
A mechanistic study on the SHi reaction at tin atoms in a radical cascade reaction†
Akio
Kamimura
*a,
Tatsuro
Yoshinaga
b,
Fumiaki
Noguchi
a,
Koichiro
Miyazaki
a and
Hidemitsu
Uno
c
aDepartment of Applied Molecular Bioscience, Graduate School of Medicine, Yamaguchi University, Ube 755-8611, Japan. E-mail: ak10@yamaguchi-u.ac.jp; Fax: +81 836 85 9231; Tel: +81 836 85 9231
bDepartment of Applied Chemistry, Faculty of Engineering, Yamaguchi University, Ube 755-8611, Japan
cDepartment of Chemistry, Graduate School of Science and Engineering, Ehime University, Matsuyama 790-8577, Japan
First published on 13th April 2015
Abstract
A kinetic study on radical cascade reactions of 1,6-enyne compounds was undertaken. The efficiency of the reaction depended on the presence of an ester group at the alkene unit, which clearly suggests that the addition of a tin radical was accelerated by the α,β-unsaturated system. Stannolane formation progressed very quickly after a short induction period and completed within 12 min in the presence of Bu3SnH at 10−2 M concentration at 110 °C. Product ratios between stannolane and exo-methylene compound depended on the concentration of Bu3SnH; a linear relationship was observed between the ratios of the two and the concentration of Bu3SnH. These results clearly indicate that the SHi process is irreversible. The slope of the graph allowed us to estimate the ks value, the SHi reaction rate at the tin atom, as 4.23 × 108 s−1 at 303 K. The use of Bu2PhSnH for the reaction suggests that the SHi process partially progresses by the frontside attack of the vinyl radical, which generates a methylene radical that undergoes neophyl rearrangement to give methylene piperidine in a cis-selective manner.
Introduction
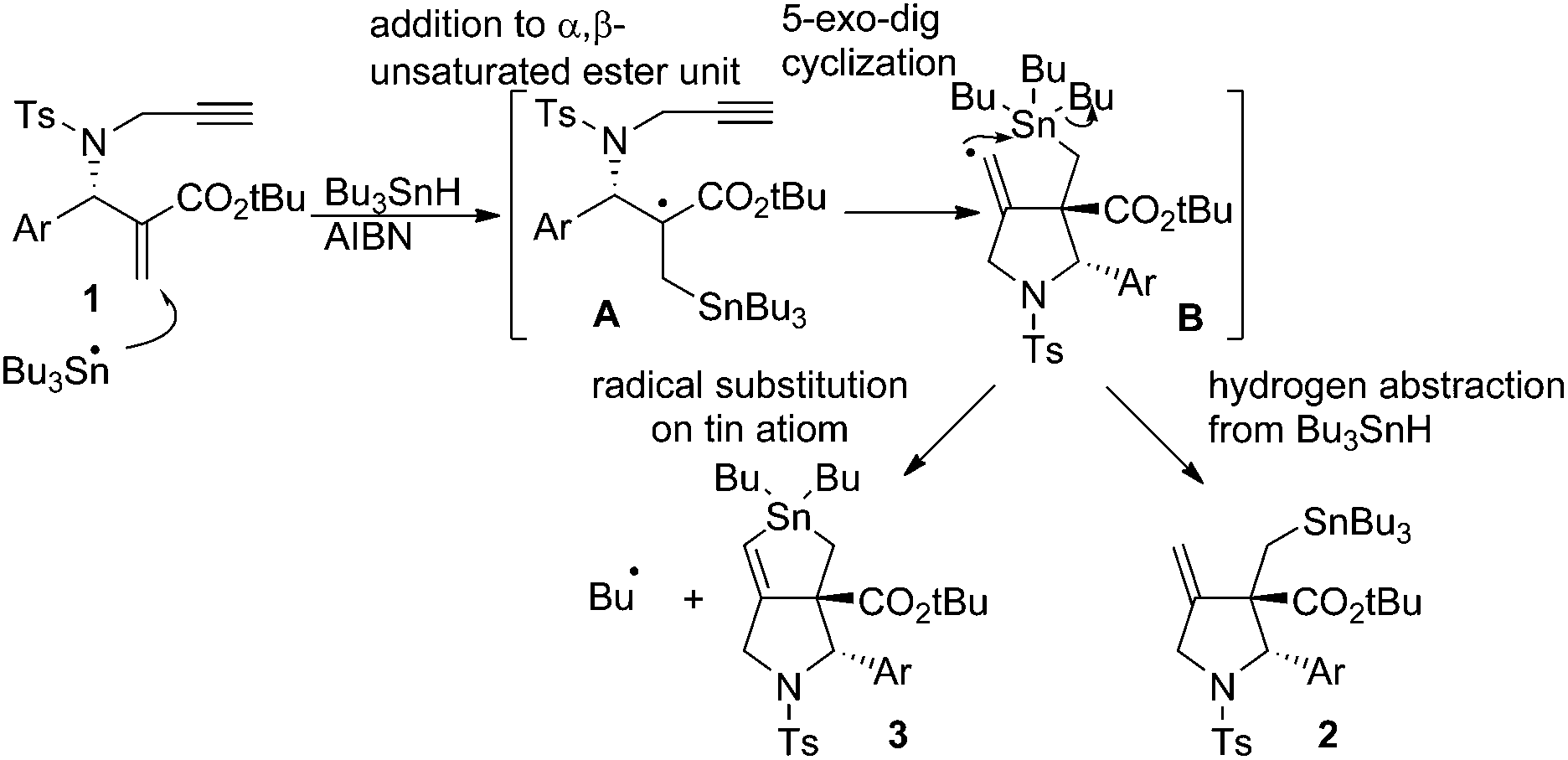
Radical reactions are regarded as a useful method in organic synthesis.1 A carbon-centered radical usually undergoes radical cyclisation,2 rearrangement,3 and fragmentation4 reactions that give new carbon frameworks. The intramolecular substitution reaction of radicals, SHi reaction, occurs on heteroatoms such as sulfur, silicon and tin, which release a heteroatom-centered radical as a leaving group in the substitution reaction.5 For example, Studer reported that the acyl radical attacks tin atoms in an SHi manner and generates a silyl radical as a leaving radical.6 Through ab initio and DFT calculations Schiesser reported that SHi and SH2 processes can take place at tin, germanium and silicon atoms resulting in the formation of alkyl radicals.7 Although an alkyl radical serves as a leaving group in the SHi reaction on a sulfur atom,8 a similar reaction on a tin atom is rather rare.5e–g Recently, we found that 1,6-enyne compounds undergo a new type of radical cascade reaction, in which tin radicals progressed addition, cyclization and SHi substitution reaction on the tin atom in one step to give bicyclic stannolane in high yields.9 The reaction progressed in a stereoselective manner, forming only one diastereomer. Note that an alkyl radical is released as a leaving group from a trialkyltin atom in a highly efficient manner. The products, stannolanes, serve as a good precursor for the double Stille coupling reaction, giving benzodihydroisoindoles and pyrrolidinoindanes in short steps.10We assume that the stannolane formation reaction progresses through the following mechanism (Scheme 1). The reaction starts by the addition reaction of a tin radical to the α,β-unsaturated ester part of compound 1 to generate α-carbonyl radical A. Then the α-carbonyl radical A attacks the terminal alkyne unit in a 5-dig-exo manner to generate the vinyl radical B. Vinyl radicals are usually regarded as highly reactive radicals. Therefore the vinylic radical in intermediate B attacks the tin atom in an intramolecular manner to give stannolane 3 in good yields. However, when the reaction is performed without a solvent, i.e., with a very high concentration of Bu3SnH, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of compound 3 and exo-methylene pyrrolidine 2 is obtained. These results clearly support the reaction mechanism shown in Scheme 1. The rate determining step of the cascade reaction should be the addition process (i.e., 1 to A) or substitution process (i.e., B to 3). The cyclization step must be faster than these two processes, because we could not have detected any products of the simple addition of Bu3SnH to the α,β-unsaturated ester unit of 1.
:1 mixture of compound 3 and exo-methylene pyrrolidine 2 is obtained. These results clearly support the reaction mechanism shown in Scheme 1. The rate determining step of the cascade reaction should be the addition process (i.e., 1 to A) or substitution process (i.e., B to 3). The cyclization step must be faster than these two processes, because we could not have detected any products of the simple addition of Bu3SnH to the α,β-unsaturated ester unit of 1.
 | ||
| Scheme 1 | ||
During our investigation of this chemistry, we were interested in the rate constant of the radical substitution reaction from intermediate B to compound 3. Although some kinetic studies on the SHi reaction have been reported, to the best of our knowledge, these reports are related only to heteroatom-centered radicals as leaving groups, and there seems to be no mechanistic study on SHi processes using an alkyl radical as a leaving group. In this paper we report the kinetic estimation of the radical substitution reaction on tin atoms. We also found that the presence of an ester group in the α,β-unsaturated system is crucial for an efficient progress of the cascade reaction. The use of PhBu2SnH provides another reaction pathway and clearly suggests that the SHi process partially occurs via a front-side attack of the tin radical to give a methylene radical. This radical undergoes neophyl rearrangement, giving piperidine derivatives as a minor product.
Results and discussion
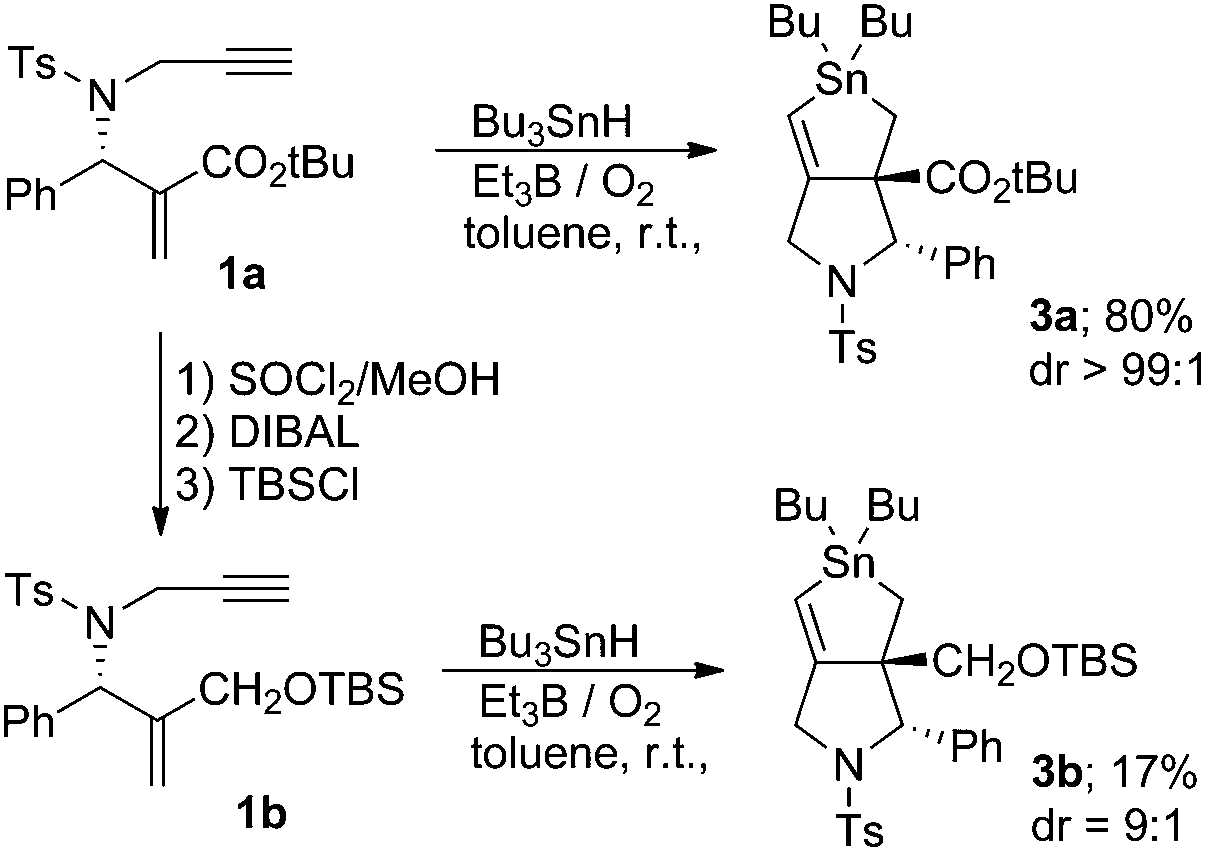
Compound 1a was prepared using a previously reported method.11 We checked the role of the ester group in the cascade reaction. We examined the reaction of compound 1b, which was prepared by the reduction of ester 1a, under the same conditions. Treatment of 1b with Bu3SnH at a concentration of 10−2 M in toluene at room temperature resulted in a sluggish reaction; all of the starting material was consumed, and compound 3b was formed in only 17% as an approximately 9:1 mixture of two diastereoisomers (Scheme 2).
 | ||
| Scheme 2 | ||
The structure of compound 3b was confirmed by chemical conversion from compound 3a. This is in sharp contrast to the reaction of 1a, in which a single isomer of stannolane 3a was produced in 80% yield in a highly selective manner.10 These results suggest that the α,β-unsaturated ester unit in 1 is important in promoting the radical addition, the first step of the cascade process. Thus, the reaction rate of the addition of the Bu3Sn radical becomes slow in the absence of the ester group.
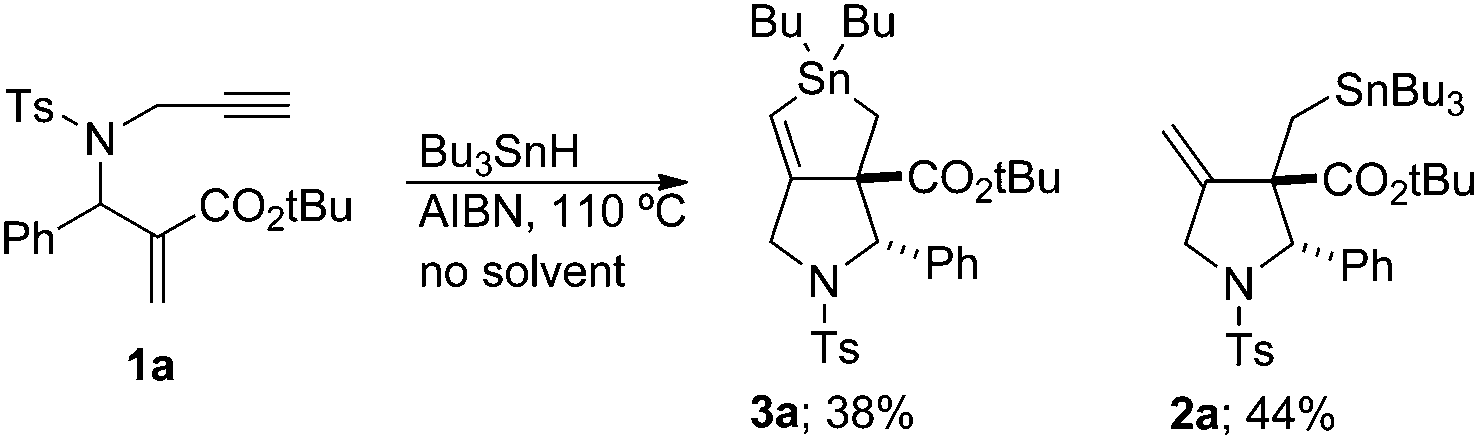
We next examined the reaction kinetics based on the product ratio. Before performing the investigation, we prepared compound 2a under solvent-free conditions (Scheme 3). Compound 2a was obtained in 44% yield along with 3a in 38% yield. Thus, the direct radical substitution at the tin atom by the vinyl radical became competitive with hydrogen abstraction from external Bu3SnH when its concentration was very high.
 | ||
| Scheme 3 | ||
The time course of the reaction between 1a and Bu3SnH (10−2 M) at 110 °C was examined (Fig. 1). The reaction started after a short induction period (approximately 4 min). The yields of the products increased in proportion to the reaction time and the reaction was complete in 12 min.
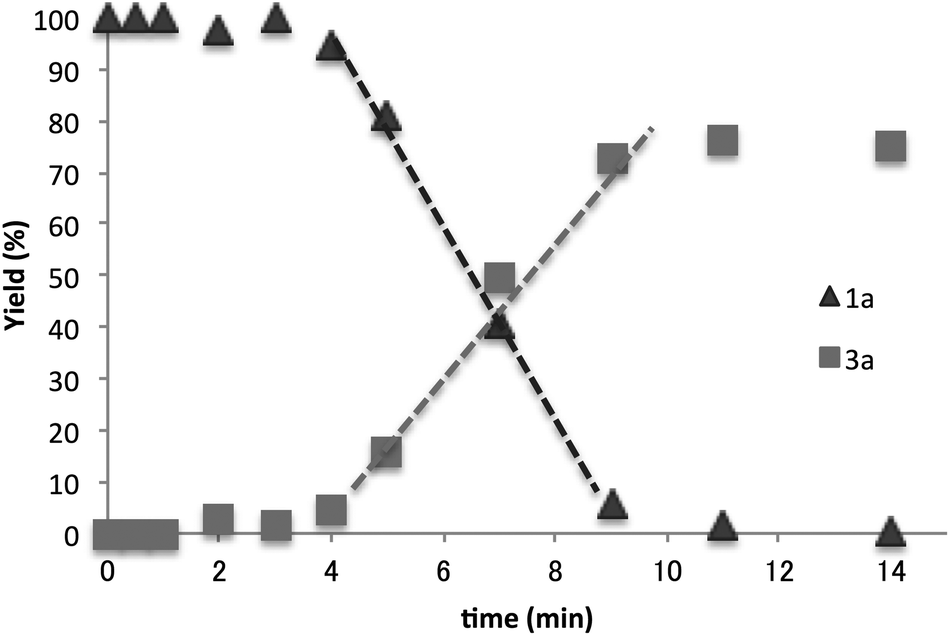 | ||
| Fig. 1 Time course of the reaction of 1a at 110 °C. | ||
To obtain the kinetic parameter of the SHi process in the cascade reaction, we examined the reaction of 1 in the presence of various concentrations of Bu3SnH (Scheme 4). We performed the reaction at 30 °C using approximately 5 equiv. of Bu3SnH. V70 was employed as a radical initiator. The yields of compound 2a and 3a after 30 min were estimated by NMR proton integration using anisole as an internal standard. The yields of 2a and 3a are summarized in Table 1.
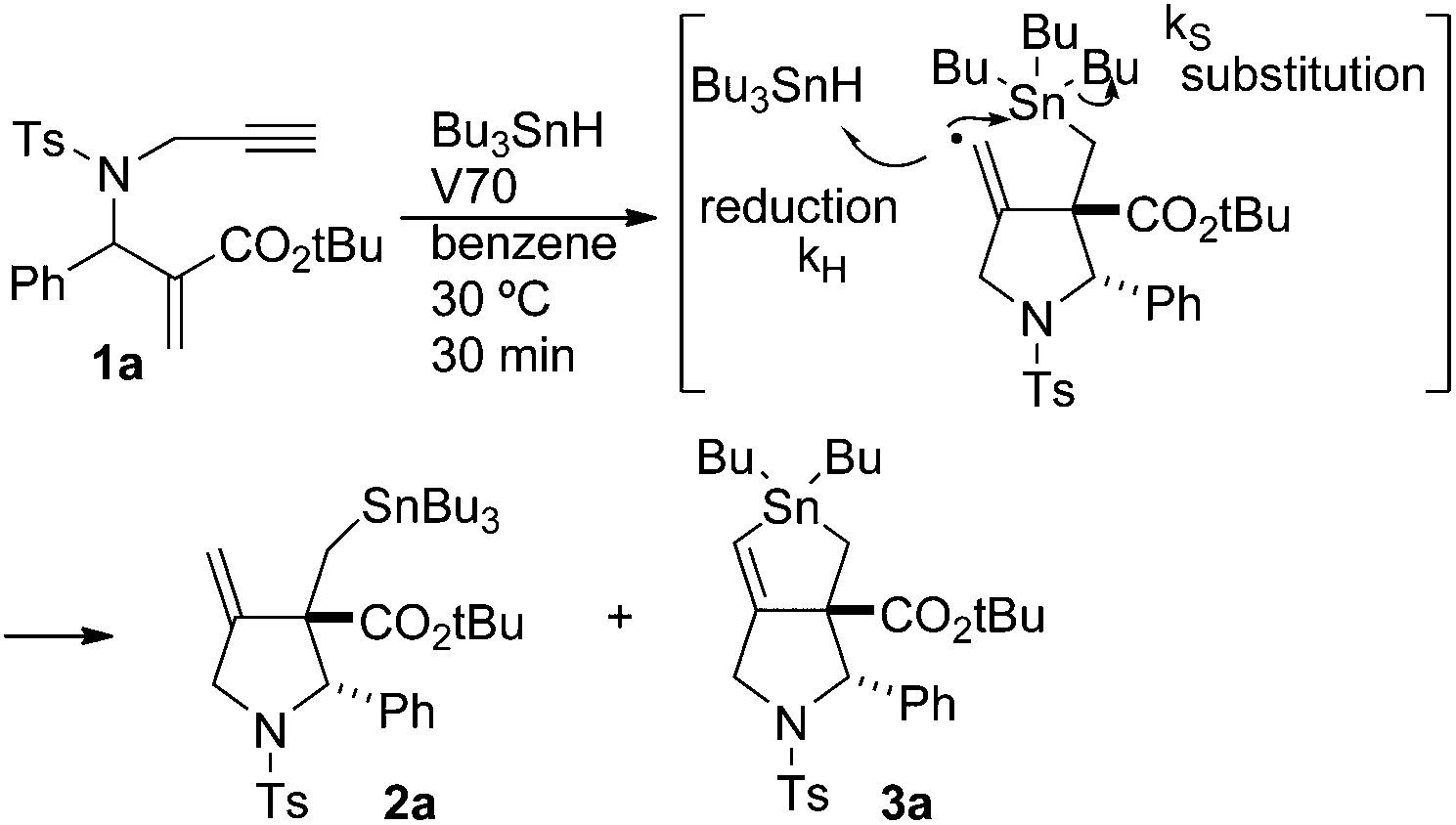 | ||
| Scheme 4 | ||
The product ratio of compound 2a/3a depended on the concentration of Bu3SnH. As the Bu3SnH concentration increased, the yield of compound 2a increased. This contrasts with the reaction performed with a 10−2 M concentration of Bu3SnH, in which the formation of compound 2a was almost non-detectable, i.e., the yield of 2a was almost zero under such conditions. Fig. 2 shows the relationship between the ratios of compound 2a/3a and the concentration of Bu3SnH.
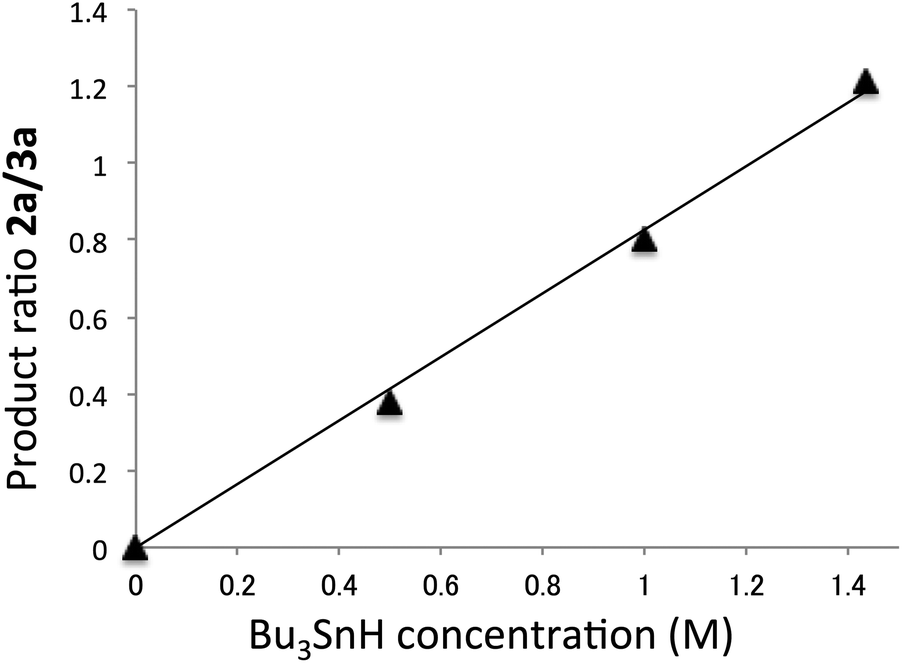 | ||
| Fig. 2 Relationship between the ratio of compound 2a/3a and Bu3SnH concentration for the reaction at 30 °C. | ||
The plot shows a strong linear relationship between the ratios and the concentration of Bu3SnH. The line passed through the origin, indicating that the radical substitution reaction is irreversible.12 The slope was calculated to be 0.828, where the correlation coefficient, R2, was 0.994. The absolute rate constant of hydrogen abstraction from Bu3SnH by the vinyl radical at 30 °C was estimated by Ingold and co-workers to be kH = 3.5 × 108 M−1 s−1.13 This value allows us to estimate the rate constant of the direct substitution reaction at the tin atom, ks, at 30 °C as follows:
| ks = kH/slope = 3.5 × 108/0.828 = 4.23 × 108 s−1 |
These results suggest that the SHi process in this cascade reaction is relatively fast. This is probably due to the configuration of the vinyl radical intermediate B, which places the vinylic radical center close to the position of the tin atom that undergoes the SHi reaction smoothly. Thus, we estimate the rate constant for the tin atom SHi reaction that forms bicyclic stannolane to release an alkyl radical.
To further explore the reaction mechanism, we employed differently substituted tin hydride. Phenyldibutyltin hydride 4a was prepared from dibutyltin dichloride by treatment with a Grignard reagent, and subsequent disproportionation reaction and reduction (Scheme 5).14 Dibutyltin dichloride underwent diphenylation by exposure to 2.5 equiv. of phenyl Grignard reagent to give dibutyldiphenyltin 4b in 82% yield. Compound 4b was converted to dibutylphenyltin chloride 4c in 58% yield by heating a 1:1 mixture of 4b and dibutyltin dichloride. Reduction of 4c was performed by treatment with NaBH4 and the desired dibutylphenyltin hydride 4a was obtained in 39% yield.
 | ||
| Scheme 5 | ||
The reaction of 1c, prepared from the corresponding tert-butyl ester,11 and 4a was examined under the same conditions as the radical cascade process. Thus, a mixture of 1c and 4a in toluene at a concentration of 10−2 M was stirred at room temperature. Compound 1c was smoothly consumed after 30 min. Then a typical workup provided compound 3c in 62% yield. Note that we could not observe any trace of 3d in the reaction mixture. This is reasonable because the phenyl radical is highly reactive and should not be generated in this substitution. Compound 3c contained two diastereomers whose ratio was approximately 1:1. Thus, no stereoselectivity on the radical substitution reaction at the tin atom was observed. The diastereomers were separated by careful chromatography, and one of the isomers, 3c-A, afforded a good crystal for which X-ray crystallographic analysis unambiguously showed the expected structure.15 Comparison of 1H NMR spectra revealed that the other isomer, 3c-B, showed a downfield shift at the CO2Me signal, indicating that the difference in structure between 3c-A and 3c-B was the difference in the configuration at the tin atom (Scheme 6).
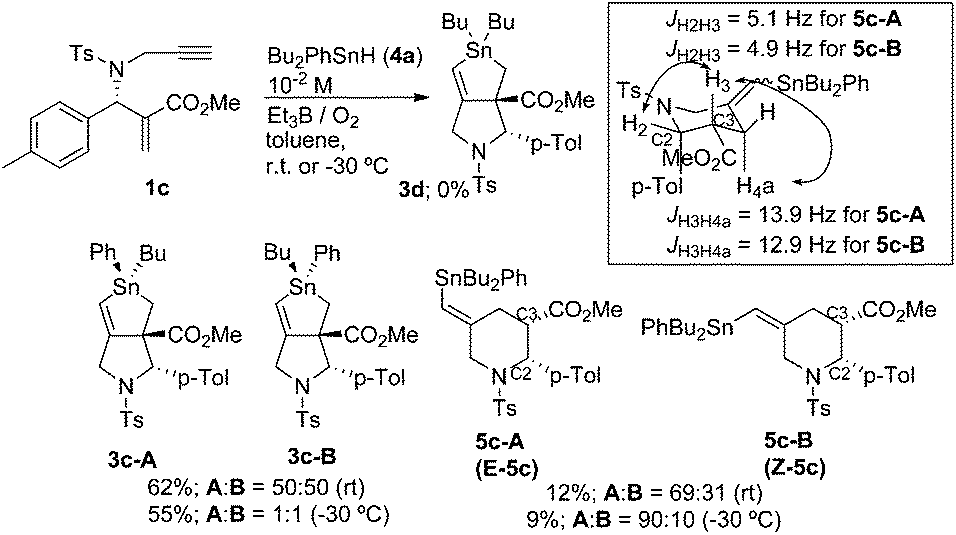 | ||
| Scheme 6 | ||
We attempted to identify minor products of this reaction. We isolated a minor product 5c in 12% yield. Compound 5c contained the two diastereomers, 5c-A and 5c-B, and their ratio was 69:31. Careful chromatographic separation of 5c-A and 5c-B allowed us to determine their structures. NMR spectra of these compounds revealed a methylenepiperidine structure. Interestingly, the difference in stereochemistry between these two isomers was the geometry of the alkene unit, and both isomers contained cis-configuration between C2 and C3. This was clearly supported by the observation of large coupling constants (12.9 and 13.9 Hz) between H4a and H3. Note that the H2 proton shows a relatively small coupling constant in cis- and trans-2,3-substituted-N-tosyl-2-arylpiperidines.16 NOESY spectra of 5c-A and 5c-B showed a cross peak between H6 and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHSnPhBu2 in 5c-A, and a cross peak between H3 and CHSnPhBu2 in 5c-B. Thus, we concluded that 5c-A had an E-configuration and 5c-B had a Z-configuration, and the E/Z ratio of 5c was approximately 2:1. This E/Z ratio was improved to approximately 10:1 when the reaction was performed at −30 °C.
CHSnPhBu2 in 5c-A, and a cross peak between H3 and CHSnPhBu2 in 5c-B. Thus, we concluded that 5c-A had an E-configuration and 5c-B had a Z-configuration, and the E/Z ratio of 5c was approximately 2:1. This E/Z ratio was improved to approximately 10:1 when the reaction was performed at −30 °C.
We next examined the radical cascade reaction with a high concentration of Bu2PhSnH (Scheme 7). Treatment of compound 1c with Bu2PhSnH under solvent-free conditions at 30 °C resulted in the quick consumption of 1c. The reaction was initiated by catalytic amounts of V70. We observed two products in the reaction mixture: one was stannolane 3c in 10% yield, and the other was exo-methylene pyrrolidine 2c, which was unambiguously determined by NMR analysis. Compound 2c was the major product of the reaction and its yield reached 57%. Note that neither 3d nor 5c was observed in the reaction mixture under these conditions.
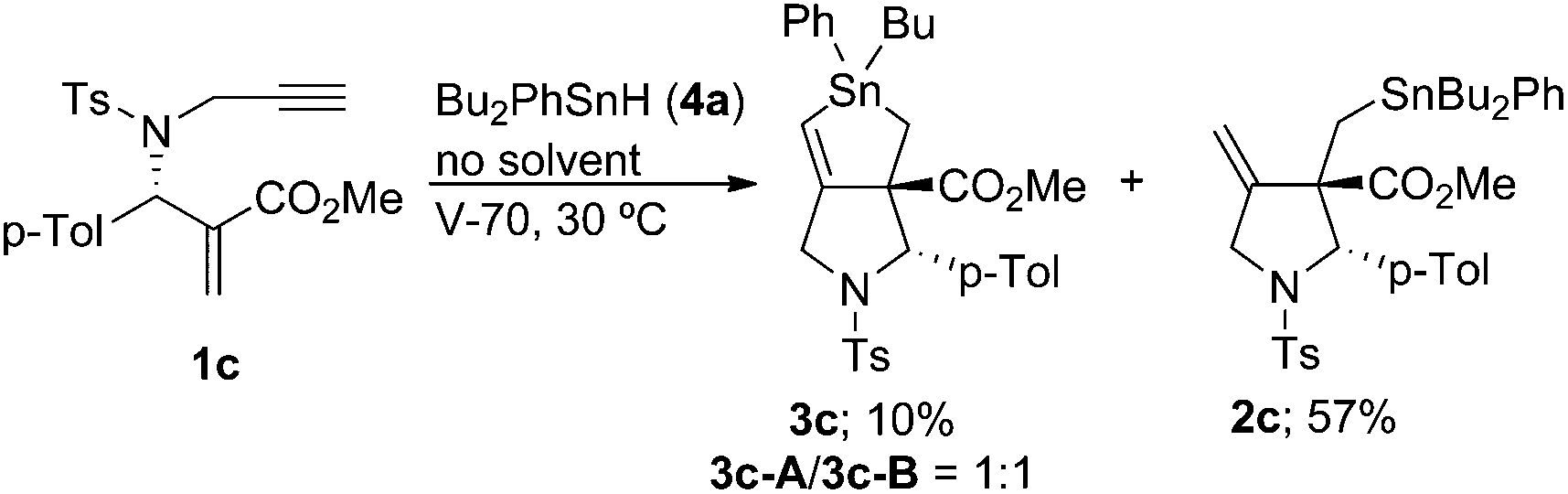 | ||
| Scheme 7 | ||
With the results of our experiments using Bu2PhSnH, we assumed the reaction pathway shown in Scheme 8. Thus, the PhBu2Sn radical attacks the terminal carbon–carbon double bond to give radical A. This is supported by the fact that the presence of the ester group attracts the addition of the tin radical to the alkene terminal. Then, cyclization yields vinyl radical B, which mainly releases the butyl radical through path a as a major process to give 3c. However, in this case there is a phenyl substituent present on the tin atom that does not serve as a leaving group during the substitution reaction. Alternatively, methylene radical C was released, which undergoes neophyl rearrangement through cyclopropyl radical D, to give 3-piperidino radical E. Due to the steric bias introduced by the phenyl group at the C2 position, cis-selective hydrogen abstraction from E occurs to give cis-5c. Note that radical E should be a mixture of geometrical isomers of the vinyltin unit because a rotation around the carbon–carbon single bond is possible in radical D. This rotation is slow and suppressed under low temperature conditions. When the reaction was performed under solvent-free conditions, the intermediate radical B is trapped by PhBu2SnH due to its high concentration. This result also indicates that the radical addition was initiated by the attack of the tin radical to the α,β-unsaturated ester unit, not to the terminal alkyne unit.
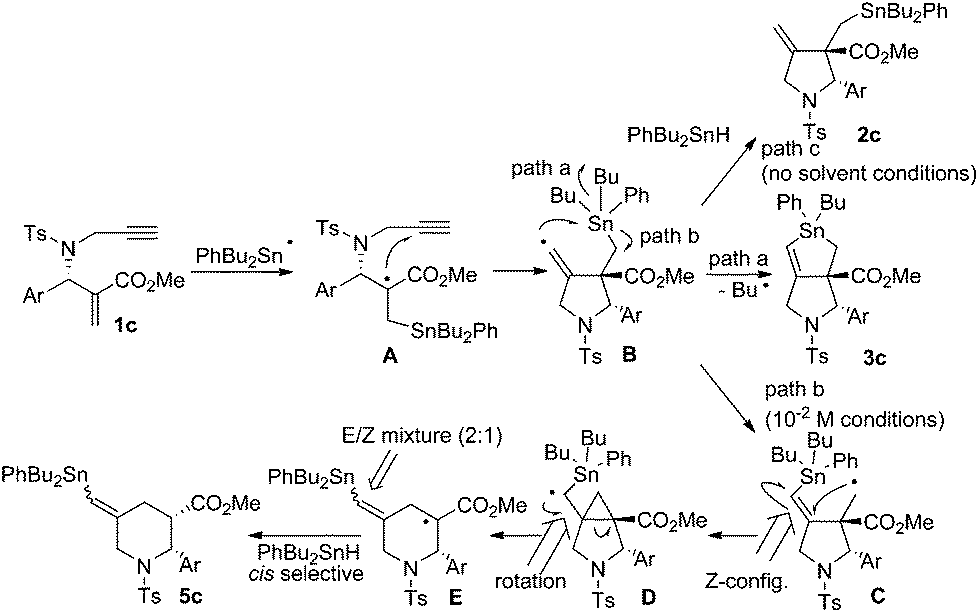 | ||
| Scheme 8 | ||
In conclusion, we have explored the reaction mechanism for the radical cascade reaction, including direct radical substitution on the tin atom. For the cascade reaction to progress efficiently, the presence of an ester group is crucial for promoting the radical addition of the tin radical, the initial process of the cascade. The reaction profile was examined and the cascade reaction is a very fast reaction that is complete within 12 min after an approximately 4 min induction period. The kinetic parameter for the direct radical substitution reaction on the tin atom, the SHi reaction, was estimated. The rate constant was determined to be 4.23 × 108 s−1, which is regarded as a relatively fast process when compared with the hydrogen abstraction reaction for tin hydride by an alkyl radical. The use of dibutylphenyltin hydride revealed a side reaction for the cascade process, which showed no diastereoselectivity on the substitution at the tin atom. Note that there is a possibility of neophyl rearrangement of the reaction intermediate to give small amounts of methylene piperidine 5a. We are currently investigating the reaction mechanism using the MO calculation method and the results will be published in due course.
Experimental
General
All 1H and 13C NMR spectra were recorded on a JEOL Delta-500 or Lambda-500 (500 MHz for 1H and 126 MHz for 13C) spectrometer. High-resolution mass spectra (HRMS) were recorded on a JEOL JMS-T100 LP spectrometer. Compound 1a and (S)-tert-butyl 2-((4-methyl-N-(prop-2-yn-1-yl)phenyl-sulfonamido)(p-tolyl)methyl)acrylate 1d were prepared by the reported methods.9,12 All reactions in this paper were performed under a nitrogen atmosphere unless otherwise mentioned. Other compounds were purchased from Aldrich Co. Ltd and were used without further purification.:1, then 7:1 v/v) to give methyl ester in 88% yield (781.3 mg, 2.04 mmol). White solid, mp 82.5–83.5 °C, [α]D +146.3 (c 1.07, CHCl3), 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 7.25–7.20 (m, 3H), 7.06–7.00 (m, 2H), 6.49 (d, J = 1.3 Hz, 1H), 6.12 (s, 1H), 5.98 (d, J = 1.8 Hz, 1H), 4.09 (dd, J = 18.4, 2.5 Hz, 1H), 3.81 (dd, J = 18.4, 2.5 Hz, 1H), 3.59 (s, 3H), 2.43 (s, 3H), 1.99 (t, J = 2.5 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 166.3, 143.6, 138.9, 137.4, 136.3, 129.5 (2C), 128.7 (2C), 128.7 (2C), 128.3, 127.8 (2C), 79.2, 72.5, 61.9, 52.1, 35.2, 21.7; HRMS (ESI-TOF): calcd for C21H21NNaO4S 406.1079 [M + Na+], found 406.1089.
Methyl ester (715.0 mg, 1.87 mmol) was dissolved in dry toluene (40 mL) and DIBAL-H (1.0 M in toluene, 5.0 mL, 5 mmol) was added to the resulting solution at −78 °C. The reaction mixture was stirred at the same temperature for 2.5 h, and then warmed to 0 °C. The reaction was quenched by adding saturated aqueous solution of Rochelle salt (15 mL). The reaction mixture was then extracted with ether (4 × 20 mL). The organic phase was combined, washed with brine (20 mL) and dried over Na2SO4. After filtration, the filtrate was concentrated in vacuo and the residue was subjected to flash chromatography (silica gel, hexane–EtOAc 10:1, then 2:1 v/v) to give the corresponding alcohol in 57% yield (376.2 mg, 1.06 mmol). Colorless oil, [α]D +55.9 (c 1.03, CHCl3), 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 8.2 Hz, 2H), 7.22–7.18 (m, 5H), 7.04 (dd, J = 6.3, 3.2 Hz, 2H), 5.73 (s, 1H), 5.42 (s, 1H), 5.05 (s, 1H), 4.13 (dd, J = 18.3, 2.5 Hz, 1H), 4.07 (d, J = 13.8 Hz, 1H), 4.02–3.94 (m, 2H), 2.47 (s, 1H), 2.37 (s, 3H), 2.03 (t, J = 2.5 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ146.7, 143.7, 137.2, 136.1, 129.5 (2C), 128.8 (2C), 128.5 (2C), 128.0, 127.7 (2C), 116.8, 79.3, 72.4, 64.5, 62.4, 34.7, 21.6; HRMS (ESI-TOF): calcd for C20H21NNaO3S 378.1140 [M + Na+], found 378.1148.
Alcohol (148.3 mg, 0.42 mmol) was dissolved in dry CH2Cl2 (20 mL), and imidazole (72.1 mg, 1.04 mmol) and TBSCl (83.1 mg, 0.55 mmol) in CH2Cl2 (5 mL) were added to the solution. The reaction mixture was stirred at room temperature for 2 days. Saturated aq. NaHCO3 (10 mL) and the resulting mixture were extracted with CH2Cl2 (3 × 10 mL). The organic phase was combined, washed with brine (20 mL) and dried over Na2SO4. After filtration, the filtrate was concentrated in vacuo and the residue was subjected to flash chromatography (silica gel, hexane–EtOAc 15:1, then 10:1 v/v) to give 1b in 89% yield (174.7 mg, 0.37 mmol). Colorless oil, [α]D +48.5 (c 1.04, CHCl3), 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 8.3 Hz, 2H), 7.30–7.22 (m, 5H), 7.15 (dd, J = 12.6, 3.3 Hz, 2H), 5.74 (s, 1H), 5.44 (t, J = 1.2 Hz, 1H), 5.06 (t, J = 1.3 Hz, 1H), 4.21 (dd, J = 18.3, 2.5 Hz, 1H), 4.01 (dd, J = 18.3, 2.5 Hz, 1H), 3.99–3.97 (m, 2H), 2.43 (s, 3H), 2.02 (t, J = 2.4 Hz, 1H), 0.90 (s, 9H), 0.01 (s, 3H), −0.04 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 146.0, 143.4, 137.6, 136.6, 129.3 (2C), 129.0 (2C), 128.4 (2C), 127.9, 127.8 (2C), 115.4, 79.3, 72.2, 64.7, 62.5, 34.7, 25.9, 21.6 (3C), 18.3, −5.4, −5.4; HRMS (ESI-TOF): calcd for C26H35NNaO3SSi 492.2005 [M + Na+], found 492.2016.
:1, 10:1, then 8:1 v/v) to give methyl ester in 82% yield (307.1 mg, 0.77 mmol).
Pale yellow oil, [α]D +128.2 (c 1.03, CHCl3), 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J = 8.3 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 7.04 (d, J = 8.0 Hz, 2H), 6.91 (d, J = 8.1 Hz, 2H), 6.48 (d, J = 1.1 Hz, 1H), 6.08 (s, 1H), 6.03 (d, J = 1.7 Hz, 1H), 4.09 (dd, J = 18.3, 2.5 Hz, 1H), 3.75 (dd, J = 18.3, 2.5 Hz, 1H), 3.60 (s, 3H), 2.44 (s, 3H), 2.29 (s, 3H), 2.02 (t, J = 2.4 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 166.3, 143.6, 139.0, 138.1, 137.4, 133.1, 129.5, 129.4, 128.6, 128.3, 127.7, 79.4, 72.6, 61.8, 52.1, 35.1, 21.6, 21.2; HRMS (ESI-TOF): calcd for C22H23NNaO3S 420.1239 [M + Na+], found 420.1246.
:1) to give 3b in 17% yield (54.3 mg, 0.07 mmol). Identification of the compounds was confirmed by comparison of spectra of the compound 3b prepared from the corresponding alcohol.
(3S,3aS)-tert-butyl 5,5-dibutyl-3-phenyl-2-tosyl-1,2,3,3a,4,5-hexahydrostannolo[3,4-c]-pyrrole-3a-carboxylate (301.2 mg, 0.51 mmol), which was prepared by the reported method,11 was dissolved in CH2Cl2 (20 mL) and TBSOTf (202.7 mg, 0.77 mmol) and 2,6-lutidine (0.09 mL, 0.77 mmol) was added to the solution. The reaction mixture was stirred for 24 h at room temperature. Water (10 mL) was added and the resulting mixture was extracted with CH2Cl2 (3 × 10 mL). The organic phase was combined, washed with brine (10 mL) and dried over Na2SO4. After filtration, the filtrate was concentrated in vacuo and the residue was subjected to flash chromatography (silica gel, hexane–EtOAc 15:1, then 5:1 v/v) to give 3b in 100% yield (360.3 mg, 0.51 mmol). Colorless oil, [α]D −7.0 (c 1.00, CHCl3), 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 8.3 Hz, 2H), 7.22–7.12 (m, 3H), 7.11 (d, J = 8.0 Hz, 2H), 7.04–6.83 (m, 2H), 6.40 (s, 1H, J119Sn–1H = 109.2 Hz), 5.09 (s, 1H), 4.16 (dd, J = 14.0, 2.2 Hz, 1H), 4.08 (dd, J = 13.9, 1.5 Hz, 1H), 3.22 (dd, J = 9.5, 2.5 Hz, 1H), 2.86 (d, J = 9.5 Hz, 1H), 2.34 (s, 3H), 1.52–1.43 (m, 2H), 1.30–1.06 (m, 8H), 1.04–0.97 (m, 2H), 0.95 (s, 9H), 0.85 (t, J = 7.3 Hz, 3H), 0.81–0.84 (m, 1H), 0.75 (t, J = 7.2 Hz, 3H), 0.03 (s, 3H), 0.02 (s, 3H), −0.02 (dd, J = 13.0, 2.5 Hz, 1H, J119Sn–1H = 57.4 Hz); 13C NMR (126 MHz, CDCl3) δ 160.7 (J119Sn–13C = 61.0 Hz), 142.7, 141.4, 136.2, 129.3 (2C), 128.1 (br, 3C), 127.2 (2C), 127.0 (2C), 126.1 (J117Sn–13C = 328.9 Hz, J119Sn–13C = 343.8 Hz), 68.6, 67.6 (J119Sn–13C = 38.2 Hz), 65.9 (J119Sn–13C = 17.7 Hz), 50.6 (J119Sn–13C = 61.8 Hz), 29.3 (J119Sn–13C = 21.9 Hz), 28.8 (J119Sn–13C = 22.5 Hz), 27.2 (J119Sn–13C = 55.8 Hz), 27.0 (J119Sn–13C = 55.4 Hz), 26.1 (3C), 21.6, 18.4 (J119Sn–13C = 65.6 Hz), 13.7 (2C), 13.4 (J117Sn–13C = 315.3 Hz, J119Sn–13C = 331.4 Hz), 12.1, 11.9 (J117Sn–13C = 325.4 Hz, J119Sn–13C = 339.3 Hz), −5.2, −5.3; HRMS (ESI-TOF): calcd for C34H53NNaO3SSi120Sn 726.2435 [M + Na+], found 726.2439.
:1 v/v) to give 2a in 44% yield (633.2 mg, 0.88 mmol) and 3a in 38% yield (497.4 mg, 0.75 mmol).
Compound 2a: Colorless oil, 1H NMR (500 MHz, CDCl3) δ 7.20–7.07 (m, 5H), 6.98–6.90 (m, 4H), 5.22 (s, 1H), 5.19 (t, J = 1.8 Hz, 1H), 5.12 (dd, J = 2.6, 1.5 Hz, 1H), 4.42 (dt, J = 12.9, 2.4 Hz, 1H), 3.96 (dt, J = 13.1, 1.5 Hz, 1H), 2.29 (s, 3H), 1.40–1.13 (m, 12H), 0.98 (d, J = 12.8 Hz, 1H), 0.82 (t, J = 7.1 Hz, 9H), 0.75–0.59 (m, 6H), 0.35 (d, J = 12.8 Hz, 1H, J119Sn–1H = 44.2 Hz); 13C NMR (126 MHz, CDCl3) δ 173.1, 149.8, 142.4, 138.5, 136.8, 129.0 (2C), 128.6 (br, 2C), 128.2 (2C), 127.8, 126.9 (2C), 109.4, 82.2, 70.4, 61.4, 52.1, 29.1 (3C), 27.9 (3C), 27.5 (3C), 21.5, 13.9, 13.8 (3C), 11.0 (3C, J119Sn–13C = 330.1 Hz); HRMS (ESI-TOF): calcd for C36H55NNaO4S120Sn 740.2772 [M + Na+], found 740.2761.
Compound 3a: Colorless oil, 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 7.7 Hz, 2H), 7.14 (d, J = 7.7 Hz, 3H), 7.07 (d, J = 7.9 Hz, 2H), 6.94 (s, 2H), 6.56 (s, 1H, J119Sn–1H = 115.3 Hz), 5.44 (s, 1H), 4.15 (d, J = 12.9 Hz, 1H), 4.01 (d, J = 12.9 Hz, 1H), 2.32 (s, 3H), 1.53–1.43 (m, 2H), 1.37 (s, 9H), 1.29–1.18 (m, 4H), 1.29–1.18 (m, 6H), 0.90 (d, J = 13.4 Hz, 1H), 0.84 (t, J = 7.4 Hz, 3H), 0.73 (t, J = 6.8 Hz, 3H), 0.22 (d, J = 13.2 Hz, 1H, J119Sn–1H = 53.4 Hz); 13C NMR (126 MHz, CDCl3) δ 173.8, 158.3, 142.6, 139.6, 136.5, 129.1 (2C), 128.1 (br, 3C), 128.0 (2C), 127.5, 127.3 (2C), 81.6, 69.7, 68.6, 50.6, 29.0, 28.7, 27.8 (4C), 27.1, 27.0, 21.5, 13.7, 13.7, 13.1, 12.9, 12.3; HRMS (ESI-TOF): calcd for C32H45NNaO4S120Sn 682.1989 [M + Na+], found 682.1996.
General procedure for kinetic estimation: typical procedure
Compound 1a (425.1 mg, 1 mmol) and Bu3SnH (1.4552 g, 5.0 mmol) were dissolved in benzene using a measuring flask (10 mL) to prepare a reaction solution at a 0.5 M concentration of Bu3SnH. A 2 mL of the solution was transferred to a 10 mL of flask using a measuring pipette, and V70 (6.3 mg, 0.02 mmol) was added to the mixture. The reaction mixture was heated at 30 °C for 1 h. After the reaction was complete, anisole (7.2 mg, 0.067 mmol) was added and compounds 2a and 3a were quantified using integrations of 0.54 ppm (1H for 3a), 5.12 ppm (1H for 2a), and 3.74 ppm (3H for anisole) in 1H NMR spectra.:2 to 10:1 v/v) to give 5c in 12% yield (16.7 mg, 0.024 mmol) and 3c in 68% yield (89.2 mg, 0.137 mmol). Diastereomeric ratios for 3c and 5c were determined to be 50:50 and 69:31 by HPLC analyses, respectively. The diastereomeric mixture of these compounds was separated by further careful flash chromatography.
:1 to 5:1 v/v) to give 3c in 10% yield (12.0 mg, 0.018 mmol) and 2c in 57% yield (76.8 mg, 0.108 mmol). Diastereomeric ratio for 3c was determined to be 50:50 by HPLC analyses.
Notes and references
- Recent reviews, see: (a) J. Zimmerman, A. Halloway and M. P. Sibi, in Handbook of Cyclization Reactions, ed. S. Ma, Wiley-VCH, Weinheim, 2010, vol 2, p. 1099 Search PubMed; (b) E. Yoshioka, S. Kohtani and H. Miyabe, Heterocycles, 2009, 79, 229–242 CrossRef CAS; (c) G. J. Rowlands, Tetrahedron, 2010, 66, 1593 CrossRef CAS PubMed; (d) H. Ishibashi, Chem. Rec., 2006, 6, 23–31 CrossRef CAS PubMed; (e) K. C. Majumdar, P. K. Basuy and S. K. Chattopadhyay, Tetrahedron, 2007, 63, 793 CrossRef CAS PubMed; (f) A. F. Barrero, J. F. Quílez del Moral, E. M. Sánchez and J. F. Arteaga, Eur. J. Org. Chem., 2006, 1627 CrossRef CAS PubMed; (g) K. C. Majumdar, P. K. Basu and P. P. Mukhopadhyay, Tetrahedron, 2005, 61, 10603 CrossRef CAS PubMed; (h) K. C. Majumdar, P. P. Mukhopadhyay and P. K. Basu, Heterocycles, 2004, 63, 1903 CrossRef CAS PubMed; (i) K. C. Majumdar, P. K. Basu and P. P. Mukhopadhyay, Tetrahedron, 2004, 60, 6239 CrossRef CAS PubMed; (j) G. J. Rowlands, Tetrahedron, 2009, 65, 8603 CrossRef CAS PubMed; (k) G. J. Rowlands, Tetrahedron, 2010, 66, 1593 CrossRef CAS PubMed; (l) S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563 CrossRef CAS PubMed; (m) B. Sautier and D. J. Procter, Chimia, 2012, 66, 399 CrossRef CAS PubMed.
- Recent reviews, see: (a) T. Taniguchi and H. Ishibashi, Heterocycles, 2013, 87, 527 CrossRef CAS PubMed; (b) K. C. Majumdar, G. V. Karunakar and B. Sinha, Synthesis, 2012, 2475 CrossRef CAS PubMed.
- Recent reviews, see: (a) D. M. Hodgson and L. H. Winning, Org. Biomol. Chem., 2007, 5, 3071 RSC; (b) G. Bucher, Angew. Chem., Int. Ed., 2010, 49, 6934 CrossRef CAS PubMed.
- Recent examples, see: (a) R. N. Ram and R. K. Tittal, Tetrahedron Lett., 2014, 55, 4342 CrossRef CAS PubMed; (b) Y. L. Tnay and S. Chiba, Chem. – Asia. J., 2015, 10, 873 CrossRef CAS PubMed; (c) L. Debien and S. Z. Zard, J. Am. Chem. Soc., 2013, 135, 3808 CrossRef CAS PubMed; (d) J. McNulty and J. Calzavara, RSC Adv., 2013, 3, 6771 RSC; (e) M.-G. Braun, B. Quiclet-Sire and S. Z. Zard, J. Am. Chem. Soc., 2011, 133, 15954 CrossRef CAS PubMed; (f) R. Heng and S. Z. Zard, Chem. Commun., 2011, 47, 3296 RSC.
- (a) K. Miura, K. Oshima and K. Utimoto, Chem. Lett., 1992, 21, 2477 CrossRef; (b) K. Miura, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1993, 66, 2348 CrossRef CAS; (c) J. Klaus, K. J. Kulicke, C. Chatgilialoglu, B. Kopping and B. Giese, Helv. Chim. Acta, 1992, 75, 935 CrossRef PubMed; (d) G. Rouquet, F. Robert, R. Méreau, F. Castet, P. Renaud and Y. Landais, Chem. – Eur. J., 2012, 18, 940 CrossRef CAS PubMed; (e) D. S. Hays and G. C. Fu, J. Am. Chem. Soc., 1995, 117, 7283 CrossRef CAS; (f) D. S. Hays and G. C. Fu, J. Org. Chem., 1998, 63, 6375 CrossRef CAS PubMed; (g) I. Ryu, A. Kurihara, H. Muraoka, S. Tsunoi, N. Kambe and N. Sonoda, J. Org. Chem., 1994, 59, 7570 CrossRef CAS.
- (a) A. Studer, S. Amrein, H. Matsubara, C. H. Schiesser, T. Doi, T. Kawamura, T. Fukuyama and I. Ryu, Chem. Commun., 2003, 1190 RSC; (b) A. Studer, Angew. Chem., Int. Ed., 1998, 37, 462 CrossRef CAS; (c) A. Studer and H. Steen, Chem. – Eur. J., 1999, 5, 759 CrossRef CAS.
- (a) S. M. Horvat, C. H. Schiesser and L. M. Wild, Organometallics, 2000, 19, 1239 CrossRef; (b) H. Matsubara, S. M. Horvat and C. H. Schiesser, Org. Biomol. Chem., 2003, 1, 1199 RSC; (c) H. Matsubara and C. H. Schiesser, Org. Biomol. Chem., 2003, 1, 4335 RSC; (d) D. Guerra, R. Castillo, J. Andrés, P. Fuentealba, A. Aizman and R. Contreras, Chem. Phys. Lett., 2006, 424, 437 CrossRef CAS PubMed; (e) S. M. Horvat and C. H. Schiesser, Organometallics, 2009, 28, 3311 CrossRef CAS.
- (a) G. Bencivenni, T. Lanza, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo and G. Zanardi, Org. Lett., 2008, 10, 1127 CrossRef CAS PubMed; (b) L. Benati, G. Calestani, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo and S. Strazzari, Org. Lett., 2003, 5, 1313 CrossRef CAS PubMed; (c) L. Benati, G. Bencivenni, R. Leardini, M. Minozzi, D. Nanni, R. Scialpi, P. Spagnolo and G. Zanardi, J. Org. Chem., 2006, 71, 3192 CrossRef CAS PubMed; (d) P. Carta, N. Puljic, C. Robert, A.-L. Dhimane, L. Fensterbank, E. Lacote and M. Malacria, Org. Lett., 2007, 9, 1061 CrossRef CAS PubMed; (e) P. P. Singh, A. K. Yadav, H. Ila and H. Junjappa, J. Org. Chem., 2009, 74, 5496 CrossRef CAS PubMed.
- A. Kamimura, S. Ishikawa, F. Noguchi, T. Moriyama, M. So, T. Murafuji and H. Uno, Chem. Commun., 2012, 47, 6592 RSC.
- A. Kamimura, M. So, S. Ishikawa and H. Uno, Org. Lett., 2013, 15, 1402 CrossRef CAS PubMed.
- A. Kamimura, Y. Yamane, R. Yo, T. Tanaka and H. Uno, J. Org. Chem., 2014, 79, 7696 CrossRef CAS PubMed.
- M. Newcomb, Tetrahedron, 1993, 49, 1151 CrossRef CAS.
- L. J. Johnston, J. Lusztyk, D. D. M. Wayner, A. N. Abeywickreyma, A. L. J. Beckwith, J. C. Scaiano and K. U. Ingold, J. Am. Chem. Soc., 1985, 107, 4594 CrossRef CAS.
- N. Carrera, M. H. Pérez-Temprano, A. C. Albéniz, J. A. Casares and P. Espinet, Organometallics, 2009, 28, 3957 CrossRef CAS.
- Crystallographic data (excluding structure factors) for the structures 3c-A have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 1050624.
- The phenyl group at the C2 position in N-tosyl-2-arylpiperidine such as 5c should occupy the axial position of the six-membered ring because of steric hindrance caused by the N-tosyl group. See: S. E. Denmark and H. M. Chi, J. Am. Chem. Soc., 2014, 136, 8915 CrossRef CAS PubMed; L. Li, H. Wang, D. Huang and Y. Shi, Tetrahedron, 2012, 68, 9853 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectral charts. CCDC 1050624. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00063g |
| This journal is © the Partner Organisations 2015 |