
Ruthenium complexes with an N-heterocyclic carbene NNC-pincer ligand: preparation and catalytic properties†
Carmen
Mejuto
a,
Marco A.
García-Eleno
b,
Gregorio
Guisado-Barrios
*a,
Denis
Spasyuk
c,
Dmitri
Gusev
*c and
Eduardo
Peris
*a
aInstitute of Advanced Materials (INAM), Universitat Jaume I, Av. Vicente Sos Baynat s/n, 12071-Castellón, Spain. E-mail: eperis@uji.es; Fax: +34 964 387522
bDpt. De Química Inorgánica, Instituto de Química, Circuito Exterior s/n, Ciudad Universitaria. Delegación Coyoacan, Mexico, D. F., Mexico
cDept. of Chemistry and Biochemistry, Wilfrid Laurier University, Waterloo, Ontario N2L 3C5, Canada
First published on 1st June 2015
Abstract
1-Methyl-3-(2-((pyridin-2-ylmethylene)amino)ethyl)-1H-imidazol-3-ium bromide was prepared and used as an N-heterocyclic carbene NNC-pincer ligand precursor. Depending on the coordination strategy, a monometallic [Ru(NNC)(CO3)(PPh3)] complex, or the [Ru(μ-Cl)(NNC)]2(2Cl−) dimer, was obtained. A di-silver complex in which two ligands are monocoordinated to the metal center through the NHC groups was also obtained and characterised. The dimetallic ruthenium complex reacts with alcohols yielding a monohydride species. The preliminary studies on the catalytic activity of the ruthenium dimer indicate that the complex is active in the reduction of ketones and aldehydes under transfer hydrogenation conditions.
Introduction
Transition metal complexes containing pincer-type ligands have attracted increasing attention due to the unusual properties of the metal center imparted by the mer-coordination of the tridentate ligands.1 The tridentate coordination mode of pincer ligands renders high thermal stability to the product complexes, and this is why pincer complexes are often used as catalysts for endothermic reactions, such as dehydrogenations of organic substrates.2 Ruthenium pincer complexes3 proved to be excellent catalysts for alcohol dehydrogenation and also for hydrogenation of organic carbonyl compounds.2b Reactivity patterns such as the deprotonation–dearomatization, metal–ligand cooperativity, and the unusual electronic structures of ruthenium pincer complexes make this type of compounds efficient catalysts for several synthetic methods that include important green transformations. Among the prominent examples are the hydrogenation of carboxylic acid esters,4 nitriles5 or CO2,6 where catalysts based on ruthenium have played a predominant role,4a–k although osmium4a,c and iron4l,m,7 have also proved to be useful catalysts. Some efficient hydrogenation catalysts have incorporated NNC-pincer ligands with N-heterocyclic carbenes, as in the relevant examples reported by Milstein,4f,8 Song9 and others.10 The introduction of NHCs into the structure of pincer ligands is expected to bring catalytic benefits by the presence of the strong electron-donor NHC group, and also by increased thermal stability of the resulting metal complexes,11 compared to the related complexes containing coordinated phosphines or amines (Scheme 1).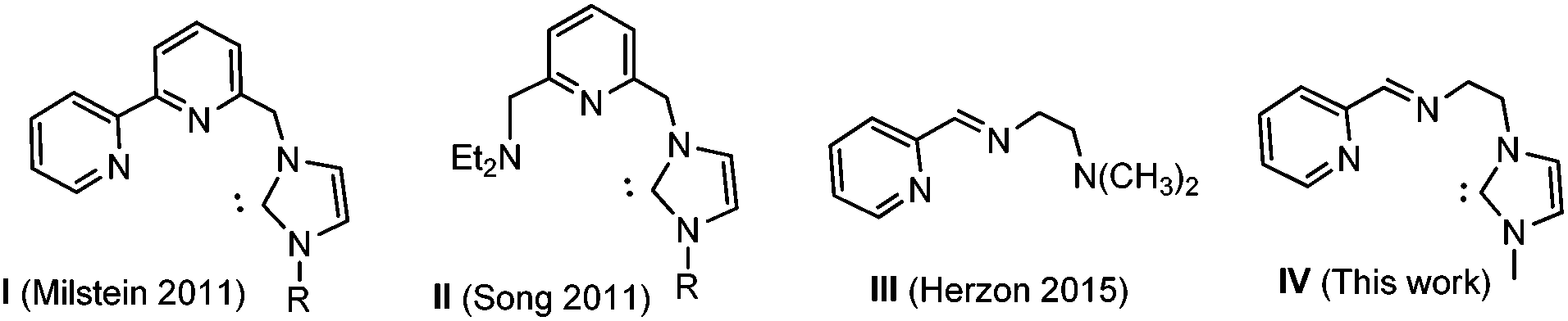 | ||
| Scheme 1 | ||
Based on these previous findings, and in our experience in the chemistry of ruthenium complexes with NHC-based pincer ligands,12 we now report the preparation of ligand IV, which we used for the syntheses of two new ruthenium-NNC pincer complexes. The new ligand IV is somewhat related to the NNN-pincer ligand III recently reported by Herzon and co-workers, which was used for making a Ru complex that was highly active in the reductive hydration of terminal alkynes.13 The coordination properties of IV will be described, together with the preliminary studies on the catalytic activity of the product ruthenium complexes in the reduction of ketones by transfer hydrogenation.
Results and discussion
Preparation of the new compounds
The imidazolium salt 1 was prepared in high yield by condensation of 3-(2-aminoethyl)-1-methylimidazolium bromide and 2-pyridinecarboxaldehyde in iPrOH at room temperature, as shown in Scheme 2. The details on the preparation of the aminoethyl-methylimidazolium salt may be found in the Experimental section of this article.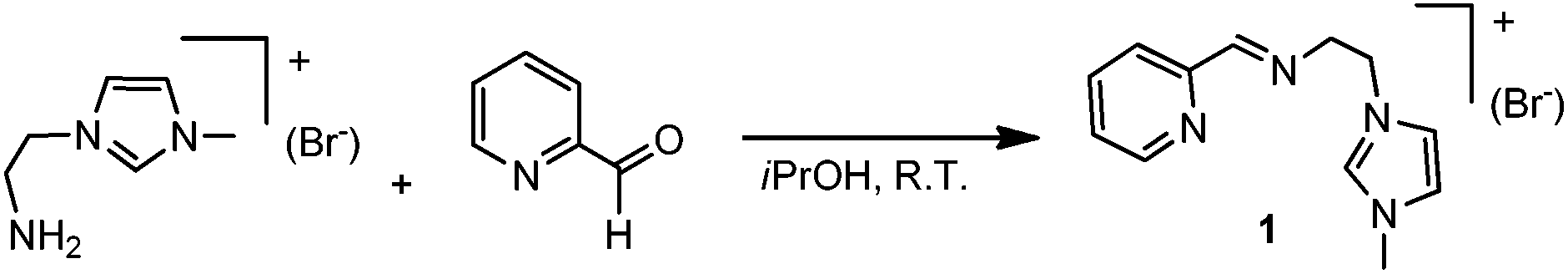 | ||
| Scheme 2 | ||
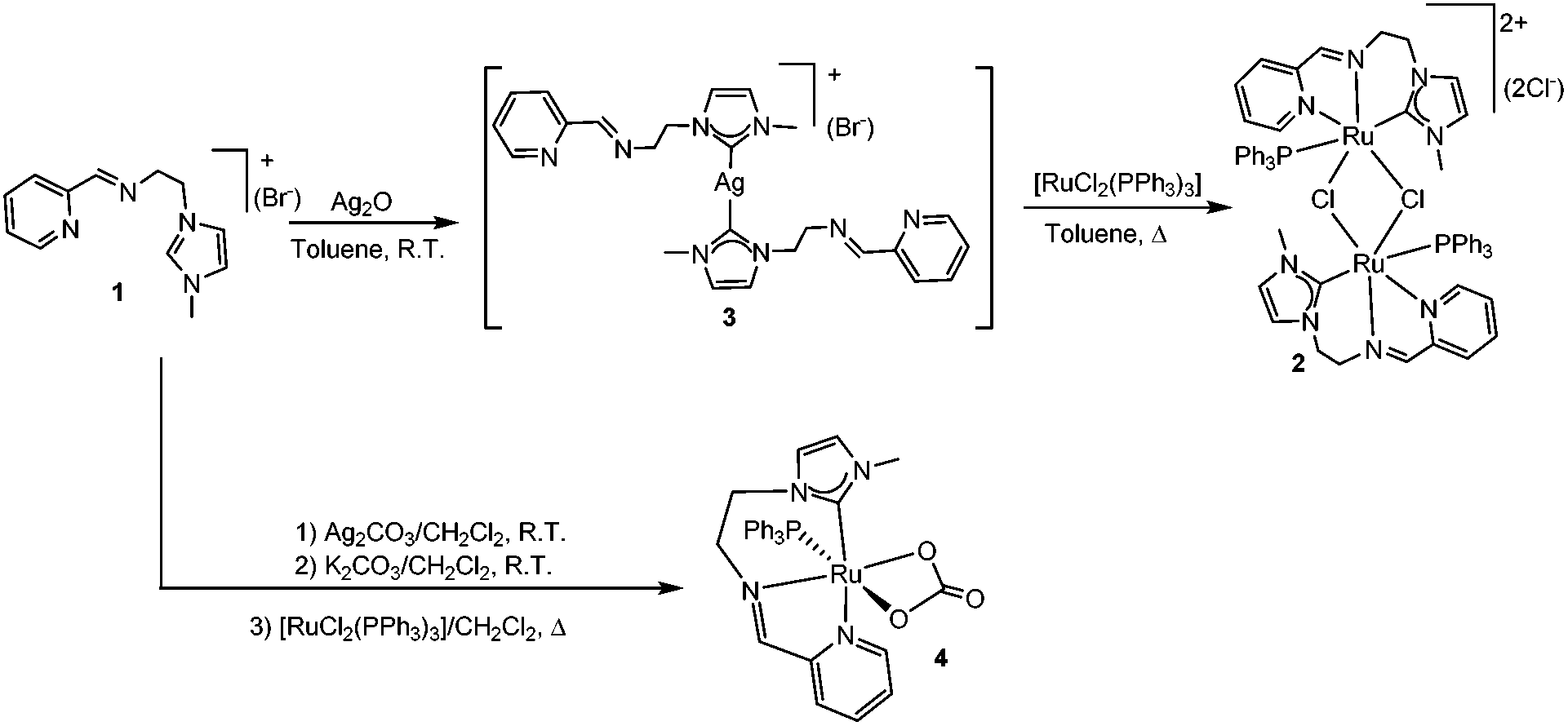
For coordination of 1 to Ru, we tried standard strategies, but all of them afforded products in low yield. For example, the transmetallation from preformed silver–NHC complexes by reaction of 1 with Ag2O and subsequent addition of [RuCl2(PPh3)3] in toluene allowed the formation of the di-ruthenium complex 2, as a red solid in ca. 30% yield (Scheme 3). The Ag–NHC intermediate could be isolated by reaction of 1 with 0.5 equivalents of Ag2O in CH2Cl2 at room temperature, yielding the [Ag(NHC)2]+(Br−) complex 3, in 50% yield.
 | ||
| Scheme 3 | ||
When the reaction is performed between 1 and [RuCl2(PPh3)3] in the presence of Ag2CO3 and K2CO3 in refluxing CH2Cl2, the monometallic complex 4 with a coordinated carbonate ligand is obtained as the only isolable complex, in 9% yield.
Complexes 2–4 were characterized by means of NMR spectroscopy and mass spectrometry, and gave satisfactory elemental analysis results. The 13C NMR spectrum of the silver di-NHC complex 3 displayed the diagnostic signal due to the carbene-carbon atoms as a singlet at 182 ppm. The 1H NMR spectra of 2 and 4 are consistent with the formation of NHC species, as seen by the disappearance of the signal due to the NCHN proton of the imidazolium fragment of 1. The 13C NMR spectrum of 2 revealed the characteristic signal due to the carbene carbon at 184.5 ppm. Although we recorded the 13C NMR spectrum of 4, we were unable to locate the signal of the carbene carbon, probably due to the low concentration at which we had to carry out the NMR study of the complex, which was obtained only in small amounts.
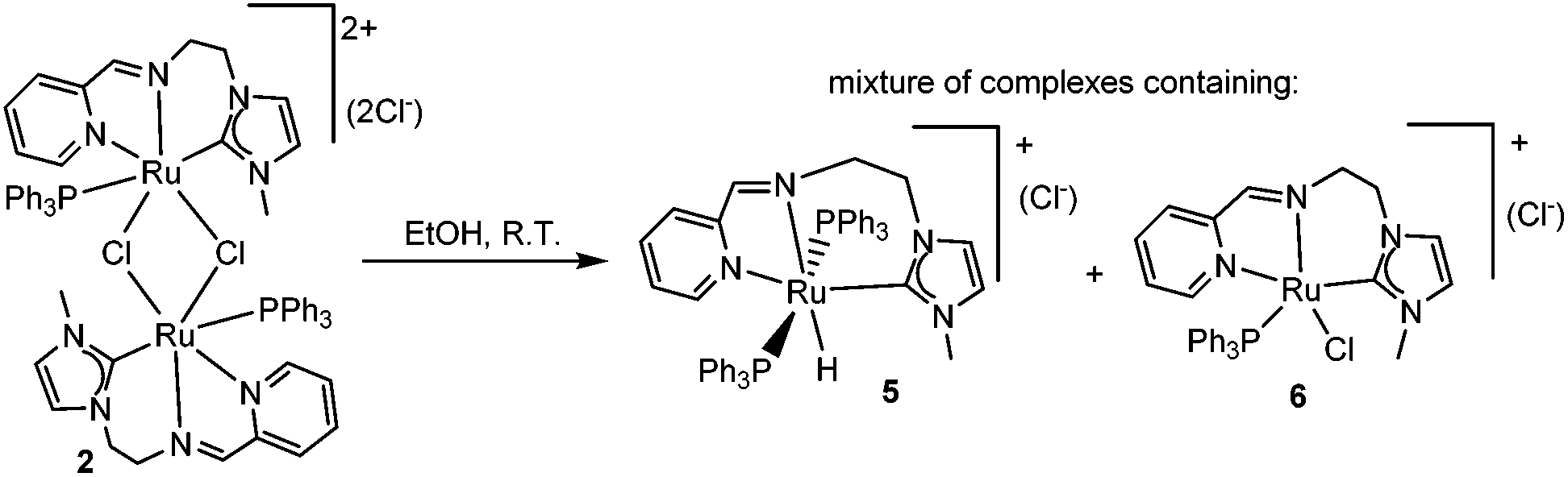
Upon prolonged standing, alcohol (EtOH or iPrOH) solutions of 2 produced a hydride species that we tentatively formulated as 5 (Scheme 4). The presence of the hydride was confirmed by 1H NMR spectroscopy, which revealed a triplet at −9.6 ppm. Although we have not been able to isolate this species, by mass spectrometry we could detect a mass peak at m/z = 841.3 assigned to [5]+, together with a peak at m/z = 613.2, which may be attributed to the ruthenium complex 6 (Scheme 4). The spontaneous formation of this ruthenium-hydride species under alcoholic conditions is an indication that complex 2 may be a good candidate for catalyzing processes involving alcohol dehydrogenation and transfer hydrogenation.14
 | ||
| Scheme 4 | ||
The molecular structures of complexes 2 and 4 were confirmed by single-crystal X-ray diffraction studies. The molecular structure of 2 (Fig. 1) exhibits two octahedral metal fragments connected by bridging chloride ligands. The NNC-pincer ligand and the phosphine complete the coordination sphere about ruthenium in 2. The PPh3 ligand is trans to a bridging chloride, and the two pincer ligands adopt a relative anti conformation, with the two imidazolylidenes (or the two pyridines) occupying opposite sites on each metal fragment. The NNC-pincer bite angle is 170.5(3)° and the distance of the Ru–Ccarbene bond is 2.041 Å. All other angles and distances lie in the expected range.
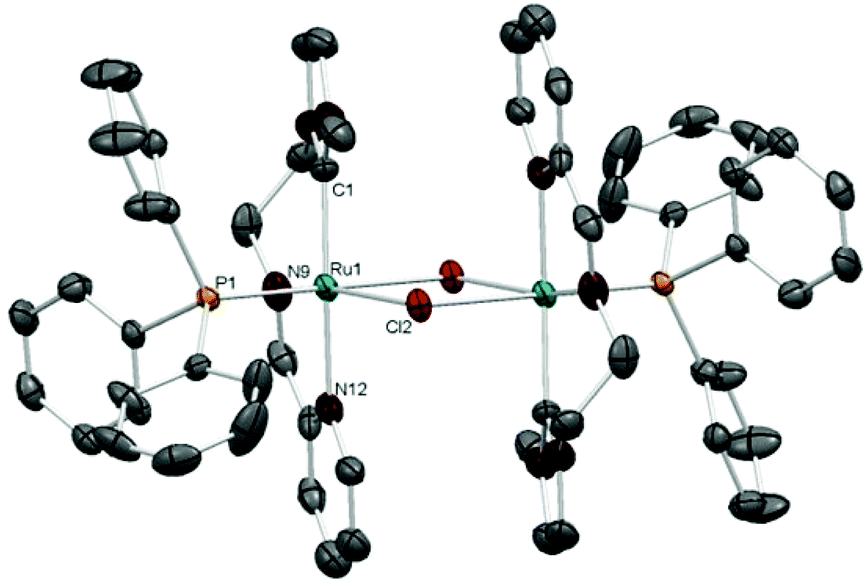 | ||
| Fig. 1 Molecular diagram of complex 2. Hydrogen atoms, counter-anions (two chlorides) and solvent (five molecules of CHCl3) have been omitted for clarity. Ellipsoids at the 30% probability level. Selected bond distances (Å) and bond angles (°): Ru(1)–C(1) = 2.041(7); Ru(1)–P(1) = 2.3066(15); Ru(1)–N(9) = 2.015(6); Ru(1)–N(12) = 2.144(6); Ru(1)–Cl(2) = 2.4527(14); C(1)–Ru(1)–P(1) = 92.3(2); C(1)–Ru(1)–Cl(2) = 95.1(2); C(1)–Ru(1)–N(9) = 94.6(3); C(1)–Ru(1)–N(12) = 170.5(3); N(9)–Ru(1)–N(12) = 77.2(3). | ||
The molecular structure of complex 4 (Fig. 2) consists of a ruthenium center in a pseudo-octahedral environment, with the mer-tridentate NNC ligand, a chelating carbonate, and PPh3 (trans to one of the oxygen atoms of the carbonate) completing the coordination sphere. The bite angle of the NNC-pincer ligand is 167.5(7)°, slightly smaller than that shown by 2. The length of the Ru–Ccarbene bond is 2.1020(2) Å. All other distances and angles are unexceptional.
 | ||
| Fig. 2 Molecular diagram of complex 4. Hydrogen atoms and solvent molecules (two molecules of H2O and one CH2Cl2) have been omitted for clarity. Thermal ellipsoids are at the 50% probability level. Selected bond distances (Å) and bond angles (°): Ru(1)–C(1) = 2.020(2); Ru(1)–P(1) = 2.272(5); Ru(1)–N(9) = 1.988(16); Ru(1)–N(12) = 2.150(16); Ru(1)–O(1) = 2.134(13); Ru(1)–O(2) = 2.120(13); C(1)–Ru(1)–P(1) = 93.3(6); C(1)–Ru(1)–O(1) = 89.4(7); C(1)–Ru(1)–O(2) = 94.7(7); C(1)–Ru(1)–N(9) = 91.8(7); C(1)–Ru(1)–N(12) = 167.5(7); N(9)–Ru(1)–N(12) = 78.6(7). | ||
Catalytic properties
Complex 2 was evaluated for activity in the transfer hydrogenation of ketones in refluxing iPrOH (82 °C). In order to optimize the reaction conditions, we first tested the reduction of acetophenone using 0.5 mol% of 2 and 10 mol% of different bases, and a 6 h reaction time. As can be seen from the results in Table 1, the best results are obtained with NaOtBu (entry 7), although the base alone is capable of reducing the ketone yielding 23% of 1-phenylethanol (entry 3). For this reason, and in order to minimize the background activity of the base,15 we considered more appropriate to use KOH, which under the same reaction conditions afforded almost quantitative yields of the alcohol (entry 5), while providing negligible amounts of the product in the absence of 2 (entry 1). Then catalyst 2 was used for the reduction of other ketones (benzophenone, cyclohexanone, 3-hexanone and phenylhexanone) and aldehydes (benzaldehyde, 4-bromobenzaldehyde and 3-phenyl-propionaldehyde) in refluxing iPrOH in the presence of KOH (entries 10–18). As can be seen from the results shown in Table 1, all five ketones were efficiently reduced under the reaction conditions used. Benzophenone as the substrate afforded lower yields of the final product, while cyclohexanone afforded quantitative yields of cyclohexanol in just ten minutes.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Base | Time | Substrate | Conv.b % | Yield % |
| a Conditions: substrate (1 mmol), base (0.1 mmol), 2 (0.5 mol%) in 3 mL of iPrOH at reflux temperature. b Conversions and yields determined by GC. c Yield determined by 1H NMR spectra. d Value in parenthesis refers to the aldol condensation product. | ||||||
| 1 | None | KOH | 6 h | Acetophenone | 9 | 4 |
| 2 | None | Cs2CO3 | 6 h | Acetophenone | 8 | 3 |
| 3 | None | NaOtBu | 6 h | Acetophenone | 36 | 23 |
| 4 | None | KOtBu | 6 h | Acetophenone | 9 | 4 |
| 5 | 2 | KOH | 6 h | Acetophenone | 94 | 86 |
| 6 | 2 | Cs2CO3 | 6 h | Acetophenone | 25 | 19 |
| 7 | 2 | NaOtBu | 6 h | Acetophenone | 94 | 90 |
| 8 | 2 | KOtBu | 6 h | Acetophenone | 85 | 79 |
| 9 | None | KOH | 1 h | Acetophenone | 1 | 1 |
| 10 | 2 | KOH | 1 h | Acetophenone | 94 | 88 |
| 11 | 2 | KOH | 1 h | Benzophenone | 99 | 68 |
| 12 | 2 | KOH | 10 min | Cyclohexanone | 99 | 99 |
| 13 | 2 | KOH | 6 h | 3-Hexanone | 85 | 82c |
| 14 | 2 | KOH | 6 h | Phenylhexanone | 93 | 93 |
| 15 | 2 | KOH | 1 h | Benzaldehyde | 98 | 33(30)d |
| 16 | 2 | KOH | 6 h | Benzaldehyde | 99 | 37(31)d |
| 17 | 2 | KOH | 6 h | 4-Bromo-benzaldehyde | 98 | 75(20)d |
| 18 | 2 | KOH | 6 h | 3-Phenyl-propionaldehyde | 99 | 38(61)d |
The reduction of the two aldehydes (benzaldehyde and 4-bromobenzaldehyde) afforded the resulting alcohols, together with the formation of the products resulting from the base-catalysed aldol condensation between the arylaldehydes and acetone. This process has been already described for the reduction of arylaldehydes by Ru(II) catalysts using similar reaction conditions.16
A study of the time-dependent reaction profiles of the reduction of acetophenone, benzophenone and cyclohexanone (Fig. 3) reveals that there is no detectable non-productive lag phase, which suggests that generation of the active catalytic species must be fast. The reaction rates corresponding to the reduction of each of the ketones are consistent with the final thermodynamic data shown in Table 1, implying that the different activities shown for each type of ketone are of kinetic nature.
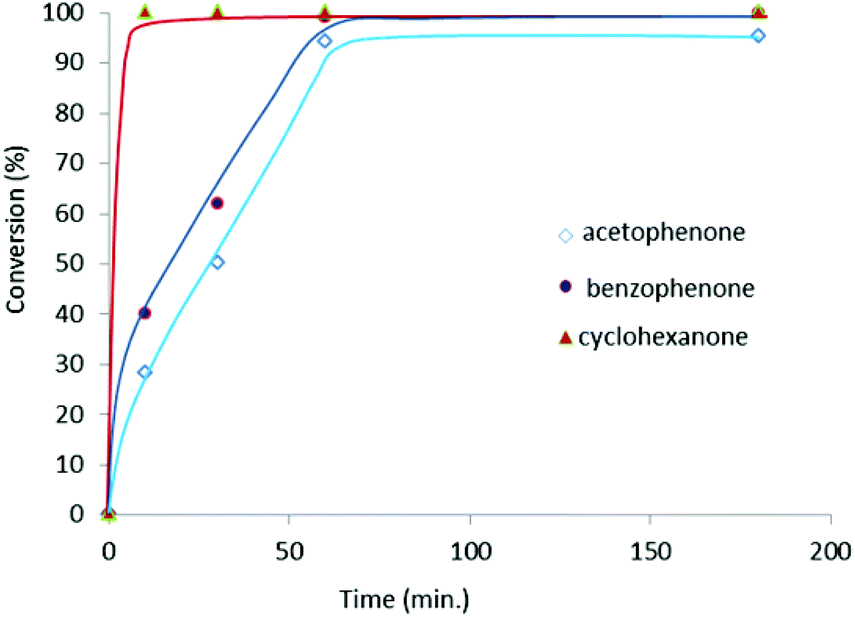 | ||
| Fig. 3 Time-dependent reaction profiles of the reduction of ketones by transfer hydrogenation. Reaction conditions: ketone (1 mmol), base (0.1 mmol), 2 (0.5 mol%) in 3 mL of iPrOH at reflux. | ||
Conclusions
We have prepared a new NNC-pincer ligand with one NHC donor group. The new ligand was coordinated to ruthenium. Depending on the coordination strategy, we were able to obtain two significantly different complexes. While the transmetallation of the ligand from a preformed silver–NHC complex to [RuCl2(PPh3)3] afforded the diruthenium complex 2 with two bridging chloride ligands, the reaction of the ligand precursor with Ag2CO3, K2CO3 and the same ruthenium source gave the monometallic carbonate pincer complex 4. The stability of the dimetallic complex 2 is high; however, it forms a ruthenium hydride species in alcoholic solutions. This reactivity indicates that the complex may easily generate monometallic species that may be active in transfer hydrogenation.The preliminary studies on the catalytic activity of 2 indicate that the complex is active in the reduction of ketones and aldehydes under transfer hydrogenation conditions. While the catalyst is very active in the reduction of ketones to the corresponding secondary alcohols, the reduction of aldehydes affords the related primary alcohols together with the product resulting from the aldol condensation between the aldehyde and acetone. The catalyst is extraordinarily active in the reduction of cyclohexanone. These promising results indicate that the catalyst may have potential for more challenging reactions, such as the reduction of carboxylic esters in the presence of molecular hydrogen. Further studies in this direction are underway.
Experimental section
General methods
All operations were carried out under a nitrogen atmosphere unless otherwise stated using standard Schlenk techniques. Solvents were dried using a solvent purification system (MBraun SPS). All reagents were used as received from the commercial suppliers. NMR spectra were recorded either on a Varian Mercury 300 or Varian NMR System 400 MHz spectrometers and referenced (1H, 13C) as follows: CD3OD (δ 3.31, 49.00), CD2Cl2 (δ 5.32, 54.00), CDCl3 (δ 7.26, 77.16) DMSO-d6 (δ 2.5, 39.52). Electrospray ionization-mass spectra (ESI-MS) were recorded on a Micromass Quatro LC instrument; nitrogen was employed as the drying and nebulizing gas. Accurate mass measurements were performed using a Q-TOF premier mass spectrometer with an electrospray source (Waters, Manchester, UK) operating at a resolution of ca. 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (fwhm). Elemental analyses were carried out on a EuroEA3000 Eurovector Analyzer.
000 (fwhm). Elemental analyses were carried out on a EuroEA3000 Eurovector Analyzer.
The NMR data of the product match the corresponding data reported in the literature.18
3-(2-Aminoethyl)-1-methylimidazolium bromide was prepared by adapting a reported method in the literature.17 A solution of 3-(2-phthalimidoethyl)-1-methylimidazolium bromide (11.78 g, 35.0 mmol) and hydrazine hydrate (7.0 g, 50%, ca. 109 mmol) in 80 mL of ethanol in a 250 mL flask was heated at 80 °C overnight. A bulky amorphous white solid formed in the flask. After cooling, 100 mL of ethanol was added into the flask and the solid was crashed into a cottage-cheese like paste; this was transferred into a fritted funnel and the product solution was vacuum-filtered into a 500 mL flask; the solid was washed with additional 2 × 50 mL portions of ethanol. The ethanol solution was rotary-evaporated, and the crude product was dried under vacuum of an oil pump at 50 °C for 30 min to yield a pale-yellow oil that was used without further purification in the next step. Yield: 7.23 g (100%). 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H, CH), 7.86 (br t, J = 1.6, 1H, CH), 7.72 (br t, J = 1.6, 1H, CH), 4.13 (t, J = 5.8, 2H, CH2), 3.86 (s, 3H, CH3), 2.89 (t, J = 5.8, 2H, CH2), 2.48 (br, NH2). 13C{1H} NMR (100 MHz, DMSO-d6) δ 136.72 (CH), 123.24 (CH), 122.47 (CH), 51.85 (CH2), 41.30 (CH2), 35.67 (CH3). According to the 1H NMR spectrum, the isolated material contained ca. 5 mol% of an aromatic impurity, presumably N,N′-phthaloylhydrazine. The NMR data of the product match the corresponding data reported in the literature.19
A solution of 3-(2-aminoethyl)-1-methylimidazolium bromide (5.87 g, 28.5 mmol) in 2-propanol (30 mL) and 2-pyridinecarboxaldehyde (3.05 g, 28.5 mol) was stirred overnight (16 h) at room temperature. Then the solvent was removed, and the resulting oil was dried under vacuum to give a grey solid. The product was recrystallized from CH2Cl2 (150 mL) to yield 7.1 g (85%) of a hygroscopic solid. 1H NMR (300 MHz, DMSO-d6) δ (ppm) 9.33 (m, 1H, CHPy), 8.62 (d, J = 4.6 Hz, 1H, CHPy), 8.34 (s, 1H, NCH), 7.92 (m, 2H, CHPy), 7.76 (s, 1H, CHPy), 7.45 (m, 2H, CHImid), 4.57 (t, J = 5.2 Hz, 2H, CH2), 4.06 (t, J = 5.2 Hz, 2H, CH2), 3.87 (s, 3H, CH3), 13C{1H} NMR (125 MHz, DMSO-d6): δ (ppm) 164.5 (NCH), 164.4 (NC), 153.5 (CPy), 149.3 (CPy), 136.9 (CPy), 125.4 (CPy), 123.4 (CPy), 122.6 (CImid), 120.6 (CImid), 58.5 (NCH2), 49.5 (NCH2), 35.7 (CH3). HRMS ESI-TOF-MS (positive mode): monoisotopic peak 215.1294, calcd 215.1297.
X-Ray crystallography
Single crystals of 2 and 4 suitable for X-ray crystallographic analysis were obtained as described above. Diffraction data were collected on an Agilent SuperNova diffractometer equipped with an Altas CCD detector using Cu Kα radiation (λ = 1.54184 Å). Single crystal was mounted on a MicroMount polymer tip (MiteGen) in a random orientation. The crystals were maintained at 293 K during data collection for 2 and at 290 K for 4. The structures were solved by direct methods in SHELXS-9720 and refined by the full-matrix method based on F2 with the program SHELXL-97 using the OLEX software package.20,21 Further crystallographic data may be found in the corresponding CIF files which were deposited at the Cambridge Crystallographic Data Centre CCDC, Cambridge, UK. The reference number for 2 and 4 were assigned as 1060997 and 1060998 respectively.Crystal data, data collection and structure refinement details for 2 and 4.
Compound 2: C70H66Cl34N8P2Ru2 (M = 2488.68): monoclinic, space group P21/a, a = 13.2500(2) Å, b = 16.9695(3) Å, c = 23.3436(4) Å, α = 90°, β = 103.9482(18)°, γ = 90°, V = 5093.95(17) Å3, Z = 2, T = 293(2) K, μ(CuKα) = 11.254 mm−1, Dcalc = 1.623 g cm−3, 44887 reflections measured (7.38 ≤ 2Θ ≤ 131.968), 8883 unique (Rint = 0.0512, Rsigma = 0.0321), which were used in all calculations. The final R1 was 0.0721 (I ≥ 2σ(I)) and wR2 was 0.2154 (all data).
Compound 4: C32H35Cl2N4O5PRu (M = 758.58): monoclinic, space group P21/c, a = 18.2792(3) Å, b = 11.74106(11) Å, c = 17.1049(2) Å, V = 3259.49(9) Å3, Z = 4, T = 199.95(10) K, μ(CuKα) = 1.54184 mm−1, Dcalc = 1.546 g mm−3, 27673 reflections measured (9.296 ≤ 2Θ ≤ 139.32), 6075 unique (Rint = 0.0499, Rsigma = 0.0291) which were used in all calculations. The final R1 was 0.2354 (I > 2σ(I)) and wR2 was 0.5147 (all data).
Acknowledgements
We gratefully acknowledge financial support from MINECO of Spain (CTQ2014-51999-P) and UJI (P11B2014-02). Carmen Mejuto is thankful to the FPI program for a fellowship, and Gregorio Guisado-Barrios thanks the MINECO for a postdoctoral grant (FPDI-2013-16525). The authors are grateful to the Serveis Centrals d'Instrumentació Científica (SCIC) of the Universitat Jaume I for providing with spectroscopic and X-Ray facilities.Notes and references
- (a) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS; (b) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed; (c) N. Selander and K. J. Szabo, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed.
- (a) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed; (b) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- H. A. Younus, N. Ahmad, W. Su and F. Verpoort, Coord. Chem. Rev., 2014, 276, 112–152 CrossRef CAS PubMed.
- (a) D. Spasyuk, C. Vicent and D. G. Gusev, J. Am. Chem. Soc., 2015, 137, 3743–3746 CrossRef CAS PubMed; (b) D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2013, 52, 2538–2542 CrossRef CAS PubMed; (c) D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2012, 51, 2772–2775 CrossRef CAS PubMed; (d) M. J. Hanton, S. Tin, B. Boardman and P. Miller, J. Mol. Catal. A: Chem., 2011, 346, 70–78 CrossRef CAS PubMed; (e) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (f) E. Fogler, E. Balaraman, Y. Ben-David, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2011, 30, 3826–3833 CrossRef CAS; (g) E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS PubMed; (h) W. Kuriyama, Y. Ino, O. Ogata, N. Sayo and T. Saito, Adv. Synth. Catal., 2010, 352, 92–96 CrossRef CAS PubMed; (i) Y. Ino, W. Kuriyama, O. Ogata and T. Matsumoto, Top. Catal., 2010, 53, 1019–1024 CrossRef CAS; (j) L. A. Saudan, C. M. Saudan, C. Debieux and P. Wyss, Angew. Chem., Int. Ed., 2007, 46, 7473–7476 CrossRef CAS PubMed; (k) J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed; (l) T. Zell, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 4685–4689 CrossRef CAS PubMed; (m) S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. J. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed.
- D. Srimani, M. Feller, Y. Ben-David and D. Milstein, Chem. Commun., 2012, 48, 11853–11855 RSC.
- (a) G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS PubMed; (b) C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS; (c) C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS PubMed; (d) G. A. Filonenko, M. P. Conley, C. Coperet, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2013, 3, 2522–2526 CrossRef CAS.
- S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed.
- E. Balaraman, E. Fogler and D. Milstein, Chem. Commun., 2012, 48, 1111–1113 RSC.
- Y. Sun, C. Koehler, R. Tan, V. T. Annibale and D. Song, Chem. Commun., 2011, 47, 8349–8351 RSC.
- (a) W. Du, L. Wang, P. Wu and Z. Yu, Chem. – Eur. J., 2012, 18, 11550–11554 CrossRef CAS PubMed; (b) C. del Pozo, M. Iglesias and F. Sanchez, Organometallics, 2011, 30, 2180–2188 CrossRef CAS; (c) C. del Pozo, A. Corma, M. Iglesias and F. Sanchez, Organometallics, 2010, 29, 4491–4498 CrossRef CAS; (d) M. Boronat, A. Corma, C. Gonzalez-Arellano, M. Iglesias and F. Sanchez, Organometallics, 2010, 29, 134–141 CrossRef CAS; (e) F. Zeng and Z. Yu, Organometallics, 2008, 27, 6025–6028 CrossRef CAS.
- (a) D. Pugh and A. A. Danopoulos, Coord. Chem. Rev., 2007, 251, 610–641 CrossRef CAS PubMed; (b) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS PubMed.
- M. Poyatos, J. A. Mata, E. Falomir, R. H. Crabtree and E. Peris, Organometallics, 2003, 22, 1110–1114 CrossRef CAS.
- M. Zeng, L. Li and S. B. Herzon, J. Am. Chem. Soc., 2014, 136, 7058–7067 CrossRef CAS PubMed.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS PubMed.
- U. Hintermair, J. Campos, T. P. Brewster, L. M. Pratt, N. D. Schley and R. H. Crabtree, ACS Catal., 2014, 4, 99–108 CrossRef CAS.
- M. Delgado-Rebollo, D. Canseco-Gonzalez, M. Hollering, H. Mueller-Bunz and M. Albrecht, Dalton Trans., 2014, 43, 4462–4473 RSC.
- J. R. Harjani, T. Friscic, L. R. MacGillivray and R. D. Singer, Dalton Trans., 2008, 4595–4601 RSC.
- S. L. M. Goh, M. P. Hoegerl, N. B. Jokic, A. D. Tanase, B. Bechlars, W. Baratta, J. Mink and F. E. Kuehn, Eur. J. Inorg. Chem., 2014, 2014, 1225–1230 CrossRef CAS PubMed.
- X. Zhou, T. Wu, K. Ding, B. Hu, M. Hou and B. Han, Chem. Commun., 2009, 1897–1899 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1060997 and 1060998. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00137d |
| This journal is © the Partner Organisations 2015 |