
1,3-Dipolar cycloaddition reactions of phthalic anhydrides with an azomethine ylide†‡
Hugo
Santos
a,
Amy
Distiller
a,
Asha M.
D'Souza
a,
Quentin
Arnoux
a,
Jonathan M.
White
b,
Adam G.
Meyer
*a and
John H.
Ryan
*a
aCSIRO Manufacturing Flagship, Bag 10, Clayton South, Victoria 3169, Australia. E-mail: adam.meyer@csiro.au; jack.ryan@csiro.au; Fax: +61 3 9545 2446
bSchool of Chemistry, Bio21 Institute, University of Melbourne, Parkville, Victoria 3010, Australia
First published on 13th April 2015
Abstract
A series of phthalic anhydrides underwent a 1,3-dipolar cycloaddition reaction with N-benzylazomethine ylide, formed in situ from N-(methoxymethyl)-N-(trimethylsilylmethyl)benzylamine and a catalytic amount of trifluoroacetic acid, to produce unstable spiro(isobenzofuran-1,5′-oxazolidin)-3-ones. The spiro-fused oxazolidines were reduced with sodium borohydride to afford 1(3H)-isobenzofuranones, which were generally isolated in moderate to high overall yields.
The 1,3-dipolar cycloaddition reaction1 of azomethine ylides with alkenes substituted with electron-withdrawing groups is an efficient and versatile method for the construction of pyrrolidine-containing molecules of biological2 or materials science interest.3 Less studied are the reactions with hetero multiple bonded systems; however carbonyl, thiocarbonyl, isothiocyanato, imino, isocyanato, nitrile, nitroso, and azo derivatives can also act as azomethine ylide dipolarophiles.4,5 In the case of carbonyl dipolarophiles, aldehydes and ketones readily undergo cycloaddition reactions with azomethine ylides to give oxazolidines.6 Generally carboxyl derivatives such as carboxylic acids and esters are unreactive in such reactions,6d although it has recently been demonstrated that certain activated carboxyl derivatives can act as azomethine ylide dipolarophiles.7 The ester-like carbonyl group of isatoic anhydrides 1 undergoes a facile cycloaddition reaction with the non-stabilised azomethine ylide A (derived from N-(methoxymethyl)-N-(trimethylsilylmethyl)benzylamine 2)8 to give cycloadducts, the spiro(benzo[d][1,3]oxazine-4,5′-oxazolidine) derivatives 3.7 The cycloadducts 3 proved to be unstable, leading to an unprecedented ring opening-decarboxylation-ring closing cascade sequence to afford 1,3-benzodiazepin-5-ones 4 (Scheme 1).7
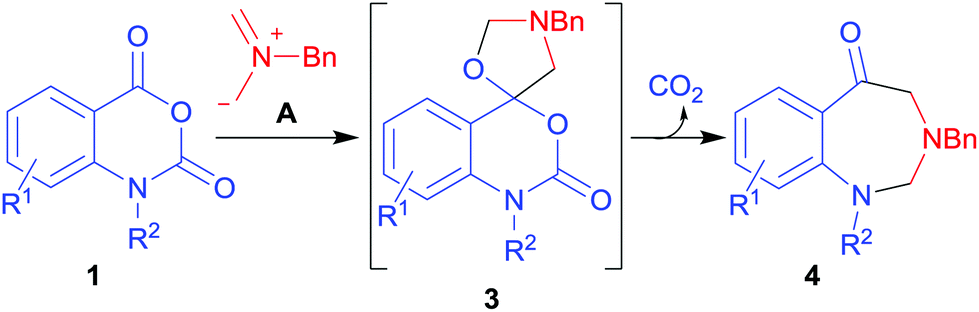 | ||
| Scheme 1 1,3-Dipolar cycloaddition reaction of isatoic anhydrides 1 with azomethine ylide A.7 | ||
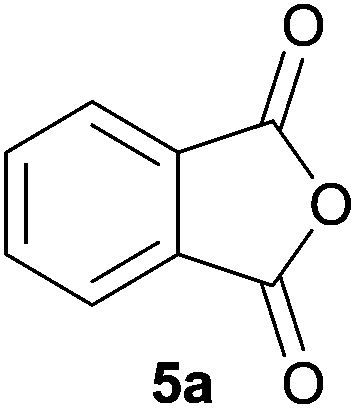
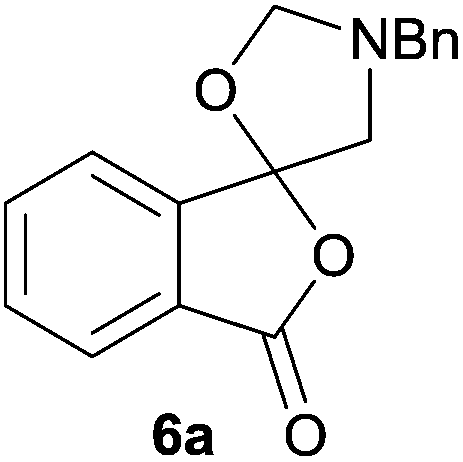
Given the fascinating reactivity of isatoic anhydrides with azomethine ylide A, we sought to expand the scope of this reaction type and identified phthalic anhydride 5a as a potential carbonyl dipolarophile. Phthalic anhydrides are readily available, inexpensive and versatile raw materials used for the manufacture of a wide range of commercial products including phenolphthalein, anthraquinones and metal phthalocyanines used in the dye industry, polyester polymers and phthalate diesters widely used as plasticizers in flexible PVC products.9,10 Phthalic anhydrides also undergo a plethora of nucleophilic ring opening reactions of the anhydride ring; however, the carbonyl group acting as a 2π-unit in cycloaddition chemistry has not been recognised in the literature. Recently, transition metal-catalysed decarbonylative cycloadditions of phthalic anhydrides with alkynes, allenes and 1,3-dienes have been reported.11 We thought that the carbonyl moieties within phthalic anhydride 5a would be sufficiently activated so as to undergo cycloaddition with azomethine ylide A to afford spiro-fused cycloadduct 6a (Scheme 2).
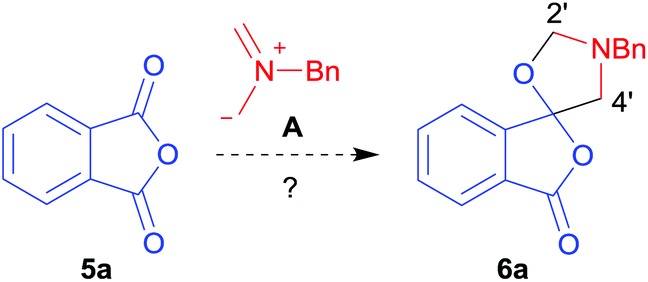 | ||
| Scheme 2 Proposed reaction of phthalic anhydride 5a with azomethine ylide A. | ||
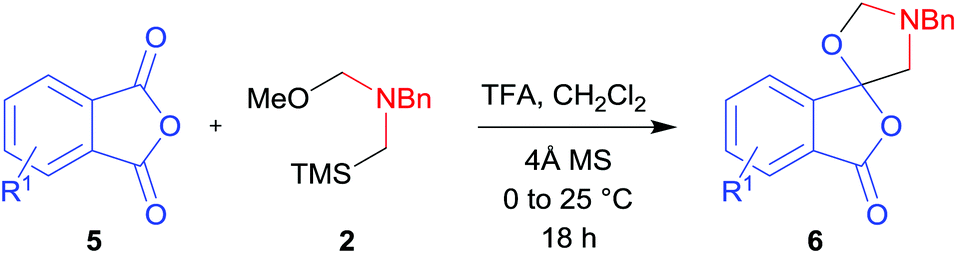
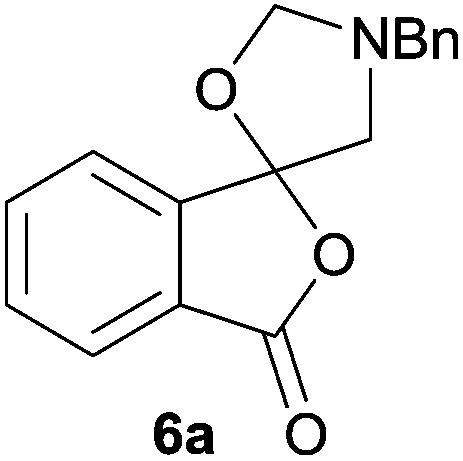
In order to investigate this cycloaddition reaction, phthalic anhydride 5a was allowed to react with azomethine ylide precursor 2 and 0.05 mole equivalents of trifluoroacetic acid (TFA)12 in the presence of 4 Å molecular sieves. The reaction was essentially complete after 18 h§ affording the anticipated spiro(isobenzofuranone-oxazolidine) 6a. Chromatographic purification was required to remove reagent-based impurities. Using silica gel chromatography, the cycloadduct could not be isolated in adequate purity due to decomposition processes that occurred during chromatography, which we assumed was due to the (hydroxylic) acidic nature of the silica gel. Consistent with this assumption, when we eluted the crude product through Florisil™, a commercially available less acidic form of silica gel, the crude cycloadduct 6a was isolated (90% crude yield) with sufficient purity to enable adequate spectroscopic characterisation.¶ The spectroscopic data was in full accord with oxazolidine 6a being a mono adduct, with a APCI MS displaying a [M + H]+ ion of m/z 282.1125, 13C NMR displaying 15 signals with one signal at δ168.1 ppm due to the resultant lactone carbonyl carbon. Additionally, the 1H NMR spectrum displayed separate AB signals at δ4.92 and δ4.85 ppm, and δ3.60 and δ3.45 ppm, assigned to the non-equivalent methylene protons at C2′ and C4′ of the oxazolidine ring system respectively. The signal at δ4.13 ppm was assigned to the diastereotopic benzylic methylene group. It is worth noting that only the product arising from a single cycloaddition of the azomethine ylide to phthalic anhydride was isolated, which suggests that, under these conditions, the remaining carbonyl group of oxazolidine 6a was significantly less reactive than the carbonyls of the starting material.
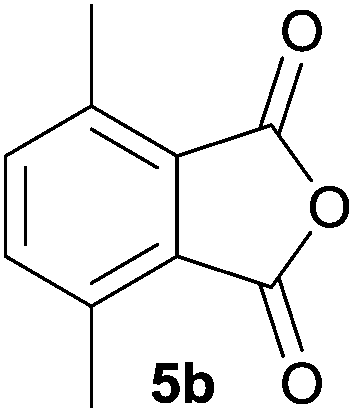
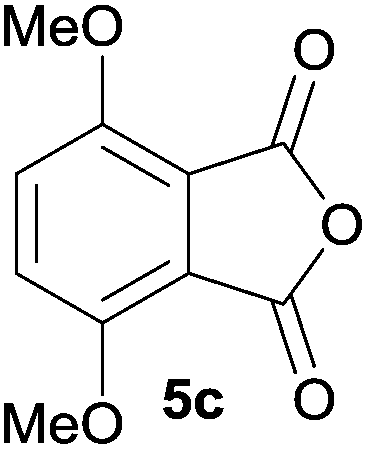
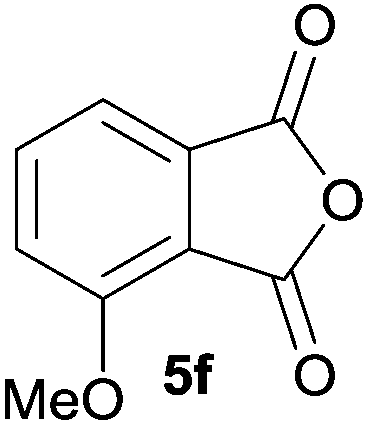
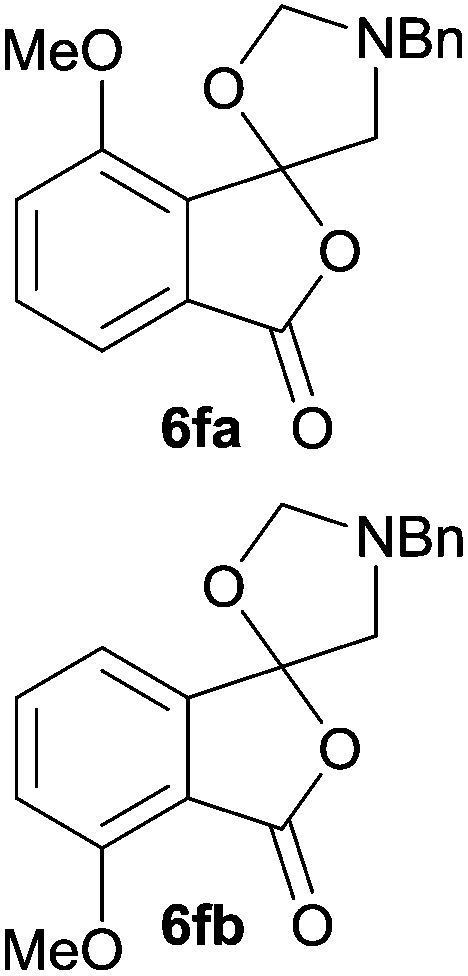
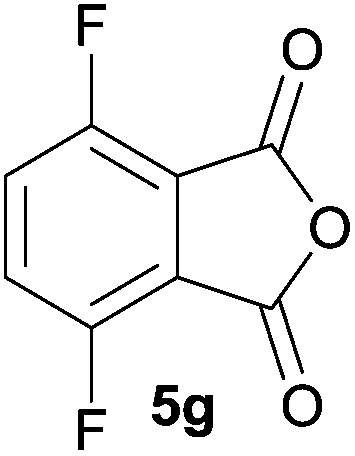
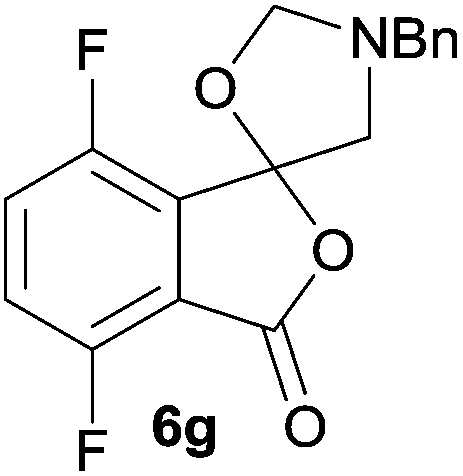
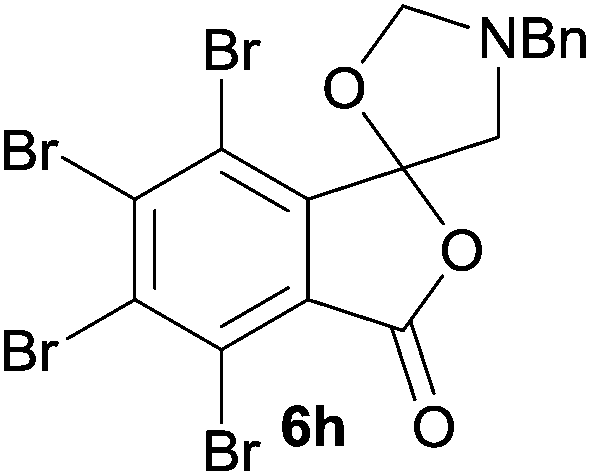
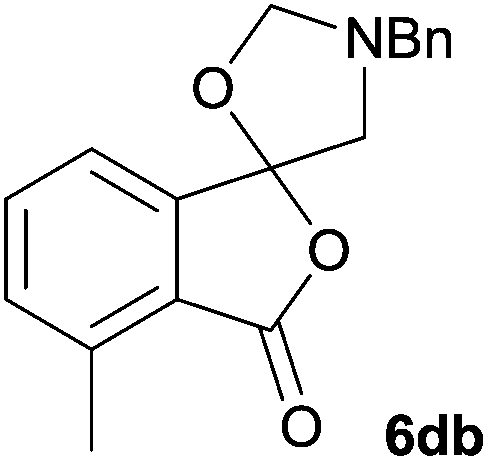
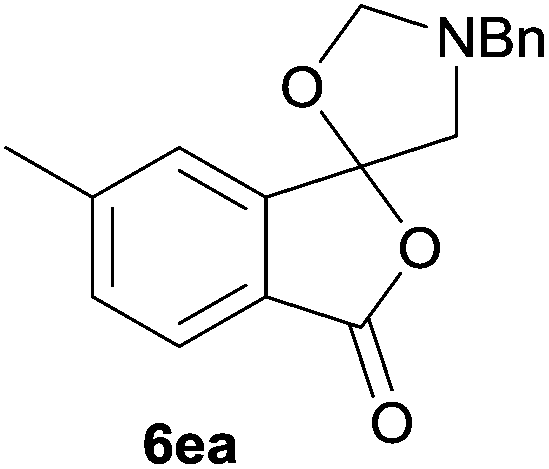
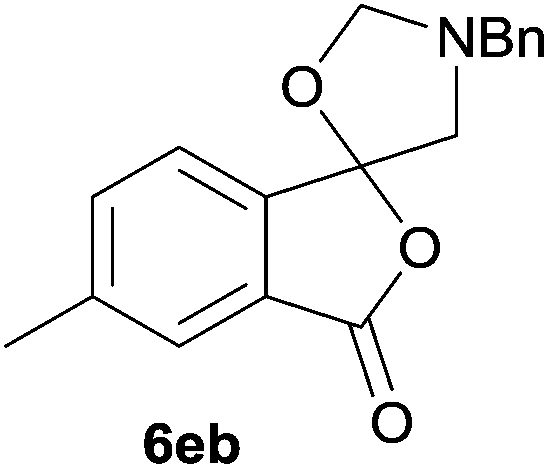
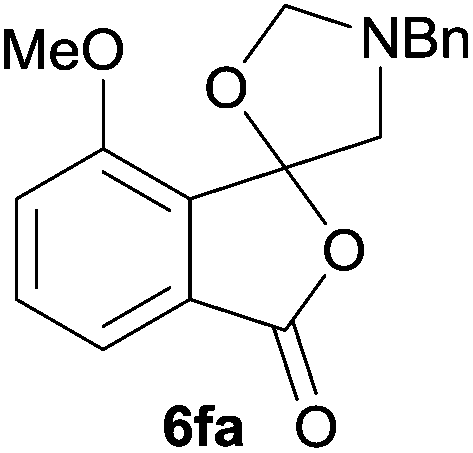
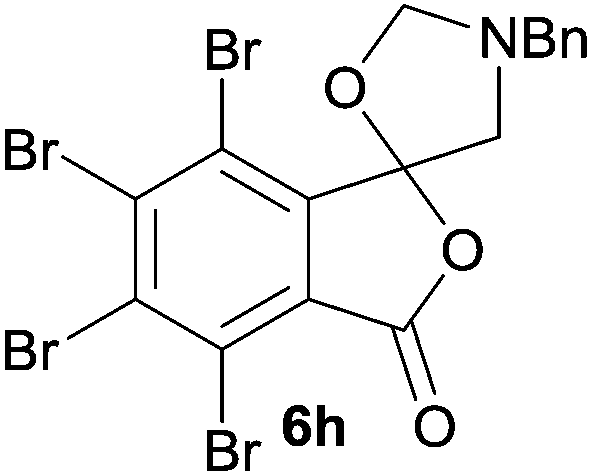
The scope of the reaction was explored by subjecting a range of substituted phthalic anhydrides 5b–h to the cycloaddition reaction conditions and isolation of the unstable crude products via Florisil™ chromatography (Table 1). The purity of products obtained using this chromatographic technique was adequate for the purpose of spectroscopic characterisation. For phthalic anhydrides 5b–f substituted with electron-donating groups, such as methyl or methoxy groups, the reaction proceeded to completion and high yields (60–100%) of the corresponding crude spiro(isobenzofuran-1,5′-oxazolidin)-3-ones 6b–f were obtained (Table 1, entries 2–6). For the symmetrical 3,6-dimethyl or dimethoxy systems 5b or 5c, the respective cycloadducts, 6b or 6c, were obtained (Table 1, entries 2 and 3). For the unsymmetrical methyl-substituted systems 5d and 5e, mixtures of the two respective cycloadducts were obtained (Table 1, entries 4 and 5). 3-Methylphthalic anhydride 5d furnished the regioisomeric cycloadducts 6da and 6db in a ratio of 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :52 (as determined by 1H NMR analysis) in excellent yield (Table 1, entry 4). When 4-methylphthalic anhydride 5e was employed, the regioisomeric cycloadducts 6ea and 6eb were obtained, again in excellent yield in a ratio of 63:37 (Table 1, entry 5). For both phthalic anhydrides 5d and 5e it was necessary to use more of the azomethine ylide precursor 2 (1.5 mole equivalent) and TFA (0.075 mol equiv.) to ensure complete consumption of the starting anhydrides. As expected, the unsymmetrical anhydride, 3-methoxyphthalic anhydride 5f, gave a 30:70 ratio of two regioisomeric cycloadducts 6fa and 6fb, obtained in good yield (Table 1, entry 6). It was not possible to separate the mixtures of regioisomeric cycloadducts 6d, 6e and 6f due to the instability of the cycloadducts to normal and reverse-phase chromatographic conditions, and lack of separation on Florisil™. Phthalic anhydrides bearing electron-withdrawing groups, 3,6-difluorophthalic anhydride 5g and 3,4,5,6-tetrabromophthalic anhydride 5h, were also subjected to the cycloaddition conditions. The reactions proceeded to completion and moderate to high crude yields (∼100% or 67%) of the corresponding crude spiro(isobenzofuran-1,5′-oxazolidin)-3-ones 6g or 6h were obtained (Table 1, entries 7 and 8).
:52 (as determined by 1H NMR analysis) in excellent yield (Table 1, entry 4). When 4-methylphthalic anhydride 5e was employed, the regioisomeric cycloadducts 6ea and 6eb were obtained, again in excellent yield in a ratio of 63:37 (Table 1, entry 5). For both phthalic anhydrides 5d and 5e it was necessary to use more of the azomethine ylide precursor 2 (1.5 mole equivalent) and TFA (0.075 mol equiv.) to ensure complete consumption of the starting anhydrides. As expected, the unsymmetrical anhydride, 3-methoxyphthalic anhydride 5f, gave a 30:70 ratio of two regioisomeric cycloadducts 6fa and 6fb, obtained in good yield (Table 1, entry 6). It was not possible to separate the mixtures of regioisomeric cycloadducts 6d, 6e and 6f due to the instability of the cycloadducts to normal and reverse-phase chromatographic conditions, and lack of separation on Florisil™. Phthalic anhydrides bearing electron-withdrawing groups, 3,6-difluorophthalic anhydride 5g and 3,4,5,6-tetrabromophthalic anhydride 5h, were also subjected to the cycloaddition conditions. The reactions proceeded to completion and moderate to high crude yields (∼100% or 67%) of the corresponding crude spiro(isobenzofuran-1,5′-oxazolidin)-3-ones 6g or 6h were obtained (Table 1, entries 7 and 8).
|
|
||||
|---|---|---|---|---|
| Entry | Anhydride | Product(s) | Ratio of regio-isomers | Crude yieldb (%) |
| a Reaction conditions: 5 (1.0 equiv.), 2 (1.1 equiv.), trifluoroacetic acid (0.05 equiv.), CH2Cl2 (7.5 mL mmol−15), 4 Å molecular sieves, N2, 0 to 25 °C, 18 h. Column chromatography on Florisil™, eluting with EtOAc. b The cycloadducts are, generally, of limited stability and decompose at ambient temperature/atmosphere. Once isolated (after filtration through Florisil™) they can be stored at 4 °C under an inert atmosphere for up to a day without significant deterioration. | ||||
| 1 |
|
|
90 | |
| 2 |
|
|
∼100 | |
| 3 |
|
|
90 | |
| 4 |
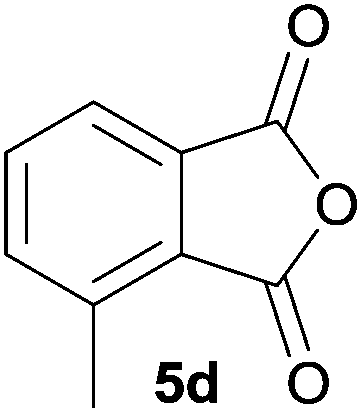
|
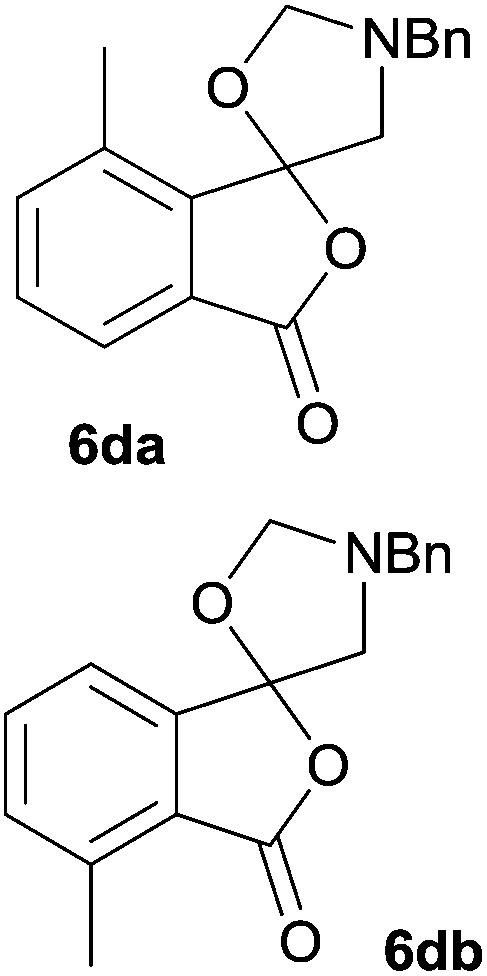
|
6da:6db = 48:52 |
∼100 |
| 5 |
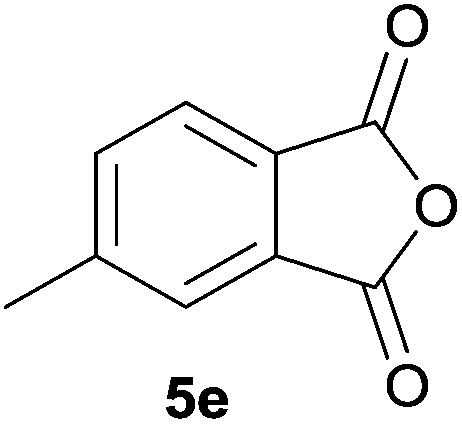
|
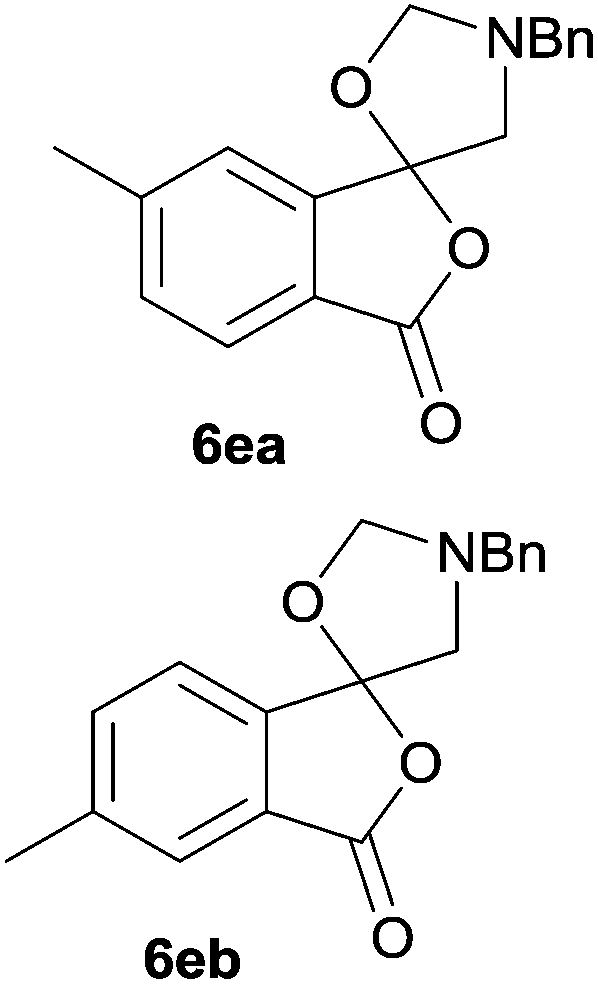
|
6ea:6eb = 63:37 |
∼100 |
| 6 |
|
|
6fa:6fb = 30:70 |
60 |
| 7 |
|
|
∼100 | |
| 8 |

|
|
67 | |
The regioselectivity trends observed for cycloadditions to the unsymmetrical systems 5e–f are in accord with the cycloaddition being a normal electron demand process and similar to those observed for the cycloadditions of azomethine ylide A with isatoic anhydrides.7 For 5e, the cycloaddition is less favoured at the carbonyl group para to methyl group, consistent with the electron-donating effect of the methyl group reducing the electrophilic character of the para carbonyl group to a greater extent that the meta carbonyl group. For 5f, the methoxy group would reduce the reactivity of the ortho-related carbonyl group relative to the meta carbonyl group by similar electronic effects and possibly additionally by steric hindrance of the incoming dipole at the ortho carbonyl group. While the lack of selectivity in the case of 5d appears anomalous, 1H NMR analysis of the crude reaction product prior to Florosil™ chromatography, indicated that 6da and 6db are formed in a ∼1:2 ratio. It appears that in the case of 5d some losses of the major cycloadduct have occurred during Florosil™ chromatography. This means that the regioselectivity for the cycloaddition of 5d is actually similar to that of 5f, which is as expected because of similar stereoelectronic effects.
The unstable nature of spiro-isobenzofuran-1,5′-oxazolidin-3-ones 6a–h is thought to be due to the sensitivity of the contiguous lactone, ketal and hemiaminal ether moieties to Brønsted or Lewis acids. Reductive ring-opening of the hemiaminal ether moiety within oxazolidines has been reported,13 and can occur by activation of the C–O bond through coordination to the oxygen atom of a reducing agent with Lewis acid properties (e.g., BH3), C–O bond fission to form an acyclic iminium ion intermediate, and reduction of this species by hydride ion. We postulated that a relatively slow ring-opening of oxazolidine 6 to give iminium ion 7 would occur,7,14 and then reduction with sodium borohydride would form the corresponding 1-(3H)-isobenzofuranones 8, which we hoped would be sufficiently stable to be isolated in high purity so as to allow chromatographic separation (Scheme 3). In the cases where mixtures of regioisomeric cycloadducts 6 were subjected to these conditions, we anticipated that the resultant regioisomeric isobenzofuranones 8 would be obtained, and if separable, would provide more robust validation of the assigned structures.
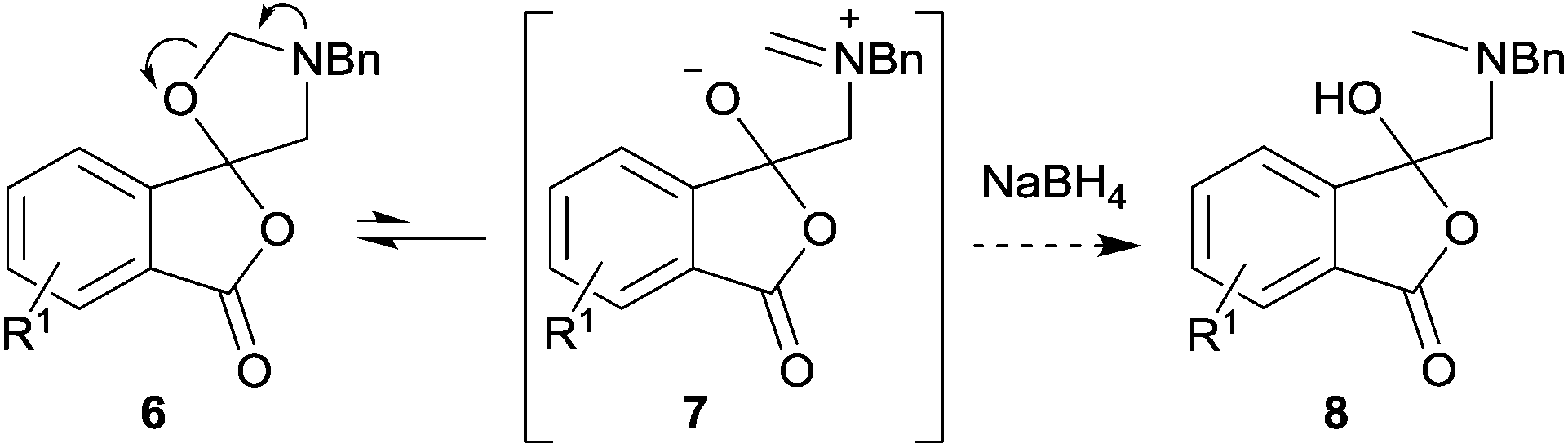 | ||
| Scheme 3 Proposed ring-opening of oxazolidines 6 with sodium borohydride. | ||
In order to investigate this reductive ring-opening reaction, spiro-fused oxazolidine 6a was treated with sodium borohydride (1.5 mole equivalent) in methanol, and after four hours at ambient temperature 1-(3H)-isobenzofuranone 8a was isolated in 76% yield after purification by silica gel chromatography (Table 2, entry 1). As far as we are aware, this is the first report describing the synthesis of this type of functionalised 1(3H)-isobenzofuranone,15 although the chemistry of 3-hydroxyisobenzofuranones (3-hydroxyphthalides) is of much interest, particularly as this moiety is found in natural products and is used as starting materials for the synthesis of bioactive compounds.15–17 The analytical and spectroscopic data obtained for the product was in full accord with the proposed structure of compound 8a; the ASAP high resolution MS showed a [M + H]+ ion at m/z 282.1283, consistent with addition of two hydrogens to 6a, the 13C NMR spectrum showed a resonance at δ168.5 ppm assigned to the carbonyl group of the lactone ring and the 1H NMR spectrum displayed three resonances at δ3.82, 2.93 and 2.55 ppm, which were assigned to the protons at the benzylic methylene group, the methylene group between the quaternary carbon centre and the nitrogen atom, and the N-methyl group, respectively. Further evidence of the structure of 1(3H)-isobenzofuranone 8a was provided by HMBC spectroscopy, which displayed two diagnostic cross-peaks associated with three-bond correlations (J3) between the N-methyl carbon atom (Cj) and both sets of methylene group protons (Hi and Hk) (Fig. 1).
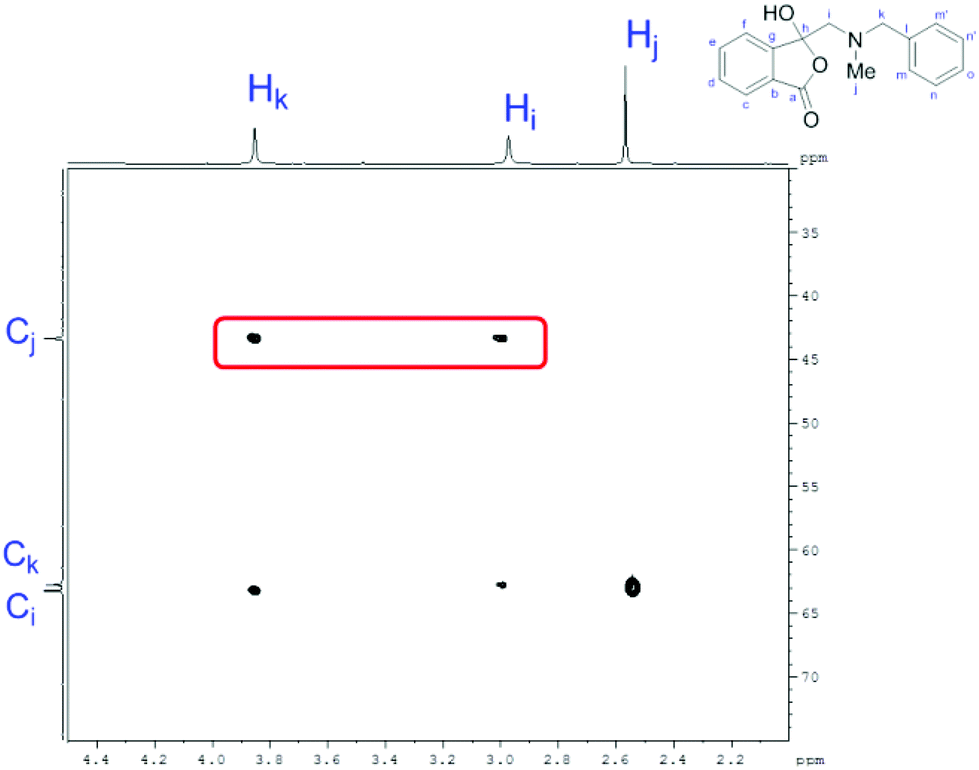 | ||
| Fig. 1 Section of the HMBC spectrum contour plot of 8a; the three-bond correlations of interest are highlighted. | ||
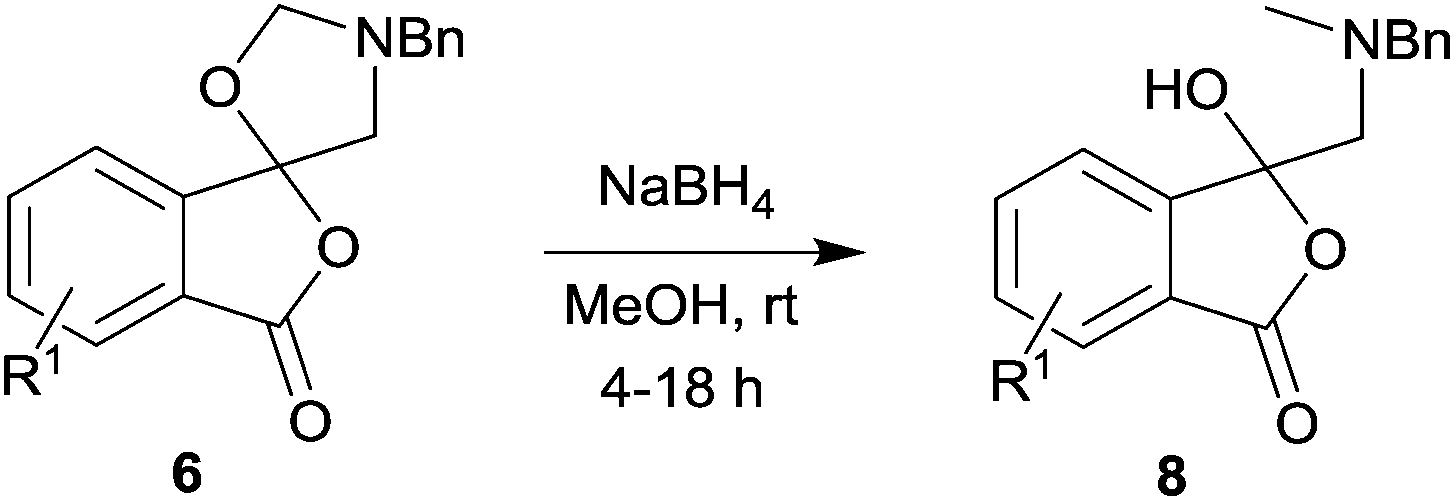
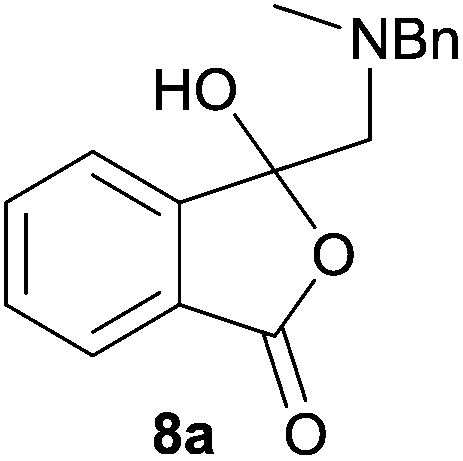
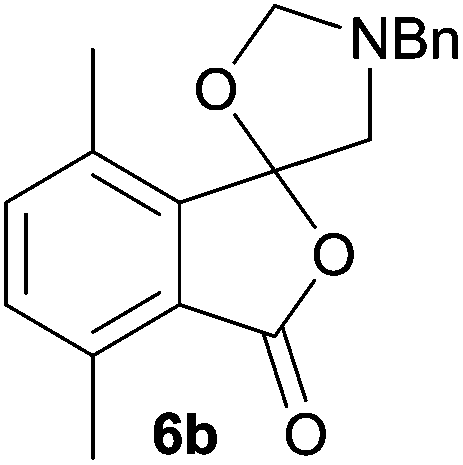
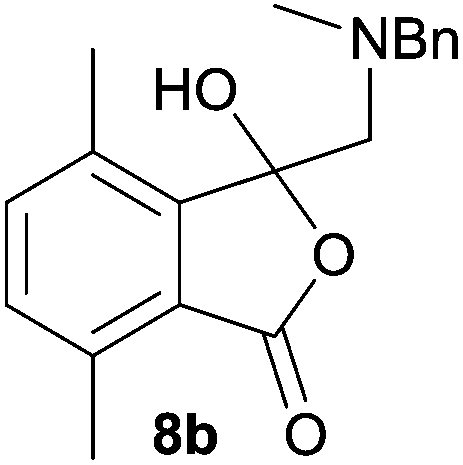
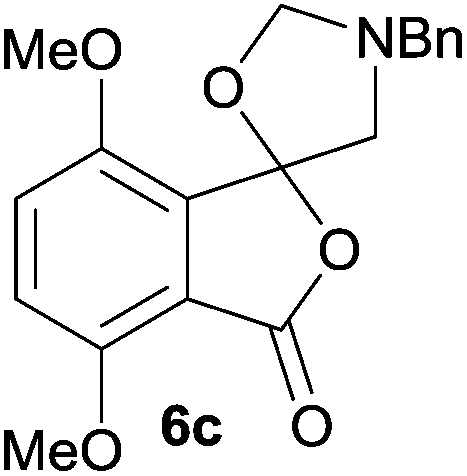
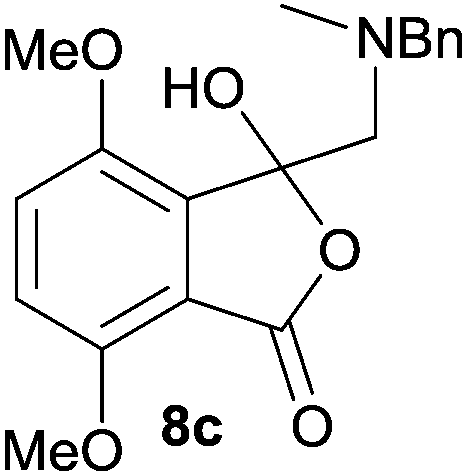
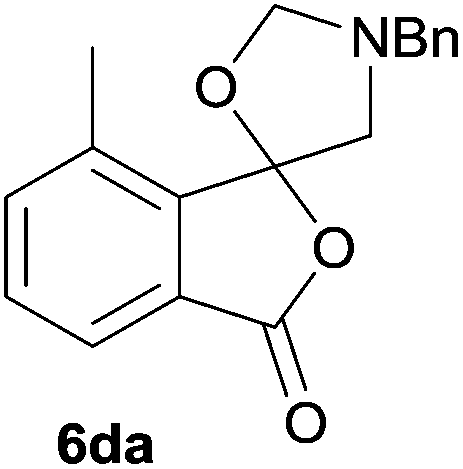
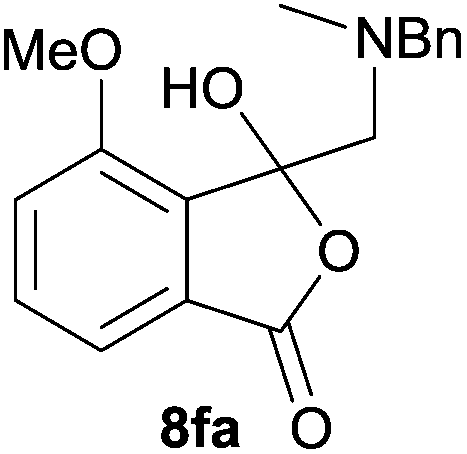
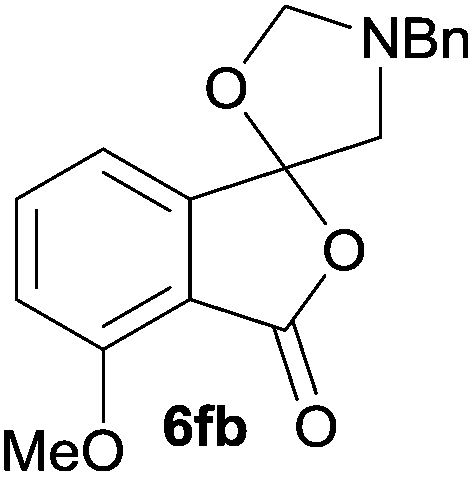
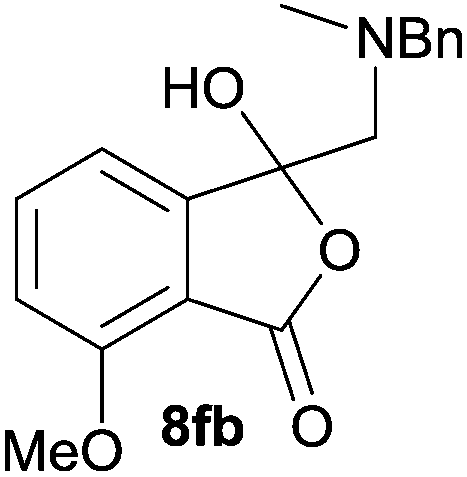
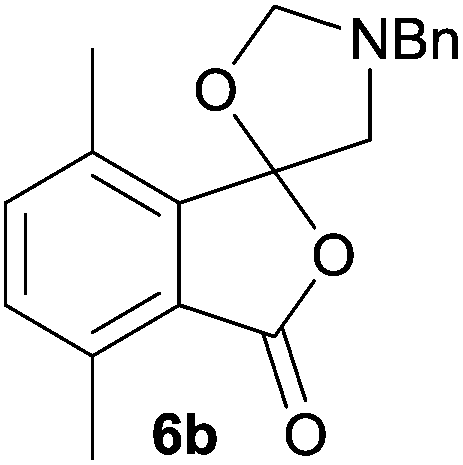
The scope of the reductive ring-opening process was explored by treating crude spiro-isobenzofuran-1,5′-oxazolidin-3-ones 6b–f, h, isolated after chromatography on Florisil™, with sodium borohydride in methanol (Table 2, entries 2–7). The 4,7-dimethyl-spiro-isobenzofuran-1,5′-oxazolidin-3-one 6b underwent reductive ring-opening smoothly to afford the corresponding 1(3H)-isobenzofuranone 8b in 69% yield (Table 2, entry 2). Whereas the two sets of diastereotopic methylene protons in the N-benzyl(methyl)aminomethyl side-chain of 8a exhibited as two singlets in the 1H NMR spectrum, the corresponding protons in 8b exhibited as four geminally coupled doublets in the 1H NMR spectrum.||
The different spectral phenomena of 8a and 8b raised some concerns about the structural assignment of 8b. Fortunately, compound 8b was a highly crystalline solid amenable to analysis by single crystal X-ray crystallography analysis (Fig. 2).** The structure exhibits an anomeric effect at the C8 hemiacetal centre and the O–H forms a weak O–H⋯N intramolecular hydrogen bond between O3 and N1 ((O⋯N = 2.6577(10) Å, O–H⋯N 128.5(14)°). As a result of this hydrogen bond the conformation about the C8–O3 bond allows for effective overlap between a lone pair on O3 with the σ* C–O antibonding orbital for the C8–O2 bond. In turn this interaction results in shortening of the C8–O3 bond distance (1.383(1) Å) and lengthening of the C8–O2 bond distance (1.477(1) Å), when compared to reported structures when this interaction is absent (mean bond length of 1.424 Å and 1.463 Å respectively).†† The anomeric effect is likely to be enhanced to a small degree by the formation of the intramolecular O–H⋯N hydrogen bond, a phenomenon that has been reported previously.18
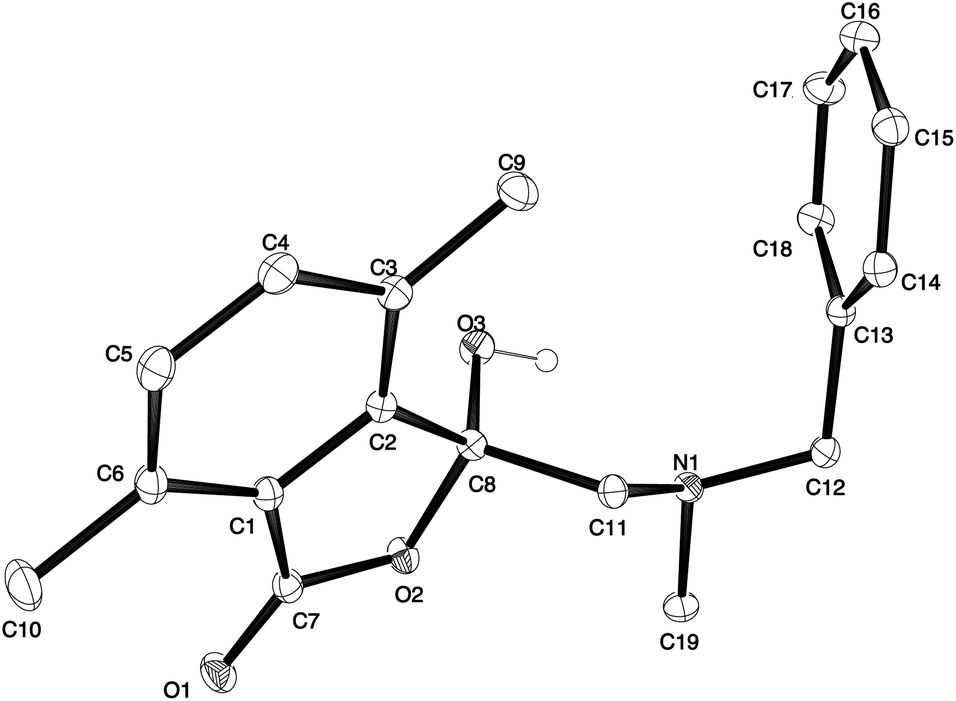 | ||
| Fig. 2 Single crystal X-ray structure of 1(3H)-isobenzofuranone 8b. | ||
The anomeric effect observed in the crystal structure of 8b suggests the possibility of equilibrium between this ring-closed form and a ring-opened ammonium-keto-carboxylate form in the solution state. Such an equilibrium would enable interconversion of the diastereotopic methylene hydrogens and could explain the differing NMR spectral phenomena observed for lactones 8a and 8b. Alternatively, the signals observed for the diastereotopic protons of 8a could be caused by rapid rotation of the less-hindered side-chain relative to 8b and coalescence of signals of the conformations involved. Further studies are required to elucidate the source of the 1H NMR phenomena observed for the side-chains methylene protons of 8a and 8b.
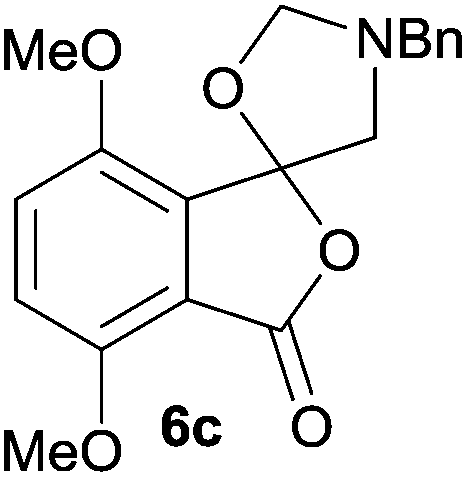
For 4,7-dimethoxy-spiro-(isobenzofuran-1,5′-oxazolidin)-3-one 6c, the reductive ring-opening with sodium borohydride proceeded to give the corresponding 1(3H)-isobenzofuranone 8c, which was obtained in pure form in a 39% yield (Table 2, entry 3).
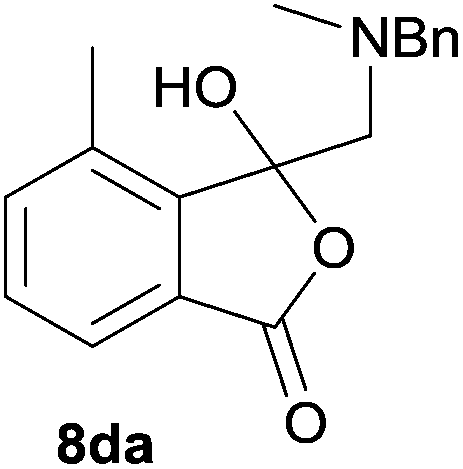
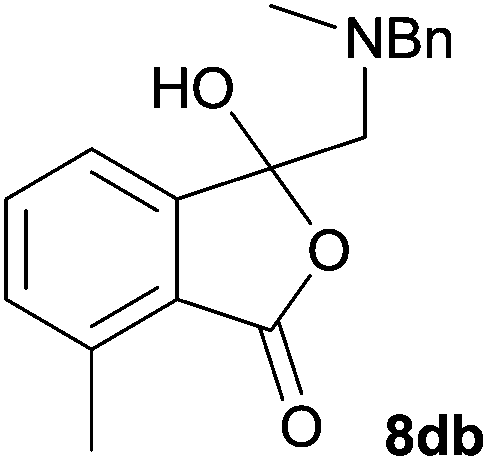
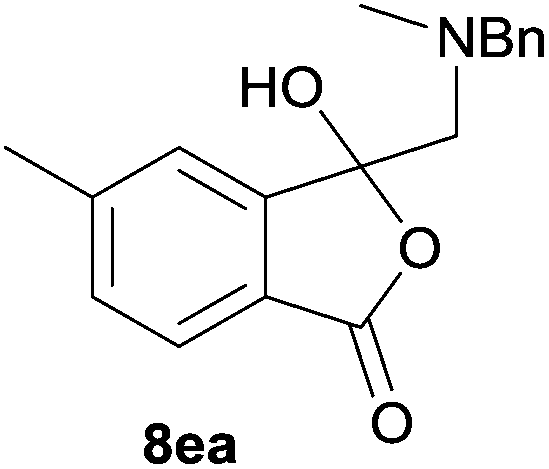
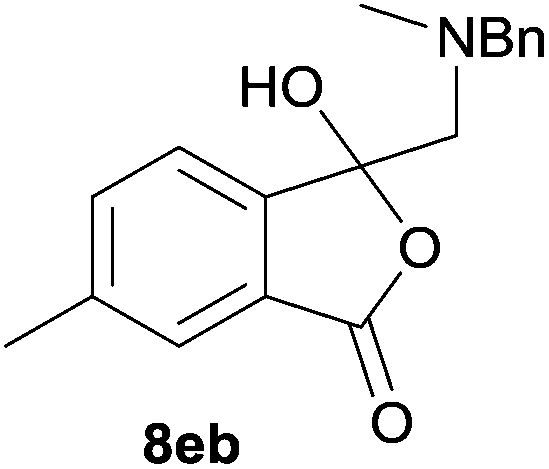
The oxazolidines obtained as mixtures of regioisomers were all reduced to the corresponding mixture of separable isobenzofuranones. The regioisomeric mixture of 6da and 6db underwent reductive ring-opening to afford a corresponding regioisomeric mixture of 1(3H)-isobenzofuranones 8da and 8db. The regioisomers were separated by chromatography to give 8da (31% yield) and 8db (31% yield) representing a combined isolated yield of 62% for the regioisomers (Table 2, entry 4). The structures of 8da and 8db were confirmed by 1H, 13C and 2D NMR experiments, and HRMS. In a similar manner, reductive ring-opening of regioisomeric mixture 6ea and 6eb afforded, after chromatographic purification, the 1(3H)-isobenzofuranones 8ea (29%) and 8eb (14%), representing a modest total overall yield of 43% (Table 2, entry 5). The regioisomeric mixture 6fa and 6fb afforded 1(3H)-isobenzofuranones 8fa (26%) and 8fb (33%), representing a combined yield of 59% (Table 2, entry 6).
Although each of these reduction reactions were judged to be complete by TLC and 1H NMR analysis, the poor to modest isolated yields in some cases is attributed to the difficulty in separating the products by chromatography.
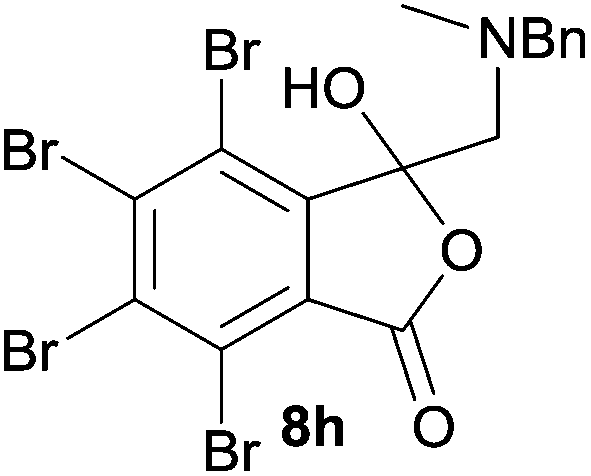
A reaction with a starting material substituted with electron-withdrawing groups was less selective leading to lower yields of the expected isobenzofuranone. The 4,5,6,7-tetrabromo-spiro-(isobenzofuran-1,5′-oxazolidin)-3-one 6h underwent reductive ring-opening to afford the corresponding 1(3H)-isobenzofuranone 8h, in only 21% yield (Table 2, entry 7). The low yield in this case was a combination of less selective reduction processes/over reduction that led to complex mixtures and the resultant difficulty in chromatographic separation of the pure product from other product contaminants during chromatography.
Conclusions
The ability of the phthalic anhydride carbonyl group to act as a 2π component in cycloaddition reactions has been demonstrated, namely in dipolar cycloaddition reactions with azomethine ylides. The product spiro(isobenzofuran)oxazolidine-3-one derivatives can be isolated in crude form via chromatography on Florisil™, and in turn selectively reduced with sodium borohydride to afford functionalised isobenzofuranone derivatives. Further derivatisation of the products is possible via deprotection of the N-Bn group or by switching the N-Bn group in the azomethine ylide to N-allyl or other N-R groups, thus expanding the utility of this method.19 Future work will be directed towards further harnessing the inherent reactivity of the spiro(isobenzofuran-oxazolidin-3-one derivatives and synthetic applications of the isobenzofuranone derivatives.Acknowledgements
HS, AD, AMD and QA thank CSIRO Australia for scholarships. We also thank Dr Roger Mulder, Dr Jo Cosgriff and Mr Carl Braybrook for assistance with collection and interpretation of NMR and/or MS data.Notes and references
- (a) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565 CrossRef PubMed. Recent examples: (b) B. M. Trost, V. Ehmke, B. M. O'Keefe and D. A. Bringley, J. Am. Chem. Soc., 2014, 136, 8213 CrossRef CAS PubMed; (c) R.-Y. Zhu, C.-S. Wang, J. Zheng, F. Shi and S.-J. Tu, J. Org. Chem., 2014, 79, 9305 CrossRef CAS PubMed; (d) C.-S. Wang, R.-Y. Zhu, J. Zheng, F. Shi and S.-J. Tu, J. Org. Chem., 2015, 80, 512 CrossRef CAS PubMed.
- (a) G. Pandey, P. Banerjee and S. R. Gadre, Chem. Rev., 2006, 16, 2047 Search PubMed; (b) I. Coldham and R. Hufton, Chem. Rev., 2005, 105, 2765 CrossRef CAS PubMed; (c) S. Husinec and V. Savic, Tetrahedron: Asymmetry, 2005, 16, 2047 CrossRef CAS PubMed; (d) C. Najera and J. M. Sansano, Angew. Chem., Int. Ed., 2005, 44, 6272 CrossRef CAS PubMed; (e) W. H. Pearson and P. Story, Synlett, 2003, 903 CrossRef CAS; (f) A. Koumbis and J. K. Gallos, Curr. Org. Chem., 2003, 7, 771 CrossRef CAS.
- N. Tagmatarchis and M. Prato, Synlett, 2003, 768 CAS.
- (a) W. Eberbach, Sci. Synth., 2004, 27, 441 CAS; (b) C. Najera and J. M. Sansano, Curr. Org. Chem., 2003, 7, 1105 CrossRef CAS; (c) L. M. Harwood and R. Vickers in Chemistry of Heterocyclic Compounds, ed. A. Padwa and W. H. Pearson, Wiley, New York, 2003, vol. 59, ch. 3, p. 169 Search PubMed; (d) O. Tsuge and S. Kanemasa, Adv. Heterocycl. Chem., 1989, 45, 231 CrossRef CAS; (e) J. W. Lown in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, 1984, vol. 1, p. 653 Search PubMed.
- (a) J. W. Lown, G. Dallas and T. W. Maloney, Can. J. Chem., 1969, 47, 3557 CrossRef CAS; (b) J. W. Lown, J. P. Moser and R. Westwood, Can. J. Chem., 1970, 48, 2227 CrossRef CAS PubMed; (c) R. Huisgen, V. Martin-Ramos and W. Scheer, Tetrahedron Lett., 1971, 477 CrossRef CAS; (d) H. W. Heine and R. P. Henzel, J. Org. Chem., 1969, 34, 171 CrossRef CAS.
- (a) J. W. Lown, R. K. Smalley, G. Dallas and T. W. Maloney, Can. J. Chem., 1970, 48, 89 CrossRef CAS PubMed; (b) G. Dallas, J. W. Lown and J. P. Moser, J. Chem. Soc. D, 1970, 278 RSC; (c) K. A. Khistiaev, M. S. Novikov, A. F. Khlebnikov and J. Magull, Tetrahedron Lett., 2008, 49, 1237 CrossRef CAS PubMed; (d) J. H. Ryan, N. Spiccia, L. S.-M. Wong and A. B. Holmes, Aust. J. Chem., 2007, 60, 898 CrossRef CAS.
- A. M. D'Souza, N. Spiccia, J. Basutto, P. Jocisz, L. S.-M. Wong, A. G. Meyer, A. B. Holmes, J. M. White and J. H. Ryan, Org. Lett., 2011, 13, 486 CrossRef PubMed.
- (a) A. Hosomi, Y. Sakata and H. Sakurai, Chem. Lett., 1984, 1117 CrossRef CAS; (b) A. Padwa and W. Dent, Org. Synth., 1989, 67, 133 CrossRef CASA. Padwa and W. Dent, Org. Synth., 1993, 231 Search PubMed , Coll. Vol. 8.
- P. K. Dubey, S. M. G. Mohuddin and R. Kumar, Asian J. Chem., 1996, 8, 341 CAS.
- E. Mahmoud, D. A. Watson and R. F. Lobo, Green Chem., 2014, 16, 167 RSC.
- (a) Y. Kajita, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2008, 130, 17226 CrossRef CAS PubMed; (b) F. Jafarpour, H. Hazrati and S. Nouraldinmousa, Org. Lett., 2013, 15, 3816 CrossRef CAS PubMed; (c) Y. Ochi, T. Kurahashi and S. Matsubara, Org. Lett., 2011, 13, 1374 CrossRef CAS PubMed; (d) K. Fujiwara, T. Kurahashi and S. Matsubara, Chem. Lett., 2011, 40, 322 CrossRef CAS.
- (a) Y. Terao, H. Kotaki, N. Imai and K. Achiwa, Chem. Pharm. Bull., 1985, 33, 896 CrossRef CAS; (b) Y. Terao, H. Kotaki, N. Imai and K. Achiwa, Chem. Pharm. Bull., 1985, 33, 2762 CrossRef CAS.
- (a) S. W. Pelletier, N. V. Mody, A. P. Venkov and H. K. Desai, Tetrahedron Lett., 1979, 20, 4939 CrossRef; (b) J. Sélambarom, S. Monge, F. Carré, J. Roquea and A. A. Pavia, Tetrahedron, 2002, 58, 9559 CrossRef; (c) Z. Kaluza, K. Bielawski, R. Cwiek, P. Niedziejko and P. Kaliski, Tetrahedron: Asymmetry, 2013, 24, 1435 CrossRef CAS PubMed.
- (a) E. D. Bergmann, Chem. Rev., 1953, 53, 309 CrossRef CAS; (b) E. D. Bergmann, E. Gil-Av and S. Pinchas, J. Am. Chem. Soc., 1953, 75, 358 CrossRef CAS; (c) P. C. Bulman Page, B. R. Buckley, M. R. J. Elsegood, C. M. Hayman, H. Heaney, G. A. Rassias, S. A. Talib and J. Liddle, Tetrahedron, 2007, 63, 10991 CrossRef CAS PubMed.
- P. M. Le, M. McCooeye and A. Windust, Anal. Bioanal. Chem., 2014, 406, 1739 CrossRef CAS PubMed.
- (a) S. Gaikwad, N. Verma, B. O. Sharma and B. C. Behera, J. Food Sci. Technol., 2014, 51, 2624 CrossRef CAS PubMed; (b) P. Seephonkai, S. G. Pyne, A. C. Willis and W. Lie, J. Nat. Prod., 2013, 76, 1358 CrossRef CAS PubMed.
- D. E. Beck, K. Agama, C. Marchand, A. Chergui, Y. Pommier and M. Cushman, J. Med. Chem., 2014, 57, 1495 CrossRef CAS PubMed.
- R. W. Alder, T. M. G. Carniero, R. W. Mowlam, A. G. Orpen, P. A. Petillo, D. J. Vachon, G. R. Weisman and J. M. White, J. Chem. Soc., Perkin Trans. 2, 1999, 589 CAS.
- (a) B. M. Fox, R. Natero, K. Richard, R. Connors, P. M. Roveto, H. Beckmann, K. Haller, J. Golde, S.-H. Xiao and F. Kayser, Bioorg. Med. Chem. Lett., 2011, 21, 2460 CrossRef CAS PubMed; (b) R. Manzano, F. Rominger and A. S. K. Hashmi, Organometallics, 2013, 32, 2199 CrossRef CAS; (c) J. Chen, T. Chen, Q. Hu, K. Püntener, Y. Ren, J. She, Z. Du and M. Scalone, Org. Process Res. Dev., 2014, 18, 1702 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Professor Ei-ichi Negishi on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds, X-ray crystallographic data for 8b (CIF) and CCDC searches. Summary of reaction optimisation of cycloaddition chemistry. CCDC 1047237. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00062a |
| § 96% conversion of starting phthalic anhydride 5a as determined by 1H NMR analysis of the reaction mixture. |
| ¶ Oxazolidine 6a decomposes under a range of conditions, including during silica chromatography, in the presence of excess trifluoroacetic acid, or when dissolved in water–methanol mixtures. |
| || A range of fluxional behaviour was observed for the diastereotopic methylene protons in the side-chains of isobenzofuranones 8. Typically, the diastereotopic protons in the products with a benzo substituent adjacent to the furanone ring (8b–d, f and h) exhibited as four distinct signals in the 1H NMR time-scale, whereas for products without substituents adjacent to the furanone ring (8a and 8e) the diastereotopic protons exhibited as two distinct signals. |
| ** Single crystals of 1(3H)-isobenzofuranone 8b were obtained by recrystallisation from dichloromethane/pentane, mounted in inert oil and transferred to the cold gas stream of the diffractometer. Crystal data. C19H21NO3, M = 311.37, T = 130.0(2) K, λ = 0.71073 Å, Monoclinic, space group P21/c a = 7.2941(2), b = 13.2249(3), c = 17.2264(4) Å, β = 96.919(2)° V = 1649.62(7) Å3, Z = 4, Dc = 1.254 Mg M−3μ(Mo-Kα) = 0.085 mm−1, F(000) = 664, crystal size 0.49 × 0.38 × 0.29 mm. θmax = 36.5°, 45689 reflections measured, 7831 independent reflections (Rint = 0.034) the final R = 0.0461 [I > 2σ(I), 6084 data] and wR(F2) = 0.10369 (all data) GOOF = 1.073. CCDC deposit code: 1047237. |
| †† The CCDC was searched for structures containing fragments that lacked the anomeric effect. The mean bond length was obtained from structures with R < 5% (see ESI†). |
| This journal is © the Partner Organisations 2015 |