
Thioether-directed acetoxylation of C(sp2)–H bonds of arenes by palladium catalysis†
Binjie
Wang
a,
Cong
Lin
a,
Yue
Liu
a,
Zili
Fan
a,
Zhanxiang
Liu
*a and
Yuhong
Zhang
*ab
aZJU-NHU United R&D Center, Department of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: liuzhanx@zju.edu.cn; yhzhang@zju.edu.cn
bState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, China
First published on 12th June 2015
Abstract
The palladium-catalyzed acetoxylation of aromatic C(sp2)–H bonds utilizing thioether as the directing group was developed. Both benzyl sulfides and phenethyl sulfides could react with PhI(OAc)2 to afford the site-selectively acetoxylated products in good yields. The directing groups could be further transformed into synthetically useful functional groups or successfully removed.
In recent years, the transition metal-catalyzed C–H bond functionalization of arenes has been widely investigated as a powerful tool for the synthesis of pharmaceuticals, bioactive molecules, and functional materials.1 In particular, the construction of C–O bonds from C–H activation has attracted much attention due to the significant importance of this class of molecules in both academic research and industry.2 However, many of these transformations suffer from poor reactivity and regioselectivity. To overcome the uncontrolled site selectivity, a number of examples of C–O bond forming reactions have been reported in recent years through the introduction of a directing group.3–9 In 2004, Sanford and co-workers described a regio- and chemoselective Pd-catalyzed acetoxylation of C(sp2)–H bonds using pyridine or other N-containing heteroarenes as directing groups.3a A copper-catalyzed ortho-acetoxylation of aryl C–H bonds was developed by Yu and coworkers using dioxygen gas as the terminal oxidant.4a Since these pioneering reports, various directing groups, such as oximes,5 sulfoximines,6 pyrimidines,7 anilides,8 amides9 and organophosphates,10 have been successfully employed as the directing groups for the selective C(sp2)–H acetoxylation using Pd, Rh or Cu catalysts. In recent years, the more challenging oxygenation of C(sp3)–H bonds has been achieved employing various N-based directing groups by several research groups.11 A majority of these successful acetoxylation reactions were limited to the efficient coordination between nitrogen and/or oxygen with the transition metals, thus expanding the scope of the directing group remains a critical challenge.
Organosulfur compounds have commonly served as important intermediates in organic synthesis and crucial precursors to the functionalized molecules. Many of the sulfur containing molecules present the unique properties and thus have found wide applications in pharmaceuticals and functional materials.12,13 Accordingly, the direct functionalization of the S-containing compounds via C–H bond activation is highly desirable. However, relevant examples using sulfur species as the directing groups for C–H bond activation are still under developed.14 Recently, our group reported the example employing thioether as a directing group for the palladium-catalyzed alkenylation and arylation of arenes.14a,b Alkenylation of benzyl thioethers and phenyl sulfoxides via Rh-catalyzed aryl C–H activation was described by Shi14d and Miura14f groups. In connection with our interest in C–H functionalization of arenes, we herein describe the palladium-catalyzed oxidative acetoxylation of C(sp2)–H bonds of arenes applying thioether as a removable directing group. Site-selective acetoxylated products could be afforded in moderate to good yields with tolerance to a wide variety of functional groups.
We initiated the investigation by the reaction of 1.0 equiv. of benzyl sulfide 1a with 10 mol% of Pd(OAc)2 and PhI(OAc)2 (1.5 equiv.) in DCE/Ac2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 110 °C for 24 h. The expected mono-acetoxylated product was isolated in 31% yield. With this promising result, we were encouraged to optimize the reaction conditions (Table 1). No desired product was generated in the absence of a palladium catalyst. The effect of oxidants was examined in detail, and PhI(OAc)2 showed the best reactivity. Other oxidants such as AgOAc, Cu(OAc)2, and K2S2O8 failed to generate the product (entries 2–4). The reaction displayed tremendous solvent dependence characteristics and was performed in acetic anhydride with 1,2-dichloroethane as the cosolvent. Other cosolvents such as acetonitrile, tetrahydrofuran, and toluene were also screened, which resulted in decreased yields (entries 5–7). The transformation performed in pure Ac2O and DCE gave 22% and 18% yields, respectively (entries 8 and 9). Moreover, the reaction efficiency could be further improved by the use of different additives.4b The employment of a stoichiometric amount of Li2CO3 gave the product in 63% yield (entry 10). Further investigation into other additives such as LiOAc, AgOAc, LiF, and K2CO3 demonstrated that the best isolated yield was achieved when 1.0 equiv. of AgOAc was used as an additive (entries 11–14). Thus the optimal reaction condition was 10 mol% of Pd(OAc)2 with 1.5 equiv. of PhI(OAc)2 as the oxidant, AgOAc as the additive, and Ac2O/DCE (1:1) as the solvent at 110 °C.
:1) at 110 °C for 24 h. The expected mono-acetoxylated product was isolated in 31% yield. With this promising result, we were encouraged to optimize the reaction conditions (Table 1). No desired product was generated in the absence of a palladium catalyst. The effect of oxidants was examined in detail, and PhI(OAc)2 showed the best reactivity. Other oxidants such as AgOAc, Cu(OAc)2, and K2S2O8 failed to generate the product (entries 2–4). The reaction displayed tremendous solvent dependence characteristics and was performed in acetic anhydride with 1,2-dichloroethane as the cosolvent. Other cosolvents such as acetonitrile, tetrahydrofuran, and toluene were also screened, which resulted in decreased yields (entries 5–7). The transformation performed in pure Ac2O and DCE gave 22% and 18% yields, respectively (entries 8 and 9). Moreover, the reaction efficiency could be further improved by the use of different additives.4b The employment of a stoichiometric amount of Li2CO3 gave the product in 63% yield (entry 10). Further investigation into other additives such as LiOAc, AgOAc, LiF, and K2CO3 demonstrated that the best isolated yield was achieved when 1.0 equiv. of AgOAc was used as an additive (entries 11–14). Thus the optimal reaction condition was 10 mol% of Pd(OAc)2 with 1.5 equiv. of PhI(OAc)2 as the oxidant, AgOAc as the additive, and Ac2O/DCE (1:1) as the solvent at 110 °C.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Oxidant | Additive | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), catalyst (10 mol%, 0.02 mmol), oxidant (1.5 eq., 0.3 mmol), additive (1.0 eq., 0.2 mmol), solvent (2 mL), 110 °C, 24 h, in a sealed tube. b Isolated yields. | |||||
| 1 | Pd(OAc)2 | DCE/Ac2O (1:1) |
PhI(OAc)2 | — | 31 |
| 2 | Pd(OAc)2 | DCE/Ac2O (1:1) |
AgOAc | — | N.R. |
| 3 | Pd(OAc)2 | DCE/Ac2O (1:1) |
Cu(OAc)2 | — | N.R. |
| 4 | Pd(OAc)2 | DCE/Ac2O (1:1) |
K2S2O8 | — | N.R. |
| 5 | Pd(OAc)2 | CH3CN/Ac2O (1:1) |
PhI(OAc)2 | — | 15 |
| 6 | Pd(OAc)2 | THF/Ac2O (1:1) |
PhI(OAc)2 | — | 14 |
| 7 | Pd(OAc)2 | Toluene/Ac2O (1:1) |
PhI(OAc)2 | — | N.R. |
| 8 | Pd(OAc)2 | DCE | PhI(OAc)2 | — | 22 |
| 9 | Pd(OAc)2 | Ac2O | PhI(OAc)2 | — | 18 |
| 10 | Pd(OAc)2 | DCE/Ac2O (1:1) |
PhI(OAc)2 | Li2CO3 | 63 |
| 11 | Pd(OAc)2 | DCE/Ac2O (1:1) |
PhI(OAc)2 | LiOAc | 56 |
| 12 | Pd(OAc) 2 |
DCE/Ac
2
O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
PhI(OAc) 2 | AgOAc | 79 |
| 13 | Pd(OAc)2 | DCE/Ac2O (1:1) |
PhI(OAc)2 | LiF | N.R. |
| 14 | Pd(OAc)2 | DCE/Ac2O (1:1) |
PhI(OAc)2 | K2CO3 | N.R. |

With the optimized reaction conditions in hand, we examined the reactivity of various substrates of benzyl(p-tolyl)sulfane in Scheme 1. Replacement of p-tolyl thioether with 4-methoxyphenyl thioether led to a decreased yield and the benzyl(methyl)sulfane failed to react, which indicated that the electronic nature of sulfur played an important role during the C–H activation (Scheme 1, 2a–2b). Different substituents in an aryl ring played an important role on the efficiency of the acetoxylation reaction. For example, arenes with a methyl group at the ortho, meta and para positions afforded the product in good yields (Scheme 1, 2c–2e). The electron-donating group such as methoxyl could afford the corresponding product in 81% yield (Scheme 1, 2f). Substrates bearing halides on the arene such as bromo, chloro, and fluoro gave the desired products in moderate yields (Scheme 1, 2g–2i), which were consistent with an electrophilic palladation process. It should be noted that a wide range of functional groups such as CF3, OCF3, CN and COOMe were compatible with this protocol, showing a relatively broad functionality tolerance to the transformation (Scheme 1, 2j–2m).
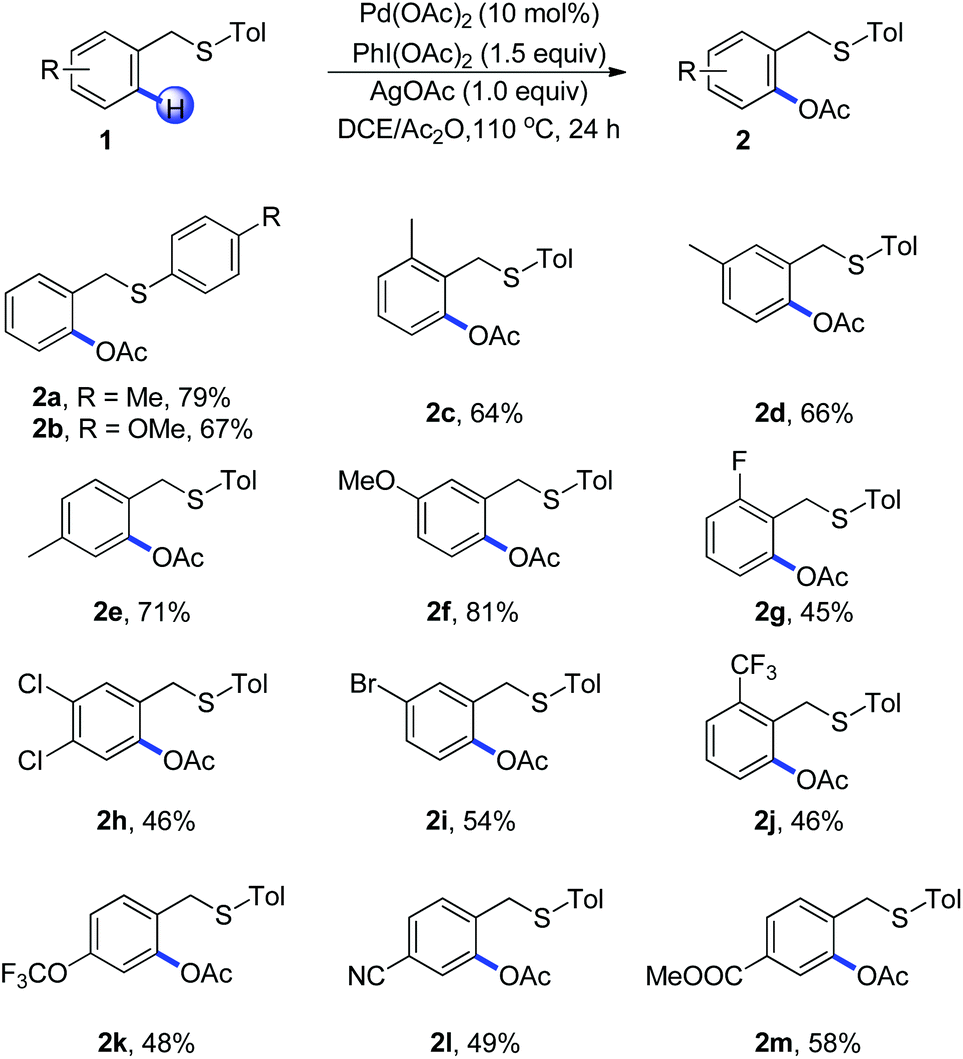 | ||
| Scheme 1 Thioether-directed acetoxylation of C(sp2)–H bonds of benzyl (p-tolyl)sulfane. Reaction conditions: 1 (0.2 mmol), Pd(OAc)2 (10 mol%, 0.02 mmol), PhI(OAc)2 (1.5 eq., 0.3 mmol), AgOAc (1.0 eq., 0.2 mmol), DCE/Ac2O (2 mL), 110 °C, 24 h, in a sealed tube. | ||
Encouraged by the efficient acetoxylation reaction via a five-membered palladacycle, we further examined the substrates with a longer length tether between the sulfur and arene in Scheme 2. The phenethyl sulfides were found to be stable under these oxidative conditions and promoted the reaction in a selective fashion (Scheme 2, 4a–4i). The presence of electron-donating substituents on the aromatic ring has a positive effect on the reaction, leading to the corresponding acetoxylation products in good yields (Scheme 2, 4b–4d). When (2-phenylpropyl)(p-tolyl)sulfane 3e was used, the acetoxylated product was isolated in a low yield of 40%, in which the steric effect might be the major reason (Scheme 2, 4e). Substrates bearing halogen substituents such as fluoro, chloro and bromo were able to undergo the C–H bond acetoxylation reaction in moderate yields, providing a useful routine for further cross-coupling reactions (Scheme 2, 4f–4h). Moreover, 2-naphthalene derivative occurred smoothly, furnishing the desired product 4i as the single regioisomer.
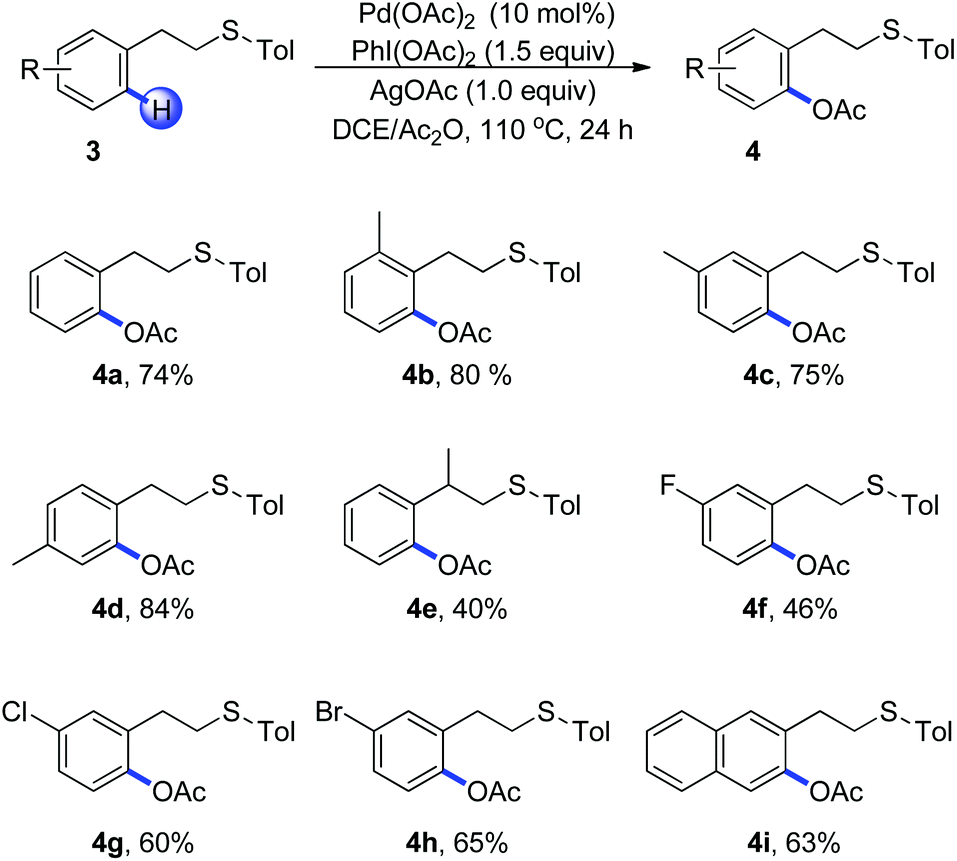 | ||
| Scheme 2 Thioether-directed acetoxylation of C(sp2)–H bonds of phenethyl sulfides. Reaction conditions: 3 (0.2 mmol), Pd(OAc)2 (10 mol%, 0.02 mmol), PhI(OAc)2 (1.5 eq., 0.3 mmol), AgOAc (1.0 eq., 0.2 mmol), DCE/Ac2O (2 mL), 110 °C, 24 h, in a sealed tube. | ||
To gain insight into the mechanism, a competitive experiment using both electron-rich (1f) and electron-deficient (1i) thioether was carried out (Scheme 3). It was revealed that the electron-rich substrate (1f) made the acetoxylation proceed faster, which was consistent with an electrophilic palladation pathway.
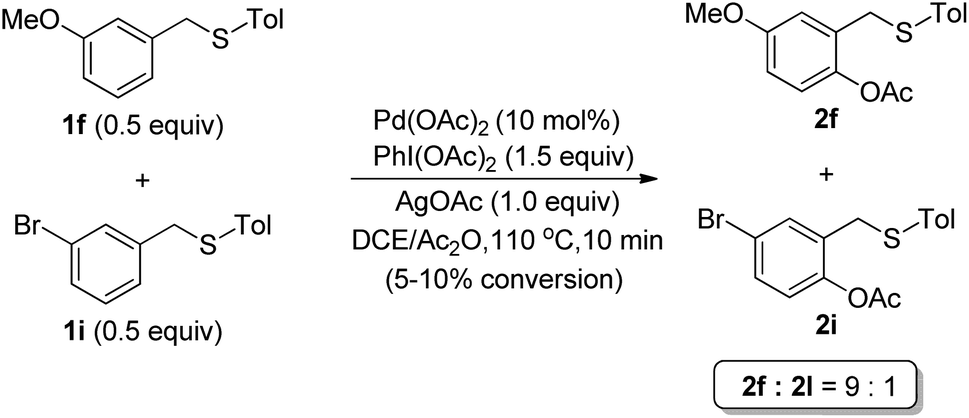 | ||
| Scheme 3 Competitive experiment. | ||
On the basis of the above results, the catalytic cycle for the chelation-controlled position-selective acetoxylation is proposed via the typical C–H activation pathway as shown in Scheme 4. The initial sulfur assisted ortho-palladation leads to the formation of a five- or six-membered palladacycle A, which is oxidized by PhI(OAc)2 in the presence of Ac2O and DCE to generate Pd(IV) intermediate B. The final reductive elimination of intermediate B could afford the ortho-acetoxylated product 2a and regenerate the active Pd(II) species. The rate-determining step might be the electrophilic palladation of the arene, which is consistent with the observed poor reactivity of arenes containing electron-withdrawing groups.
 | ||
| Scheme 4 Reaction mechanism. | ||
After developing the directed C–H acetoxylation reaction, we explored further transformations of the model product 2a (Scheme 5). For example, the oxidation of sulfide 2a by m-CPBA at 0 °C produced the corresponding synthetically useful sulfoxide 5 and sulfone 6 in high yields. The resulting sulfoxide moiety can be almost quantitatively transformed into aldehyde 7 in the presence of acetic anhydride through the Pummerer rearrangement. The cleavage of the S-tether was achieved under reductive conditions using RANEY® Ni, affording the corresponding o-tolyl acetate 8 in 72% yield. Finally, 2-((p-tolylthio)methyl)phenyl acetate 2a was successfully hydrolyzed by NaOH in EtOH/H2O to afford the product 9 in 86% yield.
 | ||
| Scheme 5 Transformation of thioethers. | ||
Conclusions
In conclusion, a palladium(II)-catalyzed thioether-directed regioselective acetoxylation of C(sp2)–H bonds of arenes is described. The ability of sulfur to direct the ortho C–H bond activation with different tether lengths is an important feature of this process. The directing group can be easily removed or converted into a myriad of functionalities, providing new synthetic methods for phenol derivatives. Further exploration of the substrate scope and synthetic utility of these transformations are in progress in our laboratory.Experimental section
General methods
The materials and solvents were purchased from common commercial sources and used without additional purification, if not otherwise noted. 1H NMR spectra were recorded at 400 MHz using TMS as the internal standard. 13C NMR spectra were recorded at 100 MHz using TMS as the internal standard. The multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublets (dd), multiplet (m), and broad resonances (br). Mass spectroscopy data were collected on an HRMS-EI and HRMS-ESI instrument.General procedure for the Pd(II)-catalyzed thioether-directed acetoxylation of C(sp2)–H bonds of arenes
A mixture of benzyl sulfide derivative (0.2 mmol), PhI(OAc)2 (76 mg, 0.3 mmol), Pd(OAc)2 (4 mg, 10 mol%), and AgOAc (33 mg, 0.2 mmol) in 2 mL DCE/Ac2O (1:1) was stirred at 110 °C for 24 h. Afterwards, the reaction mixture was allowed to cool to room temperature and filtered through a pad of Celite. The solvent was evaporated under reduced pressure and the residue was subjected to flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1:10, v/v) to obtain the desired products.
Acknowledgements
Funding from NSFC (No. 21272205, 21472165), NBRPC (No. 2011CB936003), and the Program for Zhejiang Leading Team of S&T Innovation (2011R50007) is acknowledged.Notes and references
- For selected recent reviews, see: (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (e) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (f) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (g) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (h) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (i) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (j) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (k) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (l) Y. Shang, X. Jie, J. Zhou, P. Hu, S. Huang and W. P. Su, Angew. Chem., Int. Ed., 2013, 52, 1299 CrossRef CAS PubMed.
- For selected reviews of C–O formation, see: (a) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- For selected Sanford reports on ortho-acetoxylation of sp2 C–H bonds, see: (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (b) L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef CAS PubMed; (c) D. Kalyani and M. S. Sanford, Org. Lett., 2005, 7, 4149 CrossRef CAS PubMed; (d) S. R. Neufeldt and M. S. Sanford, Org. Lett., 2010, 12, 532 CrossRef CAS PubMed. For mechanistic studies, see: (e) A. R. Dick, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 12790 CrossRef CAS PubMed; (f) K. L. Hull, E. L. Lanni and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 14047 CrossRef CAS PubMed; (g) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 9651 CrossRef CAS PubMed; (h) J. M. Racowski, A. R. Dick and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 10974 CrossRef CAS PubMed; (i) J. M. Racowski, D. B. Nicholas and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18022 CrossRef CAS PubMed; (j) D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS PubMed; (k) K. C. Amanda and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 3109 CrossRef PubMed.
- (a) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed; For pyridine derivatives: (b) N. Chernyak, A. S. Dudnik, C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2010, 132, 8270 CrossRef CAS PubMed; (c) C. Huang, N. Chernyak, A. S. Dudnik and V. Gevorgyan, Adv. Synth. Catal., 2011, 353, 1285 CrossRef CAS PubMed; (d) H. Richter, S. Beckendorf and O. G. Mancheño, Adv. Synth. Catal., 2011, 353, 295 CrossRef CAS PubMed.
- L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141 CrossRef CAS PubMed.
- (a) M. R. Yadav, R. Rit and A. Sahoo, Chem. – Eur. J., 2012, 18, 5541 CrossRef CAS PubMed; (b) R. Rit, M. R. Yadav and A. Sahoo, Org. Lett., 2014, 16, 968 CrossRef CAS PubMed.
- (a) X. Zheng, B. Song and B. Xu, Eur. J. Org. Chem., 2010, 4376 CAS; (b) S. Gu, C. Chen and W. Chen, J. Org. Chem., 2009, 74, 7203 CrossRef CAS PubMed.
- G.-W. Wang, T.-T. Yuan and X.-L. Wu, J. Org. Chem., 2008, 73, 4717 CrossRef CAS PubMed.
- C. J. Vickers, T.-S. Mei and J.-Q. Yu, Org. Lett., 2010, 12, 2511 CrossRef CAS PubMed.
- L. Y. Chan, X. Meng and S. Kim, J. Org. Chem., 2013, 78, 8826 CrossRef CAS PubMed.
- Selected examples of Pd-catalyzed C(sp3)–H acetoxylation: (a) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed; (b) P. Novak, A. Correa, J. G. Donaire and R. Martin, Angew. Chem., Int. Ed., 2011, 50, 12236 CrossRef CAS PubMed; (c) Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991 CrossRef CAS PubMed; (d) R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2012, 14, 3724 CrossRef CAS PubMed; (e) X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersen and X. Shi, Chem. Sci., 2013, 4, 3712 RSC; (f) L. Zhou and W. Lu, Org. Lett., 2014, 16, 508 CrossRef CAS PubMed; (g) Q. Gou, Z.-F. Zhang, Z.-C. Liu and J. Qin, J. Org. Chem., 2015, 80, 3176 CrossRef CAS PubMed.
- (a) S. G. Murray and F. R. Hartley, Chem. Rev., 1981, 81, 365 CrossRef CAS; (b) J. Spencer, M. Pfeffer, A. DeCian and J. Fischer, J. Org. Chem., 1995, 60, 1005 CrossRef CAS; (c) B. Koning, A. Meetsma and R. M. Kellogg, J. Org. Chem., 1998, 63, 5533 CrossRef CAS; (d) R. B. Bedford, C. S. J. Cazin, M. B. Hursthouse, M. E. Light and V. J. M. Scordia, Dalton Trans., 2004, 3864 RSC.
- (a) D. Poirier, S. Auger, Y. Mérand, J. Simard and F. Labrie, J. Med. Chem., 1994, 37, 1115 CrossRef CAS; (b) R. Cammack, Nature, 1999, 397, 214 CrossRef CAS PubMed; (c) H. Greger, T. Pacher, S. Vajrodaya, M. Bacher and O. Hofer, J. Nat. Prod., 2000, 63, 616 CrossRef CAS PubMed; (d) F. Wang, W.-G. Wang, X.-J. Wang, H.-Y. Wang, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2011, 50, 3193 CrossRef CAS PubMed.
- The thioether and sulfoxide group directed C–H functionalization reaction: (a) M. Yu, Y. Xie, C. Xie and Y. Zhang, Org. Lett., 2012, 14, 2164 CrossRef CAS PubMed; (b) J. Yao, M. Yu and Y. Zhang, Adv. Synth. Catal., 2012, 354, 3205 CrossRef CAS PubMed; (c) B. Wang, C. Shen, J. Yao, H. Yin and Y. Zhang, Org. Lett., 2014, 16, 46 CrossRef CAS PubMed; (d) X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem. – Eur. J., 2013, 19, 11898 CrossRef CAS PubMed; (e) T. Wesch, A. Berthelot-Bréhier, F. R. Leroux and F. Colobert, Org. Lett., 2013, 15, 2490 CrossRef CAS PubMed; (f) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188 CrossRef CAS PubMed; (g) T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139 CrossRef CAS PubMed; (h) R. Samanta and A. P. Antonchick, Angew. Chem., Int. Ed., 2011, 50, 5217 CrossRef CAS PubMed; (i) B. Xu, W. Liu and C. Kuang, Eur. J. Org. Chem., 2014, 2576 CrossRef CAS PubMed; (j) Y. Unoh, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2015, 17, 704 CrossRef CAS PubMed; (k) X.-S. Zhang, Y.-F. Zhang, K. Chen and Z.-J. Shi, Org. Chem. Front., 2014, 1, 1096 RSC; (l) C. K. Hazra, Q. Dherbassy, J. W. Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00144g |
| This journal is © the Partner Organisations 2015 |