
Studies on the asymmetric synthesis of pandamarilactonines: an unexpected syn-selective vinylogous Mannich reaction of N-tert-butanesulfinimines†
Jian-Liang
Ye
*,
Yu-Feng
Zhang
,
Yang
Liu
,
Jin-Yuan
Zhang
,
Yuan-Ping
Ruan
and
Pei-Qiang
Huang
*
Department of Chemistry, Fujian Provincial Key Laboratory of Chemical Biology, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, P. R. China. E-mail: pqhuang@xmu.edu.cn; yejl@xmu.edu.cn
First published on 13th April 2015
Abstract
The synthesis of pandamarilactonines in high enantiopurity is challenging due to the configurational instability of the pyrrolidin-2-yl butenolide moiety present in these alkaloids. In an attempted asymmetric synthesis of pandamarilactonine-B (2), an unanticipated syn-diastereoselective (dr = 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) asymmetric vinylogous Mannich reaction (VMR) between 3-methyl-2-(tert-butyldimethylsilyloxy)furan 9b and the Ellman (RS)-N-tert-butanesulfinimine 10a was observed. The methyl group at C-3 of TBSOF has been shown to play the role of a switch of diastereoselection in the VMR. This work ended with a concise three-pot protecting-group-free total synthesis of (−)-pandamarilactonine-A. The puzzle about the configurational instability of the pyrrolidinyl butenolide skeletons has also been disclosed by controlled experiments and with the help of computation.
:5) asymmetric vinylogous Mannich reaction (VMR) between 3-methyl-2-(tert-butyldimethylsilyloxy)furan 9b and the Ellman (RS)-N-tert-butanesulfinimine 10a was observed. The methyl group at C-3 of TBSOF has been shown to play the role of a switch of diastereoselection in the VMR. This work ended with a concise three-pot protecting-group-free total synthesis of (−)-pandamarilactonine-A. The puzzle about the configurational instability of the pyrrolidinyl butenolide skeletons has also been disclosed by controlled experiments and with the help of computation.
Introduction
The genus Pandanus (family Pandanaceae) comprises about 700 species, of which many species are used in traditional folk medicine as remedies for the treatment of diseases such as leprosy, rheumatism, epilepsy, toothache and sore throat.1 Takayama2 and co-workers have isolated several alkaloids from the genus Pandanus. Most of them feature a characteristic pyrrolidinyl butenolide/γ-lactone core structure as exemplified by pandamarilactonines-A, -B,2a -C, -D2d (cf.1–4 in Fig. 1),3 norpandamarilactonines-A (5), -B (6),2c and dubiusamine-B (7).2e Some of them are partially or fully saturated such as dubiusamine-B (7) and pandamarilactonine-E (8),2f others are either N-oxides,2e,4 or further cyclization products.4 The pyrrolidinyl γ-lactone motifs are also found in many Stemona alkaloids.5 Interestingly, Greger suggested a plausible biogenetic linkage between the Pandanus alkaloids and Stemona alkaloids.6 | ||
| Fig. 1 Structures of Pandanus alkaloids containing a pyrrolidin-2-yl butenolide/γ-lactone motif. | ||
While structurally simple, and several racemic syntheses of the Pandanus alkaloids have been reported,2a,c,d,7 the enantioselective total synthesis of pandamarilactonines in high enantiopurity is challenging due to the presence of the pyrrolidin-2-yl butenolide moiety, which is prone to either epimerization at the O-α position of butenolide or to racemization.8 This can account for the fact that the natural isolate of pandamarilactonine-A (1) exhibited a low enantiopurity (26% ee),2a while those of pandamarilactonines-B (2), -D (4) and norpandamarilactonines-A (5), -B (6) are racemates.2a,c,d The configurational instability of the pyrrolidin-2-yl butenolide-containing alkaloids has been confirmed independently by Figueredo,9 Craik,3 Takayama,10 and Honda.11
As a result, although much effort has been devoted to the enantioselective total syntheses of pandamarilactonines,2e–g,9–11 most of the syntheses of pandamarilactonines-A (1), -B (2), and norpandamarilactonines-A (5), -B (6) suffered from partial or total racemization.9–11 In the best case, during the first asymmetric total synthesis accomplished by Takayama and co-workers,10 the unnatural enantiomer (−)-pandamarilactonine-A was obtained with an estimated ee of 70%.2a,12 More recently, the same group reported the first enantioselective total syntheses of dubiusamine-B (7)2e and pandamarilactonine-E (8),2f respectively, by an alternative approach.
It is worth mentioning that Craik and co-workers have suggested that pandamarilactonines A–D were artifacts formed from pandanamine during the acid–base extraction process.3 However, Takayama and co-workers speculated that pandamarilactonine-A of high enantiopurity is naturally occurring, and the observed low optical purity could be attributed to a partial racemization occurring during the extraction/isolation process.10
In connection with our interest in the synthesis of vicinal aminol-containing compounds13 and Stemona alkaloids,14 we have developed an anti-selective asymmetric vinylogous Mannich reaction (VMR: 9a and 10) to furnish anti-5-aminoalkyl butenolides 11 (Scheme 1).15 The method is based, on one hand, on the powerful methodologies of the vinylogous Mannich reactions (VMR) developed by Casiraghi–Rassu,16 and Martin,17 respectively, and on the other hand, on the use of the Ellman N-tert-butanesulfinimines 10 (t-BS-imines)18 as versatile chiral amine templates. The method has been applied to the synthesis of (−)-deoxoprosophylline and formal synthesis of 3-epi-fagomine and 1-deoxyallonojirimycin.15 On the basis of these precedents, it was envisaged that the anti-diastereoselective method could be extended to the asymmetric synthesis of pandamarilactonine-B (2) possessing a 14,15-anti-stereochemistry. An investigation along this line has been undertaken, and the results are reported herein.
 | ||
| Scheme 1 Diastereoselective VMRs of TBSOFs (9) with Ellman sulfinimines 10. | ||
Results and discussion
On the basis of the anti-selective VMR (9a and 10) leading to anti-11 (Scheme 1),15 our retrosynthetic analysis of pandamarilactonine-B (2) is outlined in Scheme 2. (14S,15R)-Pandamarilactonine-B (2) should be available from (5S,6R)-norpandamarilactonine-A (5) and the known iodide 13.2d The former was envisioned to be synthesized from (RS)-N-tert-butanesulfinimine 10a and 3-MTBSOF (9b) by the expected anti-selective VMR.15 | ||
| Scheme 2 Retrosynthetic analysis of pandamarilactonine-B (2). | ||
Our synthesis commenced with the preparation of (RS)-t-BS-imine 10a,18h which was achieved in 94% yield by the CuSO4-mediated condensation18e of the commercially available 4-chlorobutanal 14 with (RS)-tert-butanesulfinamide 1518a,e (Scheme 3). For the key VMR, the previously established optimal conditions15a were adopted. Thus, treatment of a CH2Cl2 solution of 3-MTBSOF (9b, 1.5 mol equiv.) and t-BS-imine 10a (1.0 mol equiv.) with TMSOTf (1.0 mol equiv.) at −78 °C for 1 h yielded two separable diastereomers (16, dr = 95:5, determined by 1H NMR) in a combined yield of 97%. By analogy with our previous results,15a the major diastereomer was tentatively assigned as anti-16 with a (4R,5S)-stereochemistry.
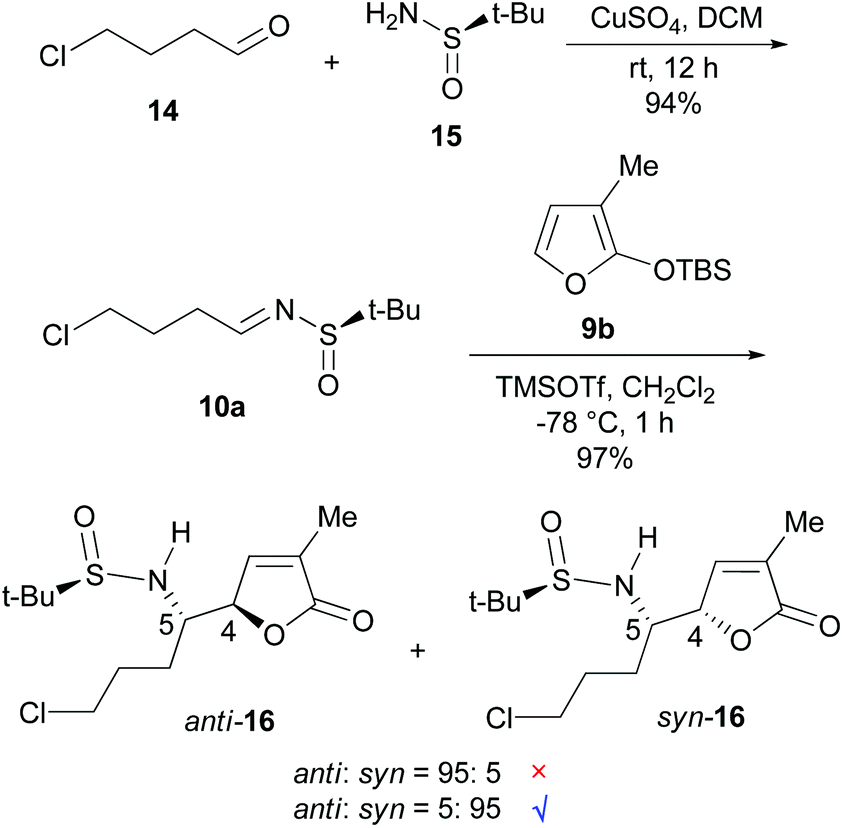 | ||
| Scheme 3 The synthesis and VMR of t-BS-imine 10a with 3-MTBSOF (9b). | ||
To confirm the stereochemistry of the major diastereomer of 16, the VMR between the known t-BS-imine 10b15a with 3-MTBSOF (9b)19 was undertaken (eqn (1) in Scheme 4). Unexpectedly, the single crystal X-ray diffraction analysis revealed that the major diastereomer of 17 (CCDC 1046182) possesses a (RS,4S,5S)-syn-stereochemistry (Fig. 2). According to this result, the stereochemistry of the major diastereomer of the VMR between 10a and 9b (Scheme 3) was revised to (4S,5S)-syn-16.
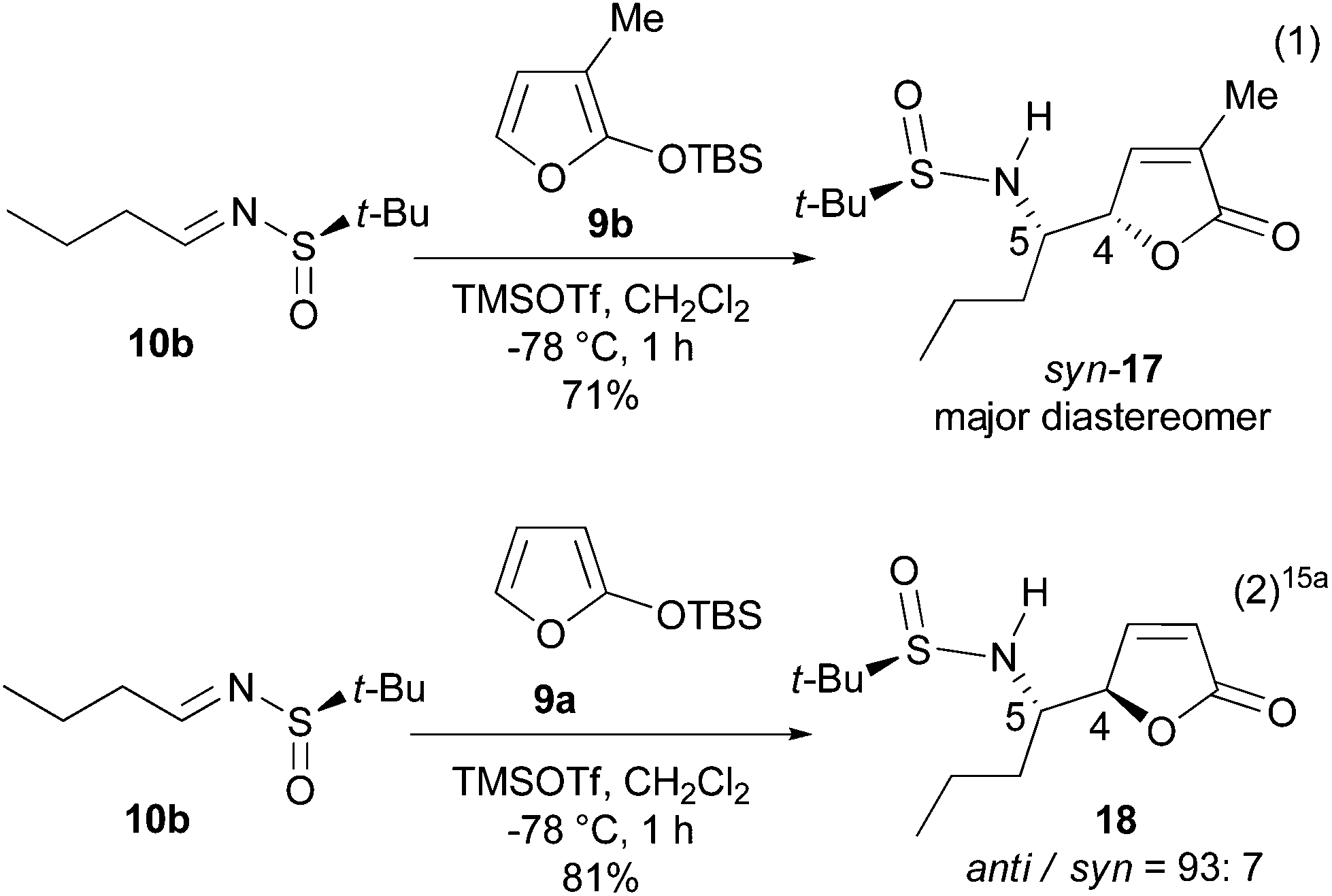 | ||
| Scheme 4 The VMRs of t-BS-imine 10b with TBSOF 9b and 9a. | ||
 | ||
| Fig. 2 X-ray single crystal structure of the major diastereomer syn-17. | ||
A comparison of the present results with those we reported previously (cf. eqn (2) in Scheme 4)15a showed that the 3-methyl group in TBSOF played the role of a diastereomeric switch. A survey of the literature showed that while a syn-selection16,17 is generally observed for the VMRs, anti-selection20 was observed for both the Davis sulfinimine21 and Ellman sulfinimine18a,d-based VMRs.15,22 Although changing of diastereoselection by varying the reaction parameters has been observed,20g,23 the inverse of diastereoselection by a modification of the substrate is rare.20e Hoveyda's results also involved modification of the reaction conditions.20e By contrast, the literature precedents showed that introducing a methyl group at the α-position of TBSOF has a minor effect on the diastereoselectivity.17a,20f,g,22,24 Thus, the observed effect of α-methyl as a switch of the diastereoselection is an interesting phenomenon which will provide a valuable tool for controlling diastereoselection of the asymmetric VMRs, and find synthetic applications.
Thus, although the stereochemical outcome of the VMR is opposed to our anticipation, and the reason is unclear at this stage, this unexpected result afforded us an opportunity to revise our synthetic target from the presumed artifact pandamarilactonine-B (2) to pandamarilactonine-A (1).
We next attempted the base-promoted cyclization of sulfinamide syn-16 to form N-t-BS-norpandamarilactonine-B (19) (Scheme 5). To our disappointment, treatment of sulfinamide syn-16 with either K2CO3/NaI, or KOH,25 or NaH (Scheme 5) did not produce the expected cyclization product 19. Thus, the N-sulfinyl group in syn-16 was cleaved on treatment with concentrated HCl in dioxane at rt for 1 h,18h and the resulting mixture was concentrated in vacuo. Without further purification, the residue was directly treated with anhydrous K2CO3 and a catalytic amount of NaI in MeCN18h at rt for 16 h under an atmosphere of argon. An analysis of the 13C NMR data of the crude product showed that the major diastereomer was identical to that of norpandamarilactonine-B (6), and the characteristic 13C NMR data of the minor diastereomer were in agreement with those reported for norpandamarilactonine-A (5).2c The diastereomeric ratio of 6:5 was determined by 1H NMR to be ca. 94:6 (cf. S13 in the ESI†). These results confirmed that the VMR between 10a and 9b is indeed syn-selective (Scheme 3), and under the reaction conditions [K2CO3, NaI (cat.) in anhydrous MeCN at rt], the reaction proceeded with only about 6% of epimerization (Scheme 5). Moreover, it was found that during the separation of two diastereomers 6 and 5, both decomposed readily on exposure to air, with the color changing from colorless to red. The configurational instability of norpandamarilactonines 6 and 5 has been noted by Figueredo,9 Takayama,10 and Honda et al.,11 respectively.
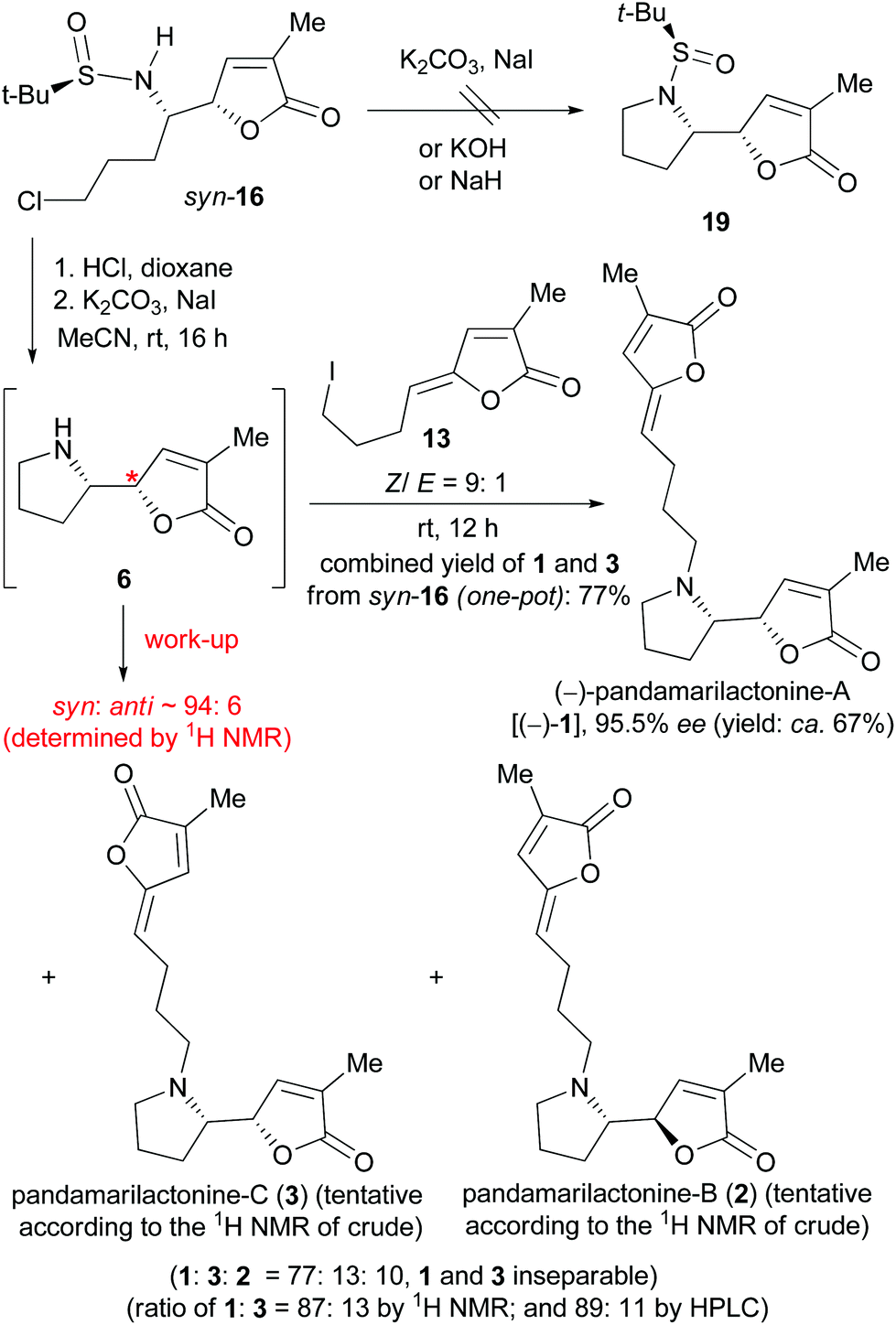 | ||
| Scheme 5 Synthesis of (−)-pandamarilactonine-A [(−)-1]. | ||
With the valuable results in hand, the synthesis of (−)-pandamarilactonine-A (1) was pursued (Scheme 5). In view of the configurational instability of norpandamarilactonine-B (6) and the mildness of the conditions [K2CO3, NaI (cat.) in anhydrous MeCN at rt] used for the transformation of syn-16 to 6, a one-pot transformation of syn-16 to 1 was envisioned.
For this purpose, the requisite Z-iodide side chain 13 was prepared according to Takayama's procedure2d and obtained as an E/Z mixture in a ratio of 1:9 (cf. S3 in the ESI†) after repeated column chromatography. Successive treatment of sulfinamide syn-16 with concentrated HCl (dioxane, rt, 1 h), K2CO3/NaI (8 mol equiv., 0.1 mol equiv., rt, 16 h, Ar), and a solution of 13 (2.5 mol equiv.) in CH2Cl2 (rt, 12 h) produced a crude product containing three diastereomers in a ratio of 77:13:11 as determined by 1H NMR (cf. S7 in the ESI†). According to the characteristic NMR data,26 the three diastereomers were tentatively assigned as pandamarilactonines-A (1), -C (3), and -B (2), respectively. Unfortunately, pandamarilactonines-A (1) and -C (3) were inseparable by chromatography. The ratio of pandamarilactonines-A (1) and -C (3) was determined as 87:13 by 1H NMR and 89:11 by HPLC (cf. ESI†). The specific optical rotation of pandamarilactonine-A (1) on being mixed with 12% of pandamarilactonine-C (3) was [α]20D −87.2 (c 0.12, CHCl3) {lit.10 [α]23D −94.0 (c 0.12, CHCl3)}. To our surprise, chiral HPLC analysis showed that the enantiomeric excess of our synthetic pandamarilactonine-A [(−)-1] was 95.5%. Since the ee of the chiral auxiliary (RS)-tert-butanesulfinamide 15 is about 98%, our synthetic approach is almost free of racemization.
Moreover, since both pandamarilactonines-A (1) and -C (3) possess a syn-stereochemistry in the vicinal amino moieties, the 14,15-syn/anti diastereoselectivity of the tandem reaction was 90:10. It can be deduced that a ca. 4% of epimerization occurred at the O-α position during the coupling reaction of 6 and 13.
On the epimerization/racemization of the δ-amino-butenolide moiety
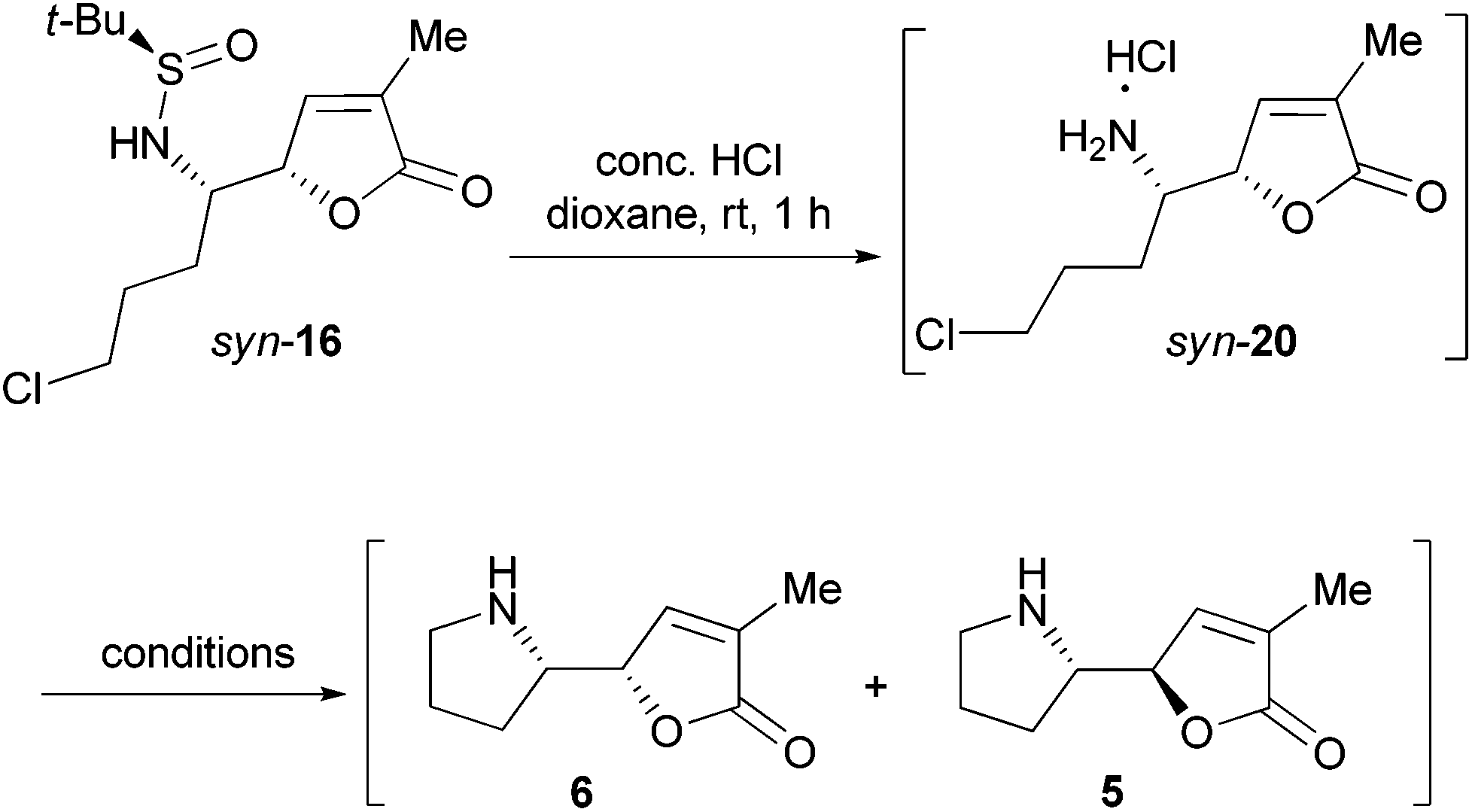
To examine the configurational stability of the δ-amino-butenolide derivatives, syn-16 was treated with 8 mol equiv. of K2CO3 in MeCN at rt under Ar for a week (Scheme 6). Under such conditions, the epimerization of syn-16 (to give anti-16) was not observed from the 1H NMR spectrum of the crude product. This result implied that the δ-amino-butenolide bearing an electron-withdrawing group at the N-atom shows a good stability. Similarly, Honda et al. have found that N-Boc-norpandamarilactonine-A possessed a much higher stability than norpandamarilactonine-A.11a Moreover, the 1H NMR spectrum of the crude product obtained from the desulfinylation (conc. HCl, dioxane, rt, 1 h) of syn-16 showed that the resulting salt is diastereomerically homogeneous (cf. S11 in the ESI†), which is presumed to be syn-20. The observed high stability of δ-amino-butenolide under acidic conditions is in agreement with those observed by Takayama et al.2a,10 | ||
| Scheme 6 Attempted epimerization of the compound syn-16. | ||
To further probe the configurational stability of the δ-amino-butenolide derivative 16 and/or norpandamarilactonine-B (6), a series of experiments have been conducted and the results are summarized in Table 1. After the desulfinylation reaction (conc. HCl, dioxane, rt, 1 h), the resulting mixture was concentrated under a reduced pressure. To the resulting concentrate, anhydrous MeCN and K2CO3 (4 mol equiv.) were added, and the mixture was stirred at rt for 12 h under Ar before being concentrated. 1H NMR analysis of the concentrate showed that the diastereomeric ratio of 6/5 was 94:6 (Table 1, entry 1, cf. S13 in the ESI†). However, when the concentrate was dissolved with CH2Cl2 and washed with NH3·H2O, the ratio of 6/5 became 88:12 (entry 2, cf. S14 in the ESI†). If the concentrate was allowed to stand with NH3·H2O for one day, a ca. 62:38 mixture of 6 and 5 was formed (entry 3, cf. S14 in the ESI†). Surprisingly, in the presence of water, treatment of the concentrate of syn-20 with K2CO3 (4 mol equiv.) also resulted in a 62:38 mixture of 9 and 8 (entry 4, cf. S15 in the ESI†). Alternatively, exposing the concentrate of syn-20 to NH3·H2O for two hours led to a ratio of 67:33 (6/5, entry 5, cf. S16 in the ESI†). Notably, Honda et al. have reported that upon standing at rt for 1 h, norpandamarilactonine-A (5) converted to a 71:29 mixture of norpandamarilactonines-A (5) and -B (6).11a Our results thus confirmed that: (1) both δ-amino-butenolide skeletons of syn-16 and syn-20 are stable under acidic conditions. This conclusion is in support of the results obtained by Takayama et al.2a,10 (2) δ-Amino-butenolide derivative 20 and norpandamarilactonine-B (6) are configurationally stable in the presence of anhydrous K2CO3 (4 mol equiv.) in MeCN. (3) Introduction of water or treating 20 and 6 with NH3·H2O resulted in substantial epimerization.
|
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Time (h) | Further treatment | 6/5a |
| a Ratios determined by analysis of 400 MHz 1H NMR spectra of unpurified reaction mixtures. | ||||
| 1 | K2CO3 (4) | 12 | No | 94:6 |
| 2 | K2CO3 (4) | 12 | Washed with NH3·H2O | 88:12 |
| 3 | K2CO3 (4) | 12 | Addition of NH3·H2O, then kept for 1 day | 62:38 |
| 4 | K2CO3 (4) & H2O | 12 | No | 62:38 |
| 5 | NH3·H2O | 2 | No | 67:33 |
To get further insight into the configurational instability of the pyrrolidinyl butenolide motif, we conducted quantum chemical calculations. The calculations were performed at the B3LYP/6-311++G (2d,p) (in MeCN with SMD solvation model)//B3LYP/6-31G* level on the desulfinylation intermediate anti-20 and its diastereomer syn-20, and norpandamarilactonines-A (5), -B (6). The results showed that the relative energies of anti-20/syn-20, and 6/5 are 0.24 kcal mol−1 and 0.27 kcal mol−1 (Fig. 3), respectively, corresponding to a ratio of 40:60 for anti-20 and syn-20, and a ratio of 61:39 for 5 and 6 under the equilibrium conditions. The calculation results match those observed for the epimerization of 5 and 6 (71:29 from 5, ref. 11a; 62:38 from 6, vide supra).
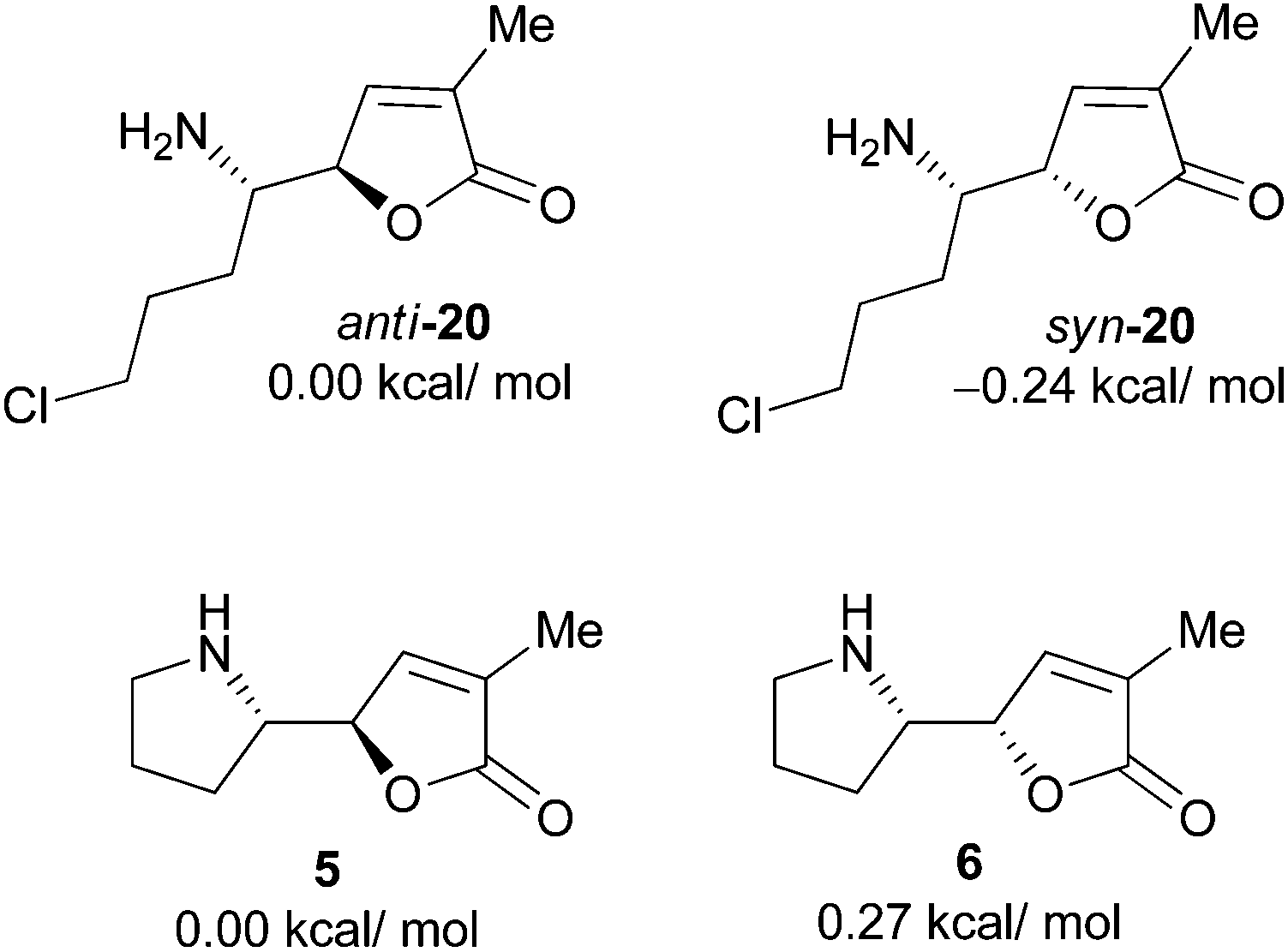 | ||
| Fig. 3 The relative energies of desulfinylated intermediates anti-20 and syn-20, norpandamarilactonine-A (5) and norpandamarilactonine-B (6) at the B3LYP/6-311++G (2d,p) (in MeCN with SMD solvation model)//B3LYP/6-31G* level. | ||
Conclusions
In summary, an unanticipated highly syn-diastereoselective asymmetric vinylogous Mannich reaction (AVMR) between the t-BS-imines 10a,b and 3-MTBSOF (9b) has been uncovered. On the basis of this reaction, a protecting-group-free asymmetric total synthesis of (−)-pandamarilactonine-A [(−)-1] has been achieved. The synthesis required only three-pot in 58% overall yield from 4-chlorobutanal 14, (RS)-tert-butanesulfinamide 15 and the known Z-iodide side chain 13, prepared in three additional steps. Although our synthetic sample of (−)-pandamarilactonine-A (6Z isomer, (−)-1) was contaminated with 13% of pandamarilactonine-C (6E isomer, 3), both syn diastereoselectivity (dr = 90:10) and the enantiomeric purity of (−)-1 (95.5% ee) are excellent. It is expectable that this approach is applicable to the asymmetric synthesis of dubiusamine-B (7)2e and pandamarilactonine-E (8)2f by using the known (3R,5R)-γ-butyrolactone iodide prepared by Takayama and co-workers.2e Moreover, by combining our results with those previously obtained by Takayama, Figueredo, Honda, Craik, and co-workers, we were able to clarify the configurational stability of the δ-amino butenolide structural motif, which is stable under the acidic and anhydrous K2CO3/MeCN conditions, while sensitive to water and basic aqueous conditions. Appropriate conditions have been defined to avoid epimerization and racemization in the total synthesis.
Experimental section
General methods
Column chromatography separation was performed on silica gel. Melting points are uncorrected. Infrared spectra were measured with a FT-IR spectrometer using the film KBr pellet technique. 1H and 13C NMR spectra were recorded in CDCl3 at 400 MHz and 100 MHz, respectively. HRMS spectra were recorded on an ESI-TOF mass spectrometer.:1) to give the known (RS)-t-BS-imine 10a18h (488 mg, yield: 94%) as a colorless oil. [α]20D −211.8 (c 1.0, CHCl3) {lit.18h [α]28D +216 (c 1.24, CHCl3)}; IR (film): 2960, 2926, 2868, 1624, 1456, 1363, 1083 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.14 (s, 9H), 2.05–2.12 (m, 2H), 2.63–2.68 (m, 2H), 3.57 (dt, J = 1.9, 6.5 Hz, 2H), 8.05 (t, J = 4.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 22.3 (3C), 27.9, 33.1, 44.0, 56.6, 167.8; HRMS calcd for C8H16ClNOS [M + Na]+: 232.0539; found: 232.0533.
:95 determined by 1H NMR). The mixture was purified by repeated flash chromatography on silica gel (eluent: hexane/EtOAc 1:2) to afford the pure compound syn-16 (2.14 g, yield: 91%) and the pure compound anti-16 (50 mg, yield: 2%). Compound syn-16 (major diastereomer): colorless oil, [α]20D −106.2 (c 1.0, CHCl3); IR (film): 3257, 2958, 2869, 1752, 1449, 1364, 1062, 912 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.12 (s, 9H), 1.68–2.15 (m, 7H including a triplet at 1.88 (dd, J = 1.6, 1.9 Hz, 3H)), 3.15 (d, J = 6.8 Hz, 1H), 3.47–3.55 (m, 1H), 3.56–3.61 (m, 2H), 4.90 (ddd, J = 4.8, 4.2, 1.9 Hz, 1H), 6.96–6.99 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 10.7, 22.7 (3C), 28.7, 30.7, 44.4, 56.5, 56.7, 82.8, 131.9, 146.3, 173.8; HRMS calcd for C13H22ClNO3S [M + Na]+: 330.0907; found: 330.0908. Compound anti-16 (minor diastereomer): colorless oil, [α]20D +71.0 (c 1.0, CHCl3); IR (film): 3243, 2959, 2925, 1752, 1062 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.19 (s, 9H), 1.49–1.70 (m, 1H), 1.82–1.99 (m, 5H including a triplet at 1.93 (dd, J = 1.6, 1.8 Hz, 3H)), 2.01–2.20 (m, 1H), 3.11 (d, J = 7.2 Hz, 1H), 3.42–3.54 (m, 1H), 3.60 (t, J = 6.0 Hz, 2H), 4.83–4.94 (m, 1H), 7.04–7.09 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 10.8, 22.6 (3C), 28.5, 29.1, 44.5, 56.4, 57.9, 83.3, 132.6, 144.6, 173.5; HRMS calcd for C13H22ClNO3S [M + Na]+: 330.0907; found: 330.0908.
:1) to afford syn-17 (384 mg, yield: 71%) as colorless crystals. mp 76–77 °C (CHCl3); [α]20D −125.2 (c 1.0, CHCl3); IR (film): 3446, 3267, 2960, 2929, 2872, 1756, 1659, 1458, 1382, 1365, 1247, 1208, 1142, 1054, 995, 974, 921 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.92 (t, J = 7.3 Hz, 3H), 1.11 (s, 9H), 1.32–1.59 (m, 2H), 1.67 (dd, J = 7.8, 15.0 Hz, 2H), 1.86 (dd, J = 1.5, 1.8 Hz, 3H), 3.06 (d, J = 6.6 Hz, 1H), 3.50 (ddd, J = 2.5, 6.9, 13.9 Hz, 1H), 4.77 − 4.83 (m, 1H), 6.98 (app t, J = 1.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 10.2, 13.3, 18.8, 22.3 (3C), 34.9, 55.8, 56.6, 82.2, 130.9, 146.6, 173.6; HRMS calcd for C13H23NO3S [M + Na]+: 296.1291; found: 296.1290.
:9 alkyl iodide 13 (prepared by Takayama's procedure in three steps, purified by repeated column chromatography and 55% overall yield from 4-chlorobutanal)2d (139 mg, 0.5 mmol) in MeCN (1 mL). The mixture was stirred at room temperature for 12 h before H2O (5 mL) and Et2O (5 mL) were added. The organic layer was separated; the aqueous layer was extracted with Et2O (5 mL × 2). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was filtered on silica gel to give the crude product including (−)-pandamarilactonines-A [(−)-1], -B (2) and -C (3) (eluent: EtOAc, dr = 77:10:13 determined by 1H NMR). The crude product was purified with repeated silica gel column chromatography (eluent: CH2Cl2/MeOH 20:1) to afford (−)-pandamarilactonines-A (1) and -C (3) as a mixture (47 mg, yield: 77%, dr = 87:13 determined by 1H NMR, yield of (−)-pandamarilactonine-A (1): 58%) as an amorphous powder. [α]20D −87.2 (c 0.12, CHCl3) {lit.10 [α]23D −94.0 (c 0.12, CHCl3) for (−)-pandamarilactonine-A (1)}; IR (film): 2923, 1759, 1091, 1048, 990 cm−1; 1H NMR (400 MHz, CDCl3) (data of the major diastereomer (−)-pandamarilactonine-A [(−)-1] read from the spectrum of the mixture) δ 1.37–1.46 (m, 1H), 1.55–1.69 (m, 3H), 1.70–1.79 (m, 2H), 1.92 (app t, J = 1.7 Hz, 3H), 1.98 (app dd, J = 0.7, 1.2 Hz, 3H), 2.16–2.25 (m, 1H), 2.39–2.47 (m, 3H), 2.75–2.96 (m, 2H), 3.11 (t, J = 7.7 Hz, 1H), 4.77–4.81 (m, 1H), 5.17 (t, J = 7.9 Hz, 1H), 6.99 (app d, J = 1.2 Hz, 1H), 7.08 (app t, J = 1.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) (data of the major diastereomer (−)-pandamarilactonine-A [(−)-1] read from the spectrum of the mixture) δ 10.5, 10.7, 23.8, 24.0, 25.7, 28.3, 54.2, 55.0, 65.3, 83.4, 114.1, 129.1, 131.2, 137.7, 146.9, 148.6, 171.1, 174.3; HRMS calcd for C18H23NO4 [M + Na]+: 318.1670; found: 318.1695.
:87:11 {lit.2a chiral HPLC analysis of natural (+)-pandamarilactonine-A (1) (Chiralcel OB, solvent 40% i-PrOH/hexane, flow rate 0.3 mL min−1, column temperature 30 °C) and exhibited two peaks at 43.2 and 51.9 min in a ratio of 63:37}.27
Acknowledgements
The authors are grateful for financial support from the NSF of China (21332007 and 21472153), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of the Ministry of Education, the National Found for Fostering Talents of Basic Science (NFFTBSno. J1310024), and the Natural Science Foundation of Fujian Province of China (no. 2014J01062, 2011J01056).Notes and references
- (a) M. Tan, Philippine Medicinal Plants in Common Use: Their Phytochemistry and Pharmacology, AKAP, Quezon City, Philipp, 1980 Search PubMed; (b) E. Quisumbing, Medicinal Plants of the Philippines, Bureau of Printing, Manila, 1978 Search PubMed.
- (a) H. Takayama, T. Ichikawa, T. Kuwajima, M. Kitajima, H. Seki, N. Aimi and M. G. Nonato, J. Am. Chem. Soc., 2000, 122, 8635–8639 CrossRef CAS; (b) H. Takayama, T. Ichikawa, M. Kitajima, N. Aimi, D. Lopez and M. G. Nonato, Tetrahedron Lett., 2001, 42, 2995–2996 CrossRef CAS; (c) H. Takayama, T. Ichikawa, M. Kitajima, M. G. Nonato and N. Aimi, J. Nat. Prod., 2001, 64, 1224–1225 CrossRef CAS PubMed; (d) H. Takayama, T. Ichikawa, M. Kitajima, M. G. Nonato and N. Aimi, Chem. Pharm. Bull., 2002, 50, 1303–1304 CrossRef CAS PubMed; (e) M. A. Tan, M. Kitajima, N. Kogure, M. G. Nonato and H. Takayama, Tetrahedron, 2010, 66, 3353–3359 CrossRef CAS; (f) M. A. Tan, M. Kitajima, N. Kogure, M. G. Nonato and H. Takayama, Tetrahedron Lett., 2010, 51, 4143–4146 CrossRef CAS; (g) M. A. Tan, M. Kitajima, N. Kogure, M. G. Nonato and H. Takayama, J. Nat. Prod., 2010, 73, 1453–1455 CrossRef CAS PubMed.
- A. A. Salim, M. J. Garson and D. J. Craik, J. Nat. Prod., 2004, 67, 54–57 CrossRef CAS PubMed.
- Note that the relative stereochemistries of pandamarilactonines-A (1) and -B (2) assigned in ref. 2a have been revised as those shown in Fig. 1 (cf.ref. 2d).
- For recent reviews on the chemistry and biological activities of Stemona alkaloids, see: (a) H. Greger, Planta Med., 2006, 72, 99 CrossRef CAS PubMed; (b) R. A. Pilli, G. B. Rosso and M. C. F. De Oliveira, Nat. Prod. Rep., 2010, 27, 1908 RSC.
- H. Greger, J. Schinnerl, S. Vajrodaya, L. Brecker and O. Hofer, Nat. Prod. Rep., 2009, 26, 1708 CrossRef PubMed.
- (a) S. Gogoi and N. P. Argade, Synthesis, 2008, 1455–1459 CAS; (b) K. Y. Seah, S. J. MacNaughton, J. W. P. Dallimore and J. Robertson, Org. Lett., 2014, 16, 884–887 CrossRef CAS PubMed.
- (a) P. Camps, J. Cardellach, J. Corbera, J. Font, R. M. Ortuño and O. Ponsatí, Tetrahedron, 1983, 39, 395–400 CrossRef CAS; (b) P. Duret, B. Figadère, R. Hocquemiller and A. Cavé, Tetrahedron Lett., 1997, 38, 8849–8852 CrossRef CAS; (c) Q. A. Yu, Y. K. Wu, Y. L. Wu, L. J. Xia and M. H. Tang, Chirality, 2000, 12, 127–129 CrossRef CAS PubMed.
- (a) P. Blanco, F. Busque, P. de March, M. Figueredo, J. Font and E. Sanfeliu, Eur. J. Org. Chem., 2004, 48–53 CrossRef CAS; (b) F. Busque, P. de March, M. Figueredo, J. Font and E. Sanfeliu, Tetrahedron Lett., 2002, 43, 5583–5585 CrossRef CAS.
- H. Takayama, R. Sudo and M. Kitajima, Tetrahedron Lett., 2005, 46, 5795–5797 CrossRef CAS.
- (a) T. Honda, M. Ushiwata and H. Mizutani, Tetrahedron Lett., 2006, 47, 6251–6254 CrossRef CAS; (b) T. Honda, M. Katoh, M. Ushiwata and H. Shigehisa, Arch. Pharm., 2008, 341, 578–583 CrossRef CAS PubMed.
- Estimated based on a comparison of the specific rotation data: [α]23D −94 (c 0.12, CHCl3) with that of the natural pandamarilactonine-A: [α]23D +35 (c 4.37, CHCl3), 26% ee (cf. ref. 2a).
- (a) A.-E Wang and P.-Q. Huang, Pure Appl. Chem., 2014, 86, 1227–1235 CAS; (b) G. Liu, T.-J. Wu, Y.-P. Ruan and P.-Q. Huang, Chem. – Eur. J., 2010, 16, 5755–5768 CrossRef CAS PubMed; (c) X.-G. Wang, A.-E Wang, Y. Hao, Y.-P. Ruan and P.-Q. Huang, J. Org. Chem., 2013, 78, 9488–9493 CrossRef CAS PubMed; (d) X.-G. Wang, A.-E Wang and P.-Q. Huang, Chin. Chem. Lett., 2014, 25, 193–196 CrossRef CAS.
- (a) S.-C. Tuo, J.-L. Ye, A.-E Wang, S.-Y. Huang and P.-Q. Huang, Org. Lett., 2011, 13, 5270–5273 CrossRef CAS PubMed; (b) X.-K. Liu, J.-L. Ye, Y.-P. Ruan, Y.-X. Li and P.-Q. Huang, J. Org. Chem., 2013, 78, 35–41 CrossRef CAS PubMed; (c) S.-C. Tuo, X.-K. Liu and P.-Q. Huang, Chin. J. Chem., 2013, 31, 55–62 CrossRef CAS.
- (a) S.-T. Ruan, J.-M. Luo, Y. Du and P.-Q. Huang, Org. Lett., 2011, 13, 4938–4941 CrossRef CAS PubMed; (b) L.-D. Guo, P. Liang, J.-F. Zheng and P.-Q. Huang, Eur. J. Org. Chem., 2013, 2230–2236 CrossRef CAS.
- For recent reviews, see: (a) G. Casiraghi, F. Zanardi, L. Battistini and G. Rassu, Synlett, 2009, 1525–1542 CrossRef CAS; (b) G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, Chem. Rev., 2011, 111, 3076–3154 CrossRef CAS PubMed; (c) G. Rassu, F. Zanardi, L. Battistini and G. Casiraghi, Chem. Soc. Rev., 2000, 29, 109–118 RSC.
- (a) S. F. Martin and J. W. Corbett, Synthesis, 1992, 55–57 CrossRef CAS. For reviews and accounts, see: (b) S. K. Bur and S. F. Martin, Tetrahedron, 2001, 57, 3221–3242 CrossRef CAS; (c) S. F. Martin, Acc. Chem. Res., 2002, 35, 895–904 CrossRef CAS PubMed. For some theoretical studies on the origins of diastereoselectivity of the VMR, see: (d) S. K. Bur and S. F. Martin, Org. Lett., 2000, 2, 3445–3447 CrossRef CAS PubMed.
- For reviews, see: (a) J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984–995 CrossRef CAS PubMed; (b) F. Ferreira, C. Botuha, F. Chemla and A. Pérez-Luna, Chem. Soc. Rev., 2009, 38, 1162–1186 RSC; (c) G. Q. Lin, M. H. Xu, Y. W. Zhong and X. W. Sun, Acc. Chem. Res., 2008, 41, 831–840 CrossRef CAS PubMed; (d) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS PubMed. For selected examples, see: (e) G. Liu, D. A. Cogan, T. D. Owens, T. P. Tang and J. A. Ellman, J. Org. Chem., 1999, 64, 1278–1284 CrossRef CAS; (f) M. T. Tang, S. K. Volkman and J. A. Ellman, J. Org. Chem., 2001, 66, 8772–8778 CrossRef PubMed; (g) Y. W. Zhong, Y. Z. Dong, K. Fang, K. Izumi, M. H. Xu and G. Q. Lin, J. Am. Chem. Soc., 2005, 127, 11956–11957 CrossRef CAS PubMed; (h) A. C. Cutter, I. R. Miller, J. F. Keily, R. K. Bellingham, M. E. Light and R. C. D. Brown, Org. Lett., 2011, 13, 3988–3991 CrossRef CAS PubMed; (i) X.-N. Wang, R. P. Hsung, R. Qi, S. K. Fox and M.-C. Lv, Org. Lett., 2013, 13, 2514–2517 CrossRef PubMed; (j) T. J. Senter, M. L. Schulte, L. C. Konkol, T. E. Wadzinski and C. W. Lindsley, Tetrahedron Lett., 2013, 54, 1645–1648 CrossRef CAS PubMed; (k) Y. Liu, J. Liu, Y. Huang and F.-L. Qing, Chem. Commun., 2013, 49, 7492–7494 RSC; (l) W. J. Morris, K. K. Muppalla, C. Cowden and R. G. Ball, J. Org. Chem., 2013, 78, 706–710 CrossRef CAS PubMed; (m) X.-N. Wang, R. P. Hsung, S. K. Fox, M.-C. Lv and R. Qi, Heterocycles, 2014, 88, 1233–1254 CrossRef CAS PubMed; (n) C. Sanaboina, S. Jana and L. Eppakayala, Synlett, 2014, 1006–1008 CAS; (o) C.-M. Si, W. Huang, Z.-T. Du, B.-G. Wei and G.-Q. Lin, Org. Lett., 2014, 16, 4328–4331 CrossRef CAS PubMed.
- C. W. Jefford, A. W. Sledeski, J. C. Rossier and J. Boukouvalas, Tetrahedron Lett., 1990, 31, 5741–5744 CrossRef CAS.
- For anti-selective VMRs, see: (a) G. Casiraghi, G. Rassu, P. Spanu, L. Pinna and F. Ulgheri, J. Org. Chem., 1993, 58, 3397–3400 CrossRef CAS; (b) L. Battistini, G. Rassu, L. Pinna, F. Zanardi and G. Casiraghi, Tetrahedron: Asymmetry, 1999, 10, 765–773 CrossRef CAS; (c) B. Dudot, J. Royer, M. Sevrin and P. George, Tetrahedron Lett., 2000, 41, 4367–4371 CrossRef CAS; (d) K. E. Harding and J. M. Southard, Tetrahedron: Asymmetry, 2005, 16, 1845–1854 CrossRef CAS; (e) E. L. Carswell, M. L. Snapper and A. H. Hoveyda, Angew. Chem., Int. Ed., 2006, 45, 7230–7233 CrossRef CAS PubMed; (f) A. Yamaguchi, S. Matsunaga and M. Shibasaki, Org. Lett., 2008, 10, 2319–2322 CrossRef CAS PubMed; (g) H. Mandai, K. Mandai, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 17961–17969 CrossRef CAS PubMed; (h) T. Akiyama, Y. Honma, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2008, 350, 399–402 CrossRef CAS; (i) H.-P. Deng, Y. Wei and M. Shi, Adv. Synth. Catal., 2009, 351, 2897–2902 CrossRef CAS; (j) A. Sartori, L. Dell'Amico, C. Curti, L. Battistini, G. Pelosi, G. Rassu, G. Casiraghi and F. Zanardi, Adv. Synth. Catal., 2011, 353, 3278–3284 CrossRef CAS; (k) B. Ranieri, C. Curti, L. Battistini, A. Sartori, L. Pinna, G. Casiraghi and F. Zanardi, J. Org. Chem., 2011, 76, 10291–10298 CrossRef CAS PubMed; (l) M. Hayashi, M. Sano, Y. Funahashi and S. Nakamura, Angew. Chem., Int. Ed., 2013, 52, 5557–5560 CrossRef CAS PubMed; (m) L.-S. Zheng, L. Li, K.-F. Yang, Z.-J. Zheng, X.-Q. Xiao and L.-W. Xu, Tetrahedron, 2013, 69, 8777–8784 CrossRef CAS; (n) A. Sartori, L. Dell'Amico, L. Battistini, C. Curti, S. Rivara, D. Pala, P. S. Kerry, G. Pelosi, G. Casiraghi, G. Rassu and F. Zanardi, Org. Biomol. Chem., 2014, 12, 1561–1569 RSC; (o) S. Janody, P. Hermange, P. Retailleau, J. Thierry and R. H. Dodd, J. Org. Chem., 2014, 79, 5673–5683 CrossRef CAS PubMed. See also: (p) G. Landelle, A. Claraz, S. Oudeyer and V. Levacher, Tetrahedron Lett., 2012, 53, 2414–2416 CrossRef CAS; (q) Q.-Y. Zhao, Z.-L. Yuan and M. Shi, Adv. Synth. Catal., 2011, 353, 637–643 CrossRef CAS; (r) Q.-Y. Zhao and M. Shi, Tetrahedron, 2011, 67, 3724–3732 CrossRef CAS.
- For reviews, see: (a) P. Zhou, B. C. Chen and F. A. Davis, Tetrahedron, 2004, 60, 8003–8030 CrossRef CAS; (b) D. Morton and R. A. Stockman, Tetrahedron, 2006, 62, 8869–8905 CrossRef CAS.
- (a) D. A. DeGoey, H.-J. Chen, W. J. Flosi, D. J. Grampovnik, C. M. Yeung, L. L. Klein and D. J. Kempf, J. Org. Chem., 2002, 67, 5445–5453 CrossRef CAS PubMed; (b) D. M. Barnes, M. A. McLaughlin, T. Oie, M. W. Rasmussen, K. D. Stewart and S. J. Wittenberger, Org. Lett., 2002, 4, 1427–1430 CrossRef CAS PubMed; (c) D. M. Barnes, L. Bhagavatula, J. DeMattei, A. Gupta, D. R. Hill, S. Manna, M. A. McLaughlin, P. Nichols, R. Premchandran, M. W. Rasmussen, Z. Tian and S. J. Wittenberger, Tetrahedron: Asymmetry, 2003, 14, 3541–3551 CrossRef CAS; (d) V. U. B. Rao, A. P. Jadhav, D. Garad and R. P. Singh, Org. Lett., 2014, 16, 648–651 CrossRef CAS PubMed.
- L.-J. Liu and J.-T. Liu, Tetrahedron, 2014, 70, 1236–1245 CrossRef CAS.
- (a) S. F. Martin, K. J. Barr, D. W. Smith and S. K. Bur, J. Am. Chem. Soc., 1999, 121, 6990–6997 CrossRef CAS; (b) S. F. Martin and O. D. Lopez, Tetrahedron Lett., 1999, 40, 8949–8953 CrossRef CAS.
- E. Leemans, S. Mangelinckx and N. De Kimpe, Chem. Commun., 2010, 46, 3122–3124 RSC.
- 1H and 13C NMR data of the major diastereomer were in accord with those of pandamarilactonine-A (1).2a The minor diastereomer of the 13% fraction was tentatively assigned to pandamarilactonine-C (3) according to the characteristic 1H NMR peaks at δ 7.31, 7.03, 5.63, which were identical to those reported for H-4, H-16 and H-6 of pandamarilactonine-C (3).2d The minor diastereomer of the 10% fraction was tentatively assigned to pandamarilactonine-B (2) according to the characteristic 1H NMR peaks at δ 7.05 and 4.71, which were identical to those reported for H-16 and H-15 of pandamarilactonine-B (2).2a.
- The retention times (tR) 43.2 and 51.9 min of natural (+)-pandamarilactonine-A (1) with a flow rate 0.3 mL min−1 correspond to 16.2 and 19.5 min with a flow rate 0.8 mL min−1. So the peaks at 16.7 and 20.0 min were assigned to the (−)-pandamarilactonine-A [(−)-1], and the enantiomeric excess of our synthetic (−)-pandamarilactonine-A
[(−)-1] was determined as 95.5%. The HPLC analysis results (2:87:11) matched to the integral ratio (87:13) afforded in 1H NMR of the mixture of (−)-pandamarilactonines-A [(−)-1] and –C (3). Hence the peak at 26.6 min was tentatively assigned to the pandamarilactonine-C (3).
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of compounds 10a, 13, anti-16, syn-16, syn-17, and the mixture of (−)-1 and 3. 1H NMR spectra on investigation of epimerization. The X-ray structure of syn-17. Chiral HPLC chromatograms of the mixture of (−)-1 and 3. Computational details for compounds 5, 6, anti-20, and syn-20. CCDC 1046182. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00098j |
| This journal is © the Partner Organisations 2015 |