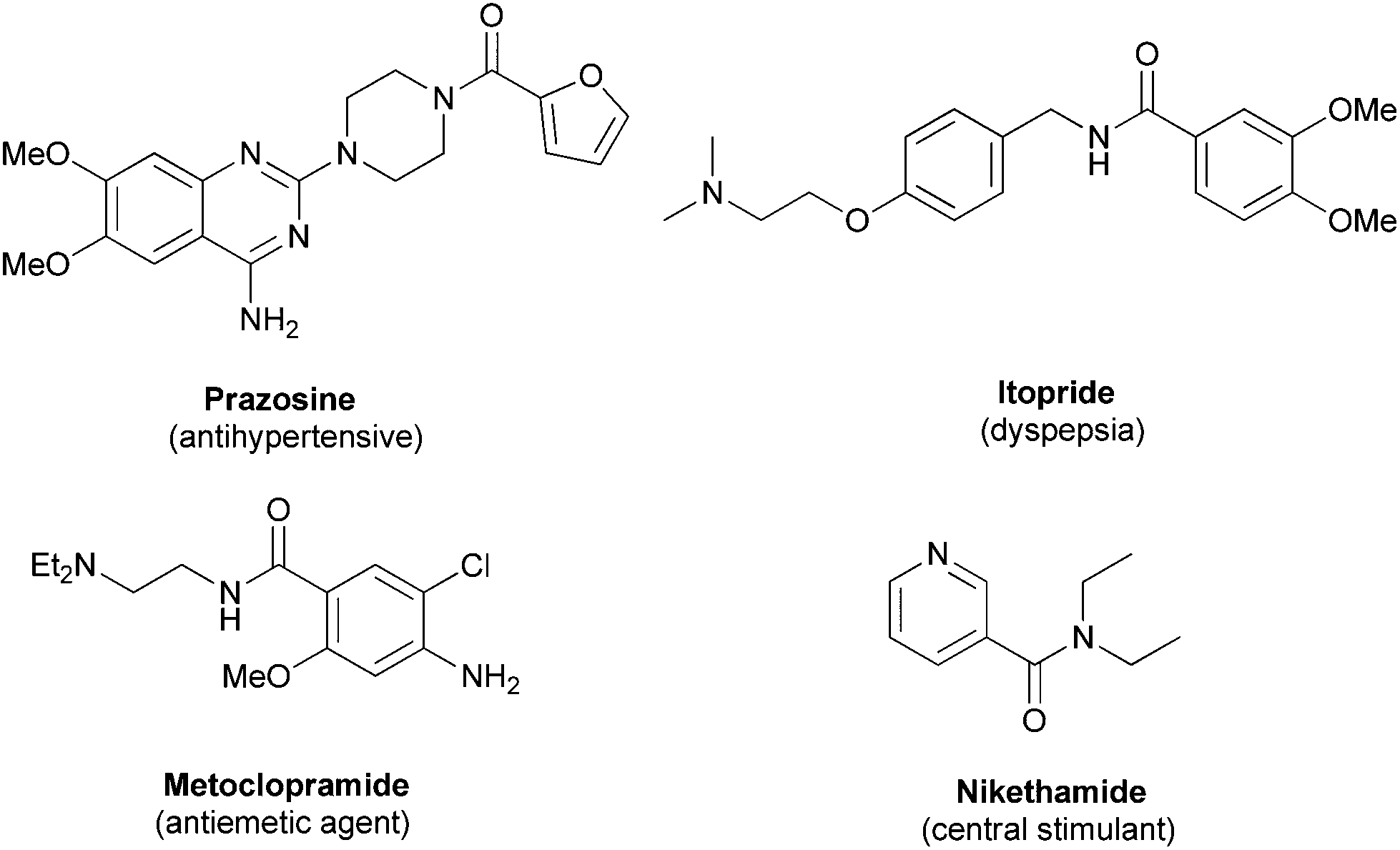 | ||
| Fig. 1 Structure of pharmaceutical compounds with an amide bond. | ||
Aromatic azo compounds are important materials and have been extensively applied in fields such as indicators, dyes, nonlinear optics and pharmaceuticals due to their unique properties.6 Due to the extensive applications, substantial quantities of toxic and carcinogenic azo compounds are dumped into the environment as industrial waste. Thus, it is vital to develop methods for the elimination of these compounds for environmental concerns. The cleavage of the NN double bond in azo compounds can be achieved through either electrolytic reduction7 or chemical reduction using reducing agents such as metal iron8 and sulfides.9 Complementary to hydrogenations are transfer hydrogenations (CTH),10 where typically alcohols especially isopropanol or formic acid–amine mixtures are usually used as hydrogen donors.11
Recently, the reaction of azobenzenes with aldehydes has been studied by the research groups of Wang12 and J. A. Ellman,13 respectively. In Wang's work, the acylation of azobenzenes by Pd-catalyzed oxidative coupling of azobenzenes with aldehydes using tert-butyl hydroperoxide (TBHP) as an oxidant via chelation-assisted ortho C–H bond activation was developed (Scheme 1a), while the J. A. Ellman group has succeeded in synthesizing 2-aryl indazoles by co-catalyzed reaction of azobenzenes with aldehydes (Scheme 1b). To the best of our knowledge, the NN double bond cleavage of aromatic azo compounds under oxidative conditions is rarely reported.14 To further explore this reaction as well as continue our research in aromatic azo compounds,15 we would like to report the first example of accessing amide compounds via TBHP-mediated reaction of azobenzenes with aldehydes or benzylamines (Scheme 1c).
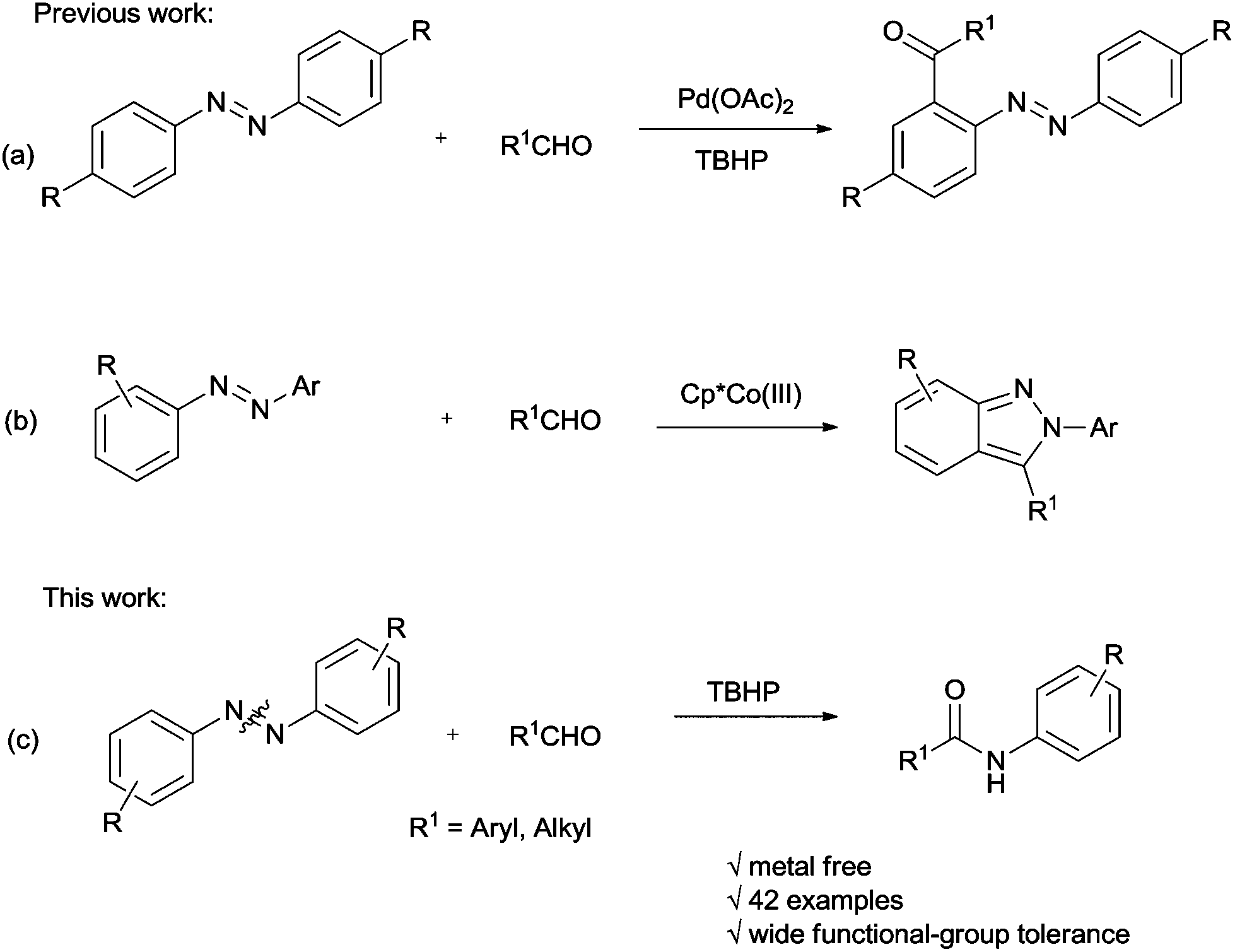 | ||
| Scheme 1 Reaction of azobenzenes with aldehydes studied by different groups. | ||
Results and discussion
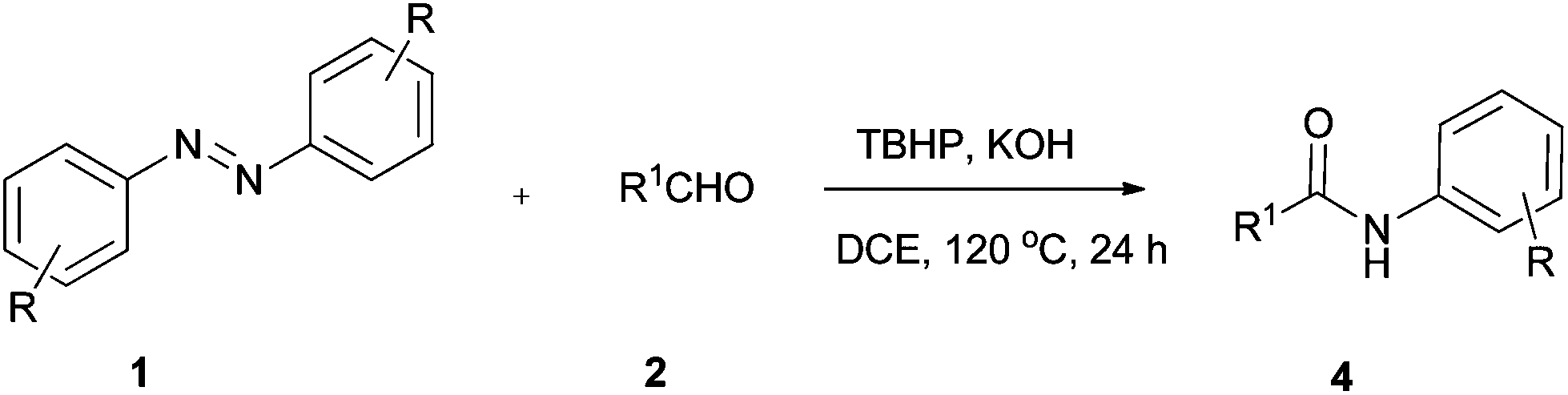
Our study started from the model reaction of azobenzene (1a) with benzaldehyde (2a) to optimize the reaction conditions. The results are summarized in Table 1. The reaction was carried out in DCE at 120 °C with tert-butyl hydroperoxide (TBHP) (4.0 equiv., 70% solution in water) as an oxidant. Gratifyingly the product was obtained in 50% yield after 24 h (Table 1, entry 1). Inspired by this result, various oxidants including di-tert-butyl peroxide (DTBP), DDQ, PhI(OAc)2, and tert-butyl peroxybenzoate (TBPB) were employed with no improvement in yield (Table 1, entries 2–5). Additives were next examined (Table 1, entries 6–11). To our delight, the desired product 4a was obtained in 81% yield in the presence of TBHP and KOH. Efforts to enhance yield by replacing DCE with other solvents such as PhCl, dioxane, DMF, CH3CN, and DMSO (Table 1, entries 12–16) proved fruitless. Additional screening revealed that the yield decreased gradually upon increasing or decreasing the temperature (Table 1, entries 17–19). A decrease in the amount of TBHP led to a decrease of the yield of the product (Table 1, entry 20). There was a slight decrease in yield when the reaction was conducted under argon (Table 1, entry 21).|
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant | Additive (equiv.) | Solvent | T (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.25 mmol), 2a (0.25 mmol), oxidant (4.0 equiv.), additive, solvent (1.0 mL), 24 h, air. b Isolated yield. c TBHP (3.0 equiv.) was used. d Under argon. | |||||
| 1 | TBHP | — | DCE | 120 | 50 |
| 2 | DTBP | — | DCE | 120 | N.P. |
| 3 | DDQ | — | DCE | 120 | N.P. |
| 4 | PhI(OAc)2 | — | DCE | 120 | N.P. |
| 5 | TBPB | — | DCE | 120 | 46 |
| 6 | TBHP | TBAB (1.0) | DCE | 120 | 75 |
| 7 | TBHP | I2 (0.2) | DCE | 120 | 8 |
| 8 | TBHP | KI (0.2) | DCE | 120 | N.P. |
| 9 | TBHP | FeCl2 (0.1) | DCE | 120 | Trace |
| 10 | TBHP | KOH (1.0) | DCE | 120 | 81 |
| 11 | TBHP | PivOH (2.0) | DCE | 120 | 54 |
| 12 | TBHP | KOH (1.0) | PhCl | 120 | 61 |
| 13 | TBHP | KOH (1.0) | CH3CN | 120 | 43 |
| 14 | TBHP | KOH (1.0) | Dioxane | 120 | 63 |
| 15 | TBHP | KOH (1.0) | DMF | 120 | Trace |
| 16 | TBHP | KOH (1.0) | DMSO | 120 | 33 |
| 17 | TBHP | KOH (1.0) | DCE | 110 | 56 |
| 18 | TBHP | KOH (1.0) | DCE | rt | N.P. |
| 19 | TBHP | KOH (1.0) | DCE | 140 | 77 |
| 20c | TBHP | KOH (1.0) | DCE | 120 | 60 |
| 21d | TBHP | KOH (1.0) | DCE | 120 | 62 |

With the optimized conditions in hand, we explored the scope of this novel TBHP-mediated reaction of aldehydes with azobenzenes, and the results are summarized in Table 2. Generally, electron-poor azobenzenes are more reactive than electron-rich aromatic azo compounds. Azobenzenes with electron-withdrawing groups (such as 4-F, 4-Cl, 4-Br, 3-Cl, 3-Br, 4-COOEt, and 4-OCF3) on the aromatic ring worked well with benzaldehyde giving the corresponding products in good to excellent yields (Table 2, entries 6–12), while electron-donating groups (such as 4-OMe, 2-Me, 3-Me, and 4-Me) on the aromatic ring had a slightly negative effect on the yield of the reaction (Table 2, entries 2–5). The fact that the ortho-substituted and 2,4-disubstituted azobenzenes afforded a relatively lower yield than para- or meta-substituted azobenzenes showed that steric hindrance largely affected the efficiency of this reaction (Table 2, entries 2 and 13). Next, the scope of aldehydes was explored. Contrary to the electronic effect observed in substituted azobenzenes, the aromatic aldehydes with electron-donating groups at ortho, meta and para positions of the aromatic ring afforded the corresponding products in 43–72% yields (Table 2, entries 14–20), while electron-withdrawing groups (such as 4-F, 4-Cl, 4-Br, 3-F, 4-NO2, 4-CN, 4-CF3, and 2-CF3) on the aromatic ring gave slightly lower yields (Table 2, entries 21–28). Notably, meta-F benzaldehyde worked well with azobenzene affording the corresponding product 4x in 79% yield (Table 2, entry 24). To our delight, aliphatic aldehydes such as cyclohexanecarbaldehyde, butyraldehyde could also be transformed into the corresponding amide compounds (Table 2, entries 29 and 31). When azobenzene was treated with furaldehyde, product 4ad was isolated in 38% yield (Table 2, entry 30).
|
|
||||
|---|---|---|---|---|
| Entry | 1, R = | 2, R1 = | Product 4 | Yieldb (%) |
| a Reaction conditions: azobenzenes 1 (0.25 mmol), aldehydes 2 (0.25 mmol), TBHP (4.0 equiv.), KOH (1.0 equiv.), DCE (1.0 mL), 120 °C, 24 h, air. b Isolated yield. | ||||
| 1 | H | Ph | 4a | 81 |
| 2 | 2-Me | Ph | 4b | 47 |
| 3 | 3-Me | Ph | 4c | 70 |
| 4 | 4-Me | Ph | 4d | 72 |
| 5 | 4-OMe | Ph | 4e | 41 |
| 6 | 4-F | Ph | 4f | 64 |
| 7 | 4-Cl | Ph | 4g | 75 |
| 8 | 4-Br | Ph | 4h | 91 |
| 9 | 3-Cl | Ph | 4i | 57 |
| 10 | 3-Br | Ph | 4j | 58 |
| 11 | 4-COOEt | Ph | 4k | 82 |
| 12 | 4-OCF3 | Ph | 4l | 70 |
| 13 | 2,4-Me, Me | Ph | 4m | 29 |
| 14 | H | 2-MeC6H4 | 4n | 57 |
| 15 | H | 3-MeC6H4 | 4o | 72 |
| 16 | H | 4-MeC6H4 | 4p | 68 |
| 17 | H | 2-OMeC6H4 | 4q | 46 |
| 18 | H | 3-OMeC6H4 | 4r | 64 |
| 19 | H | 4-OMeC6H4 | 4s | 66 |
| 20 | H | 4-Dimethylamino C6H4 | 4t | 43 |
| 21 | H | 4-FC6H4 | 4u | 41 |
| 22 | H | 4-ClC6H4 | 4v | 53 |
| 23 | H | 4-BrC6H4 | 4w | 39 |
| 24 | H | 3-FC6H4 | 4x | 79 |
| 25 | H | 4-NO2C6H4 | 4y | 40 |
| 26 | H | 4-CF3C6H4 | 4z | 43 |
| 27 | H | 4-CNC6H4 | 4aa | 38 |
| 28 | H | 2-CF3C6H4 | 4ab | 43 |
| 29 | H | Cyclohexyl | 4ac | 34 |
| 30 | H | Furan-2-yl | 4ad | 38 |
| 31 | H | n-Propyl | 4ae | 47 |
Proceeding further toward the substrate exploration of this protocol, a broad range of readily available benzylamines were also screened for this reaction protocol, which are summarized in Table 3. The optimized reaction parameters were azobenzene (0.25 mmol), benzylamines (0.5 mmol), TBHP (4.0 equiv.), and DCE as a solvent, at 120 °C for 24 h (see the ESI† for the optimization table). It was found that no obvious decrease in yield was observed when aldehydes were replaced with the corresponding benzylamines, and the electronic effect of benzylamine observed was similar with substituted aldehydes. The reaction of azobenzene with benzylamines with substituents such as Me, OMe, F, Cl, and Br on different positions of the phenyl ring delivered the corresponding products in 47–80% yields (Table 3, entries 2–10). Furthermore, when benzylamine hydrochloride was employed, the corresponding product was also formed as long as KOH was added as an additive (Table 3, entry 11).
|
|
||||
|---|---|---|---|---|
| Entry | 1, R = | 3, R2 = | Product 4 | Yieldb (%) |
| a Reaction conditions: azobenzene 1a (0.25 mmol), benzylamines 3 (0.5 mmol), TBHP (4.0 equiv.), DCE (1.0 mL), 120 °C, 24 h, air. b Isolated yield. c p-Nitrobenzylamine hydrochloride was used; KOH (1.0 equiv.) was added. | ||||
| 1 | H | Ph | 4a′ | 72 |
| 2 | H | 2-MeC6H4 | 4n′ | 54 |
| 3 | H | 3-MeC6H4 | 4o′ | 74 |
| 4 | H | 4-MeC6H4 | 4p′ | 66 |
| 5 | H | 4-OMeC6H4 | 4s′ | 61 |
| 6 | H | 4-FC6H4 | 4u′ | 53 |
| 7 | H | 4-ClC6H4 | 4v′ | 56 |
| 8 | H | 4-BrC6H4 | 4w′ | 47 |
| 9 | H | 3-FC6H4 | 4x′ | 80 |
| 10 | H | 2-FC6H4 | 4af | 57 |
| 11 | H | 4-NO2C6H4 | 4y′ | 37c |

To probe the mechanism of this reaction, several control experiments were performed (Scheme 2). When a typical radical scavenger tetramethylpiperidine N-oxide (TEMPO) was added to the reaction of azobenzene (1a) with benzaldehyde (2a) under the optimized conditions, no corresponding product was detected, which indicates that a radical process may be involved in this reaction (Scheme 2a). Subsequently, benzoic acid was employed to react with aniline under standard conditions, and no product was observed either, which excludes the possibility that formal disproportionation reaction between azobenzene and benzaldehyde was involved (Scheme 2b). As for the unsymmetrically substituted azobenzene, two amidation products, 4a and 4k, were isolated in 38% and 22% yields, respectively (Scheme 2c). On the other hand, there was only a trace of the corresponding product detected when aniline reacted with benzaldehyde (Scheme 2d) and no corresponding product was furnished for ethyl 4-aminobenzoate under standard conditions (Scheme 2e). Moreover, more than 90% of azobenzene survived in the absence of aldehyde (Scheme 2f). These results indicate that amines may not be the intermediates in the transformation, and the amidation reaction may start from the attack of aldehydes to the NN double bond of azobenzenes and the acyl radical can attack the different positions of the NN double bond when unsymmetrically substituted azobenzene is used.
 | ||
| Scheme 2 Control experiments for investigation of the mechanism. | ||
On the basis of the previous reports12,16–19 and the above results, a tentative mechanism for TBHP-mediated reaction of aldehydes or benzylamines with aromatic azo compounds is depicted in Scheme 3. Benzylamine (3a) first underwent oxidation, hydrolysis and the second radical oxidation with a solution of TBHP in water to form the acyl radical.12,17 Then, the NN double bond of azobenzene (1a) was attacked by this acyl radical to form the intermediate 5.14,16 This intermediate 5 may then abstract one H from tBuOH to provide 6.19 Finally, intermediate 6 could afford the product 4a and nitrosobenzene via the hydrolysis process.18 At the same time, the decomposition of the unstable nitrosobenzene led to the formation of traces of aniline and azoxybenzene (detected by HRMS, see ESI for Fig. S1†), and nitrobenzene (determined by GC-MS, see ESI for Fig. S2 and S3†).
 | ||
| Scheme 3 Tentative mechanism for the amidation of aldehydes or benzylamines. | ||
Conclusions
In conclusion, we have developed a novel method for the synthesis of amide compounds by reaction of aldehydes or benzylamines with aromatic azo compounds using TBHP as an oxidant. Appreciable aldehydes including aliphatic aldehydes can be used in this reaction allowing a direct preparation of the corresponding amide compounds in 29–91% yields. Based on the extensive experimental data, we propose a plausible mechanism. Further studies to refine the mechanism and to extend the application of azobenzenes as synthons are currently underway in our laboratory.Acknowledgements
This research was financially supported by the National Nature Science Foundation of China (21272069, 20672035) and the Fundamental Research Funds for the Central Universities and Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences.Notes and references
- (a) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (b) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (c) D. W. Zhang, X. Zhao, J. L. Hou and Z. T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed; (d) R. Garcia-Alvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46–66 RSC; (e) T. Cupido, J. Tulla-Puche, J. Spengler and F. Albericio, Curr. Opin. Drug Discovery Dev., 2007, 10, 768–783 CAS; (f) V. Onnis, M. T. Cocco and R. Fadda, Bioorg. Med. Chem., 2009, 17, 6158–6165 CrossRef CAS PubMed.
- (a) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599–3601 CrossRef CAS PubMed; (b) M. Hashimoto, Y. Obora, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2008, 73, 2894–2897 CrossRef CAS PubMed.
- (a) E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS; (b) F. Damkaci and P. DeShong, J. Am. Chem. Soc., 2003, 125, 4408–4409 CrossRef CAS PubMed.
- J. J. Ritter and P. P. Minieri, J. Am. Chem. Soc., 1948, 70, 4045–4048 CrossRef CAS.
- (a) W. J. Yoo and C. J. Li, J. Am. Chem. Soc., 2006, 128, 13064–13065 CrossRef CAS PubMed; (b) S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. L. L. Chai and A. Q. Chen, J. Org. Chem., 2012, 77, 8007–8015 CrossRef CAS PubMed; (c) S. C. Ghosh, J. S. Y. Ngiam, C. L. L. Chai, A. M. Seayad, T. T. Dang and A. Q. Chen, Adv. Synth. Catal., 2012, 354, 1407–1412 CrossRef CAS PubMed; (d) M. Zhang and X. F. Wu, Tetrahedron Lett., 2013, 54, 1059–1062 CrossRef CAS PubMed; (e) Y. Suto, N. Yamagiwa and Y. Torisawa, Tetrahedron Lett., 2008, 49, 5732–5735 CrossRef CAS PubMed; (f) Y. Z. Ding, X. Zhang, D. Y. Zhang, Y. T. Chen, Z. B. Wu and S. Yang, Tetrahedron Lett., 2015, 56, 831–833 CrossRef CAS PubMed; (g) K. E. Kovi and C. Wolf, Org. Lett., 2007, 9, 3429–3432 CrossRef PubMed.
- (a) A. Bafana, S. S. Devi and T. Chakrabarti, Environ. Rev., 2011, 19, 350 CrossRef CAS PubMed; (b) G. J. Gainsford, M. D. H. Bhuiyan, I. Asselberghs and K. Clays, Dyes Pigm., 2012, 95, 455 CrossRef PubMed; (c) D. H. Qu and H. Tian, Adv. Mater., 2006, 18, 2035 CrossRef CAS PubMed; (d) A. J. Harvey and A. D. Abell, Tetrahedron, 2000, 56, 9763 CrossRef CAS; (e) M. R. Banghart, A. Mourot, D. L. Fortin, J. Z. Yao, R. H. Kramer and D. Trauner, Angew. Chem., Int. Ed., 2009, 48, 9097 CrossRef CAS PubMed; (f) F. Puntoriero, P. Ceroni, V. Balzani, G. Bergamini and F. Voegtle, J. Am. Chem. Soc., 2007, 129, 10714 CrossRef CAS PubMed; (g) Y. Zhou, D. S. Wang, S. L. Huang, G. Auernhammer, Y. J. He, H. J. Butt and S. W, Chem. Commun., 2015, 51, 2725–2727 RSC.
- R. Jain, M. Bhargava and N. Sharma, J. Sci. Ind. Res., 2003, 62, 813–819 CAS.
- C. Cao, L. P. Wei, Q. G. Huang, L. S. Wang and S. K. Han, Chemosphere, 1999, 38, 565–571 CrossRef.
- F. P. van der Zee, G. Lettinga and J. A. Field, Water Sci. Technol., 2000, 42, 301–308 CAS.
- For reviews on transfer hydrogenations, see: (a) S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC; (b) J. S. M. Samec, J. E. Backvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- (a) S. Gowda, K. Abraj and D. C. Gowda, Tetrahedron Lett., 2002, 43, 1329–1331 CrossRef CAS; (b) F. K. Khan, J. Dash, C. Sudheer and R. K. Gupta, Tetrahedron Lett., 2003, 44, 7783–7787 CrossRef CAS PubMed.
- H. J. Li, P. H. Li and L. Wang, Org. Lett., 2013, 15, 629 Search PubMed.
- J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490–498 CrossRef CAS PubMed.
- (a) A. S. Ozen and V. Aviyente, J. Phys. Chem. A, 2004, 108, 5990–6000 CrossRef; (b) A. S. Ozen and V. Aviyente, J. Phys. Chem. A, 2003, 107, 4898–4907 CrossRef.
- G. Hong, D. Mao, S. Y. Wu and L. M. Wang, J. Org. Chem., 2014, 79, 10629–10635 CrossRef CAS PubMed.
- K. Selvam, S. Balachandran, R. Velmurugan and M. Swaminathan, Appl. Catal., A., 2012, 413–414, 213–222 CrossRef CAS PubMed.
- For literatures on the generation of the acyl radical, see: (a) Q. Zhang, F. Yang and Y. J. Wu, Chem. Commun., 2013, 49, 6837 RSC; (b) A. B. Khemnar and B. M. Bhanage, Org. Biomol. Chem., 2014, 12, 9631–9637 RSC; (c) O. Basle, J. Bidange, Q. Shuai and C. J. Li, Adv. Synth. Catal., 2010, 352, 1145–1149 CrossRef CAS PubMed; (d) C. L. Li, L. Wang, P. H. Li and W. Zhou, Chem. – Eur. J., 2011, 17, 10208 CrossRef CAS PubMed; (e) Y. N. Wu, B. Z. Li, F. Mao, X. S. Li and F. Y. Kwong, Org. Lett., 2011, 13, 3258 CrossRef CAS PubMed.
- G. W. Rong, D. F. Liu, Y. Hong, J. Chen, Y. Zheng, G. Q. Zhang and J. C. Mao, Adv. Synth. Catal., 2015, 357, 71–76 CrossRef CAS PubMed.
- H. B. Peng, J. T. Yu, Y. Jiang, H. T. Yang and J. Cheng, J. Org. Chem., 2014, 79, 9847–9853 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, optimization of reaction conditions, analytical data and NMR spectra for products. See DOI: 10.1039/c5qo00125k |
| This journal is © the Partner Organisations 2015 |