
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclohexene oxide/carbon dioxide copolymerization by chromium(III) amino-bis(phenolato) complexes and MALDI-TOF MS analysis of the polycarbonates†
Katalin
Devaine-Pressing
a,
Louise N.
Dawe‡
ab and
Christopher M.
Kozak
*a
aDepartment of Chemistry, Memorial University of Newfoundland, St. John's, Newfoundland A1B 3X7, Canada. E-mail: ckozak@mun.ca; Tel: +1-709-864-8082
bC-CART X-ray Diffraction Laboratory, Department of Chemistry, Memorial University of Newfoundland, St. John's, Newfoundland A1B 3X7, Canada
First published on 27th July 2015
Abstract
Amine-bis(phenolate) chromium(III) chloride complexes 1·THF, 2·THF and 1·DMAP catalyze the copolymerization of cyclohexene oxide and carbon dioxide. These catalysts incorporate tetradentate amine-bis(phenolate) ligands [L1] and [L2], (where [L1] = 2-pyridyl-N,N-bis(2-methylene-4-methoxy-6-tert-butylphenolato) and [L2] = dimethylaminoethylamino-N,N-bis(2-methylene-4-methoxy-6-tert-butylphenolato)) and when combined with 4-(N,N-dimethylamino)pyridine (DMAP) or bis(triphenylphosphoranylidene)ammonium chloride or azide (PPNCl or PPNN3), yield low molecular weight polycarbonate with narrow dispersities. The structure of 1·DMAP incorporates one molecule of 4-(N,N-dimethylamino)pyridine (DMAP) and can be used as a single-component catalyst precursor. Polymer end group analysis by MALDI-TOF mass spectrometry reveals possible initiation pathways.
Introduction
The copolymerization of carbon dioxide (CO2) and epoxides has recently become a growing area of interest for several reasons. Most importantly, the reaction is an appealing alternative to the traditional method of polycarbonate synthesis, which involves the use of the endocrine disruptor Bisphenol-A and highly toxic phosgene.1–4 Furthermore, the high free energy of epoxides drives the reaction forward to convert the very stable CO2.5 Another advantage is that CO2 is incorporated into the product, which is favorable not only with respect to the atom economy of the reaction, but also because CO2 is a readily available, non-toxic and low-cost feedstock.6 Also, CO2 can be considered a renewable resource,7 so its utilization is preferred over dwindling fossil fuels that are still the main basis for commercial polycarbonate synthesis.8In order to carry out and control the CO2/epoxide copolymerization reaction, an efficient catalyst system is needed.9 Recently, a large number of complexes have been developed as active catalyst precursors for the copolymerization of CO2 and epoxides. For example, complexes with Zn,10–12 Al,13 Co,14–21 Fe,22,23 Mg,24 and Cr25–42 have proven to be active in CO2/epoxide copolymerization. Many catalyst systems require a suitable ionic or neutral nucleophilic co-catalyst, the most broadly used include methylimidazole (N-MeIm), 4-(N,N-dimethylamino)pyridine (DMAP) and bis(triphenylphosphoranylidene)ammonium (PPN+) salts, such as PPN-chloride, azide, or 2,4-dinitrophenolate (PPNCl, PPNN3 PPN(2,4-DNP)).
The mechanism of CO2/epoxide coupling and copolymerization, particularly the role of the co-catalyst, has been studied by several groups.34,43–48 For chromium salen complexes, Darensbourg found that the anionic nucleophiles of PPN+ salts do not exhibit initiation periods, unlike the neutral co-catalysts studied (N-heterocyclic amines or phosphines). This was proposed to be due to the fast formation of active, anionic six-coordinate [(salen)Cr(N3)X]− derivatives.34 Studies of the binding of DMAP to salen and salan Cr(III) complexes using electrospray ionization mass spectrometry showed that coordination of two DMAP molecules to the Cr center of the salen complex is possible, even under low DMAP to Cr ratios.44 It was proposed that the stability of the six-coordinate [salenCr(DMAP)2]+ ions is a cause for the long initiation time for this catalyst system. In comparison, no induction period was observed for the salan analogue, which also showed a much lower propensity for bis-DMAP adduct formation, requiring much higher DMAP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr ratios to observe the presence of [salanCr(DMAP)2]+ ions. The difference in the geometry of the two complexes was speculated as the reason for the difference in DMAP binding. The time needed for the active species to form was ascribed to the slow dissociation of DMAP from the Cr-center of the salen complex to generate a vacant site allowing coordination of the epoxide for subsequent ring opening.
:Cr ratios to observe the presence of [salanCr(DMAP)2]+ ions. The difference in the geometry of the two complexes was speculated as the reason for the difference in DMAP binding. The time needed for the active species to form was ascribed to the slow dissociation of DMAP from the Cr-center of the salen complex to generate a vacant site allowing coordination of the epoxide for subsequent ring opening.
We previously reported the activity of amine-bis(phenolato) chromium(III) complexes for the copolymerization of epoxides and CO249–51 and the binding ability of DMAP to several derivatives of these ligands using matrix assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry.52 When used with DMAP, our Cr(III) complexes showed good activity with over 80% conversion of cyclohexene oxide to poly(cyclohexene carbonate) (PCHC) with nearly quantitative carbonate linkages giving polymers with molecular weights up to 13.1 kg mol−1. The polymer dispersities were generally low showing good control of the reaction.
Next, we sought to investigate whether amine-bis(phenolato) chromium(III) complexes bearing methoxy groups para to the phenolate oxygen instead of the tert-butyl substituents present in our previously reported compounds will exhibit an enhanced catalytic activity due to the increased electron donating ability of the phenolate, which was a positive effect observed by Darensbourg and co-workers.33 Herein, we report the synthesis and structure of three new amine-bis(phenolato) chromium(III) complexes including an amine-bis(phenolato) chromium(III)-DMAP adduct and their activity for the copolymerization of CO2 and cyclohexene oxide. Furthermore, the effect of three co-catalysts (DMAP, PPNCl and PPNN3) is also studied in detail by end-group analysis of the resulting polymers using MALDI-TOF MS, revealing further insights of the initiation of the polymerization.
Results and discussion
Synthesis and characterization of chromium complexes
The protio ligands 2-pyridylmethylamino-N,N-bis(2-hydroxy-3-tert-butyl-5-methoxyphenol) (H2[L1])53–55 and dimethylethyleneamino-N,N-bis(2-hydroxy-3-tert-butyl-5-methoxyphenol)56 (H2[L2]) (Scheme 1) were prepared via modified Mannich condensation and using water in place of methanol as solvent. The amine-bis(phenolato) chromium(III) complexes 1 and 2 (or their THF adducts, 1·THF and 2·THF) can be synthesized by salt metathesis using the alkali-metallated amine-bis(phenolate)s. The protonated ligands H2[L1] and H2[L2] were reacted with either nBuLi or NaH at −78 °C in THF to afford the corresponding Li or Na salts, respectively, which were subsequently reacted with CrCl3(THF)3 at −78 °C in THF (Scheme 1).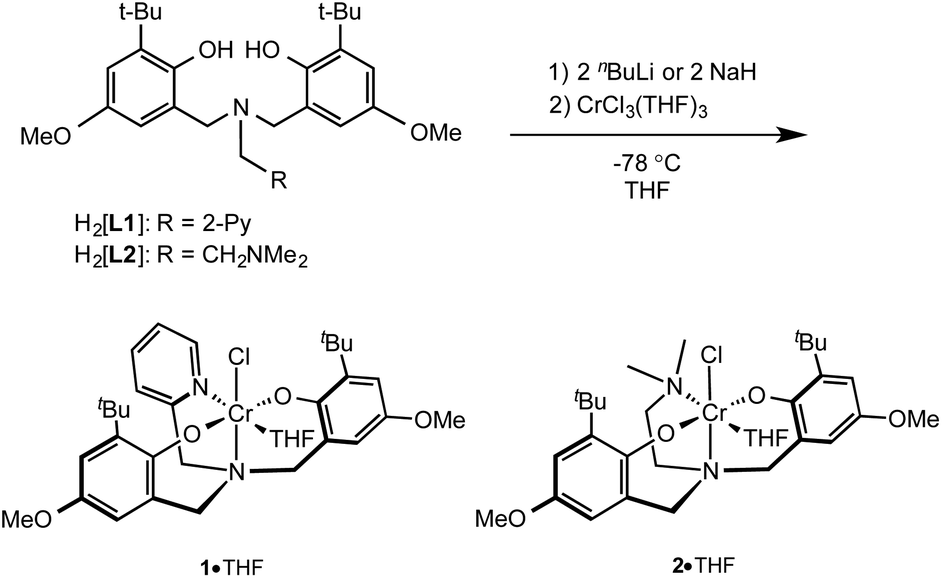 | ||
| Scheme 1 Synthesis of 1·THF and 2·THF. | ||
Dark green and purple solids of 1 and 2, respectively, were obtained in good yields, regardless of the alkali metal used. Elemental analyses of the purified amorphous materials were most consistent with the THF-free compounds (see Experimental), however the structures obtained by single crystal X-ray diffraction showed the crystalline materials are THF adducts (see below). The complexes were further characterized by MALDI-TOF MS, UV-Vis and IR spectroscopy and magnetic susceptibility measurement. The MALDI-TOF mass spectrum of 1 shows a fragment at m/z 577.17 corresponding to the [CrClL1]+˙ ion. The fragment at m/z 542.22 represents the [CrL1]+ ion after the loss of the chloride.
The isotopic distribution of the experimental [CrClL1]+˙ and [CrL1]+ ions are in good agreement with the theoretical representations (see ESI Fig. S1 and S2†). Two additional peaks are observed at the higher mass region (Fig. S1†) with the peak at m/z 1156.30 corresponding to a dimeric species [Cr2L12Cl2]+˙ and the peak at m/z 1119.35 represents the fragment ion after chloride loss, [Cr2L12Cl]+. The presence of a chloride-bridged dimeric complex is very probable and we have previously reported the structure of such a species.49 The complex is probably formed as a dimer during synthesis, dissociating to monomeric species in the presence of a coordinating solvent such as THF. In the case of complex 2, fragments are observed at m/z 522.20 and 557.17, which correspond to the [CrL2]+ and [CrClL2]+˙ ions, respectively.
Crystals of 1·THF and 2·THF suitable for X-ray diffraction were grown via slow evaporation of toluene/THF solvent mixtures. The molecular structures are shown in Fig. 1 and crystallographic and structure refinement data are given in ESI Table S1.† As previously observed for other Cr(III)-complexes of amine-bis(phenolato) ligands from our group,49–51,57 the structure of 1·THF shows a distorted octahedral geometry at the chromium with one THF molecule coordinated to the metal trans to the pendant pyridyl donor. The two phenolate oxygens are coordinated trans to each other and the chloride group is trans to the amine nitrogen. The same geometry was observed with complex 2·THF.
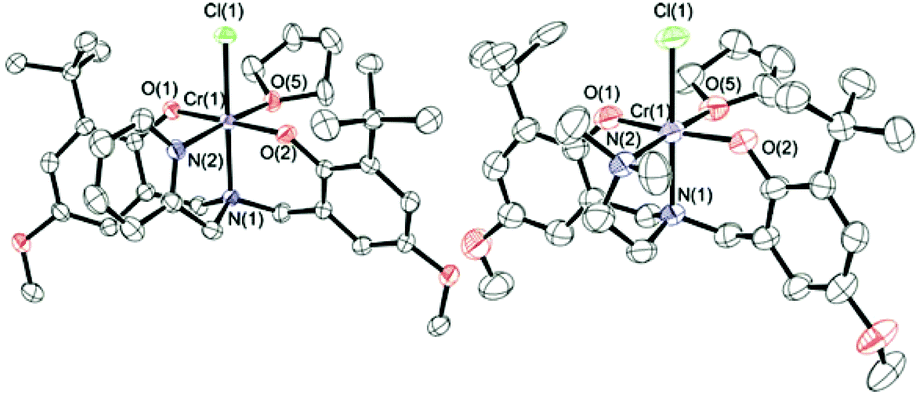 | ||
| Fig. 1 Partially labeled molecular structures of 1·THF (left) and 2·THF (right). Thermal ellipsoids are drawn at 50% probability and H atoms are excluded for clarity. | ||
Detailed mechanistic studies previously showed that the first step in obtaining the catalytically active species from the catalyst precursor complexes is the coordination of a nucleophilic co-catalyst to the metal center. Indeed, six-coordinate co-catalyst bound salen chromium(III) complexes have been observed both spectroscopically44 and structurally.26,44,58 Similarly, reaction of 1·THF with DMAP allowed isolation of the six-coordinate DMAP adduct, 1·DMAP. The MALDI-TOF mass spectrum of 1·DMAP (Fig. S4†) shows the presence of an intense peak at m/z 699.23 corresponding to the [CrClL1DMAP]+˙ ion, indicating that the DMAP is quite strongly bound to the chromium center.52 The fragment at m/z 664.27 corresponds to the [CrL1DMAP]+ ion resulting from chloride loss. The theoretical models are in good agreement with the experimentally observed peaks.
Crystals of 1·DMAP were obtained via slow evaporation of an equimolar solution of 1·THF and DMAP in dichloromethane. The molecular structure is shown in Fig. 2 and a comparison of the bond lengths and angles for the three Cr complexes is given in Table 1. 1·DMAP contains two strong σ-donor groups (the pendant pyridyl and DMAP) coordinated to the chromium. As a consequence, 1·DMAP contains the most electron-rich chromium center among these three complexes, and thus shows the longest average interatomic distances – the two exceptions being the Cr(1)–Cl(1) and the Cr(1)–N(2) interatomic distances. The Cr(1)–N(2) bond is, of course, the longest in complex 2·THF, as expected for an sp3-hybridized amino N-donor compared to the pyridine donor in 1·THF and 1·DMAP. Furthermore, the steric influence of the tertiary amine is reflected in the bond angles around the chromium. Both the O(1)–Cr(1)–N(2) and O(2)–Cr(1)–N(2) angles are greater than 90° (90.23(15) and 95.29(14) respectively) in 2·THF meaning that the phenolate oxygens are pushed away from the tertiary amine. The pushing away of the phenolates by the bulkier amine results in a modest weakening of the bond between THF and Cr, resulting in a longer Cr(1)–O(5) bond length in 2·THF than in 1·THF. Interestingly, this increased steric crowding of the chromium coordination sphere is believed to inhibit binding of more than one DMAP molecule to the metal in dimethylaminoethyl-functionalized amine,52 however, the influence of the phenolate substituents cannot be ignored.
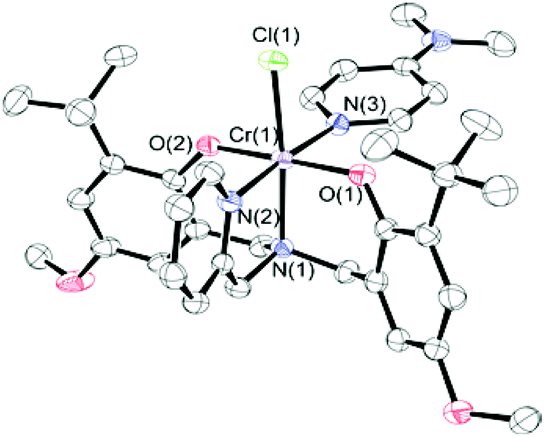 | ||
| Fig. 2 Molecular structure of 1·DMAP. Thermal ellipsoids are drawn at 50% probability. H atoms are excluded for clarity. | ||
| 1·THF | 2·THF | 1·DMAP | |
|---|---|---|---|
| Cr(1)–O(1) | 1.943(2) | 1.901(3) | 1.946(2) |
| Cr(1)–O(2) | 1.912(2) | 1.913(3) | 1.926(2) |
| Cr(1)–O(5) | 2.074(2) | 2.095(3) | — |
| Cr(1)–N(1) | 2.097(2) | 2.086(4) | 2.129(2) |
| Cr(1)–N(2) | 2.065(3) | 2.116(4) | 2.088(2) |
| Cr(1)–N(3) | — | — | 2.100(2) |
| Cr(1)–Cl(1) | 2.3463(9) | 2.319(2) | 2.3386(11) |
| O(1)–Cr(1)–O(2) | 178.17(9) | 174.22(13) | 178.84(8) |
| O(1)–Cr(1)–O(5) | 86.98(8) | 87.38(14) | — |
| O(2)–Cr(1)–O(5) | 91.88(9) | 87.26(13) | — |
| O(1)–Cr(1)–N(2) | 89.25(9) | 90.23(15) | 87.15(9) |
| O(2)–Cr(1)–N(2) | 91.74(10) | 95.29(14) | 92.86(9) |
| O(1)–Cr(1)–N(1) | 92.09(8) | 91.38(13) | 91.23(8) |
| O(2)–Cr(1)–N(1) | 86.53(8) | 90.92(13) | 87.62(8) |
| O(1)–Cr(1)–N(3) | — | — | 89.16(9) |
| O(2)–Cr(1)–N(3) | — | — | 90.78(9) |
| O(1)–Cr(1)–Cl(1) | 91.78(6) | 90.21(9) | 91.07(6) |
| O(2)–Cr(1)–Cl(1) | 89.67(6) | 87.67(9) | 90.09(6) |
| N(2)–Cr(1)–Cl(1) | 95.35(7) | 94.41(10) | 93.49(7) |
| O(5)–Cr(1)–Cl(1) | 90.70(6) | 90.52(9) | — |
| N(3)–Cr(1)–Cl(1) | — | — | 89.12(7) |
Copolymerization of cyclohexene oxide with CO2
The copolymerization of CHO and CO2 was investigated with complex 1·THF, 2·THF and 1·DMAP (Scheme 2). Nucleophilic neutral or ionic co-catalysts are essential for the reaction; therefore, the activity of the complexes was tested with DMAP, PPNCl or PPNN3. Copolymerization results show good conversion of cyclohexene oxide to PCHC employing 1·THF and 1·DMAP complexes with moderate conversions using 2·THF (Table 2). It is worth noting that the catalysts are selective toward polymer formation (Scheme 2, product a) as there is no or negligible evidence of either polyether (product b) (3.74 ppm in Fig. S13†) or cyclic carbonate (c) (4.10 ppm in Fig. S13†) formation based on 1H NMR spectroscopy (Fig. S13 and S14†). Resonances at 3.58 and 4.41 ppm correspond to the methine protons of the cyclohexane rings of the end groups. The carbonyl region of the 13C NMR spectra shows that the obtained polymers are atactic, containing both syndiotactic and isotactic PCHC (Fig. S15†). The maximum conversion achieved was 83% (Table 2, entries 5, 6 and 11) at which point only the solid polymer was found in the reaction vessel after opening the reactor. No conversion of CHO was observed in the absence of co-catalysts (Table 2, entry 1). Also, complex 1·THF with DMAP did not show any activity toward CHO homopolymerization (entry 2). Overall, very good conversions and yields were obtained at CO2 pressures of ∼40 bar, 60 °C and at 0.2 mol% catalyst loading (entries 3, 5–7, 10 and 11). The dispersities (Mw/Mn) of the polymers did not change with molecular weight and are generally narrow with values between 1.26 and 1.43. Decreasing the catalyst loading from 0.2 mol% to 0.1 mol% resulted in a decreased conversion as well as molecular weight (entry 4). This is in accordance with previously obtained CHO/CO2 copolymerization results in our group.49,51 Comparing the three co-catalysts utilized, there is no significant difference in conversions, molecular weights and dispersities obtained. PPNCl and PPNN3 proved to be only slightly better than DMAP, giving higher conversions, activities and molecular weights when 1·THF was used (entries 3, 5, 6). Carrying out the reaction at room temperature did not produce any polymer product (entry 8) and shortening the reaction time from 24 h to 5 h also produced lower conversion (entry 9). Complex 2·THF proved to be inferior to complexes 1·THF and 1·DMAP, as it showed low conversions with DMAP and PPNCl co-catalysts, and moderate activity with PPNN3 (entries 12–14).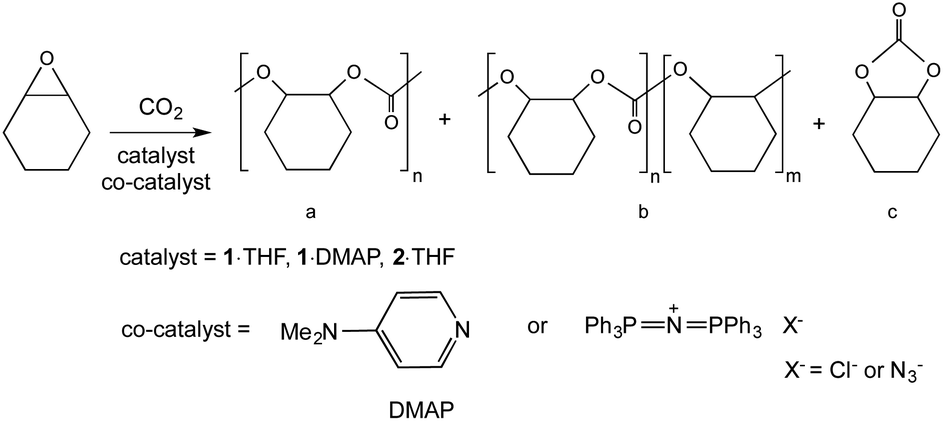 | ||
| Scheme 2 Copolymerization of CHO and CO2 with the possible products of the reaction: (a) PCHC, (b) polyether formation in the polymer chain and (c) cyclohexene carbonate. | ||
| Entrya | Cat. | [Cr]:[CHO]:[Co-cat.] |
Co-cat. | Time (h) | Temp (°C) | CO2 Pressure (bar) | % Conversionb | % Yieldc | TONd |
M
ne (kg mol−1) |
M
w/Mne |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a All copolymerization reactions were carried out in neat cyclohexene-oxide (4 mL). b Calculated by 1H-NMR. c Yield = moles of isolated product (mass of polymer/molar mass of repeating unit) divided by the moles of starting monomer. d Turnover number: moles of repeating units produced per mole of Cr present. e Determined by gel permeation chromatography (GPC) in CHCl3, calibrated with polystyrene standards. f ND = not determined due to low or no conversion. g Based on two runs. | |||||||||||
| 1 | 1·THF | 1:500:0 |
— | 24 | 60 | 40 | 0 | 0 | 0 | NDf | ND |
| 2 | 1·THF | 1:500:1 |
DMAP | 24 | 60 | 0 | 0 | 0 | 0 | ND | ND |
| 3g | 1·THF | 1:500:1 |
DMAP | 24 | 60 | 42 | 75 ± 7 | 72 ± 1 | 375 ± 35 | 7.1 | 1.36 |
| 4g | 1·THF | 1:1000:1 |
DMAP | 24 | 60 | 40 | 32 ± 4 | 29 ± 1 | 320 ± 40 | 3.7 | 1.29 |
| 5 | 1·THF | 1:500:1 |
PPNCl | 24 | 60 | 44 | 83 | 82 | 415 | 8.1 | 1.43 |
| 6 | 1·THF | 1:500:1 |
PPNN3 | 24 | 60 | 42 | 83 | 73 | 415 | 7.5 | 1.26 |
| 7 | 1·DMAP | 1:500:0 |
— | 24 | 60 | 40 | 81 | 62 | 405 | 7.3 | 1.30 |
| 8 | 1·DMAP | 1:500:0 |
— | 24 | r.t. | 40 | 0 | 0 | 0 | ND | ND |
| 9 | 1·DMAP | 1:500:0 |
— | 5 | 60 | 40 | 44 | 25 | 220 | ND | ND |
| 10 | 1·DMAP | 1:500:1 |
PPNCl | 24 | 60 | 40 | 78 | 73 | 390 | 4.9 | 1.37 |
| 11 | 1·DMAP | 1:500:1 |
PPNN3 | 24 | 60 | 40 | 83 | 73 | 415 | 5.6 | 1.32 |
| 12g | 2·THF | 1:500:1 |
DMAP | 24 | 60 | 40 | 12 ± 3 | ND | 60 | ND | ND |
| 13 | 2·THF | 1:500:1 |
PPNCl | 24 | 60 | 40 | 20 | ND | 100 | ND | ND |
| 14 | 2·THF | 1:500:1 |
PPNN3 | 24 | 60 | 40 | 62 | 46 | 310 | 4.3 | 1.32 |
Next, we investigated the effect of co-catalyst mixtures on the copolymerization, thus 1·DMAP was used together with one eq. of either PPNCl or PPNN3. Interestingly, only a slight decrease in molecular weights could be observed giving 4.9 kg mol−1 with PPNCl and 5.6 kg mol−1 with PPNN3 (entries 10 and 11) compared to 7.3 kg mol−1 using 1·DMAP alone (entry 7). Selectivity toward polymer formation was unaffected by the addition of these salts. Usually, increasing the amount of co-catalyst used can favor the formation of cyclic carbonate over polycarbonate,23,50 however, in our system cyclic carbonate formation was not observed upon addition of PPNX salts to 1·DMAP (entries 10 and 11). Potentially, molecular weights might be controlled with elevated amounts of co-catalysts. The observed lower molecular weight can be attributed to the increased concentration of co-catalyst that serves to initiate ring-opening of the epoxide fragment, hence an increased concentration of activated epoxide monomer resulting in growth of a larger number of polymer chains. The increased concentration of nucleophilic co-catalysts may also assist the displacement of the polymer chains from the metal center, which can then participate in chain transfer events leading to polymers terminated with the different initiator species.
The effect of methoxy substituents para and tert-butyl groups ortho to the phenoxide group has been investigated for chromium(III) salen complexes.33 In that study, the rate of copolymer production was increased when these strongly electron donating salen complexes were employed compared to the di-tert-butyl analogues. It was proposed that the increased electron-donating ability of the phenolates had a more positive effect on the rate of polymer formation than by modifying the diimine backbone. Where we previously reported the CHO/CO2 copolymerization activity of the di-tert-butyl substituted amine-bis(phenolate) chromium(III) complex bearing a pyridyl side-arm (i.e. a di-tert-butyl functionalized analog of complex 1),49 we compared the rate of copolymerization of that complex with 1·THF and 1·DMAP monitored by in situ attenuated total reflectance infrared spectroscopy (ATR-IR). The initial rates of CHO/CO2 copolymerization catalyzed by 1·DMAP, 1·THF and the previously reported chromium(III) amine-bis(phenolate) complex (abbreviated as CrCl[O2NN′]BuBuPy) were compared and are presented in Fig. 3. DMAP was added as co-catalyst for 1·THF and CrCl[O2NN′]BuBuPy.
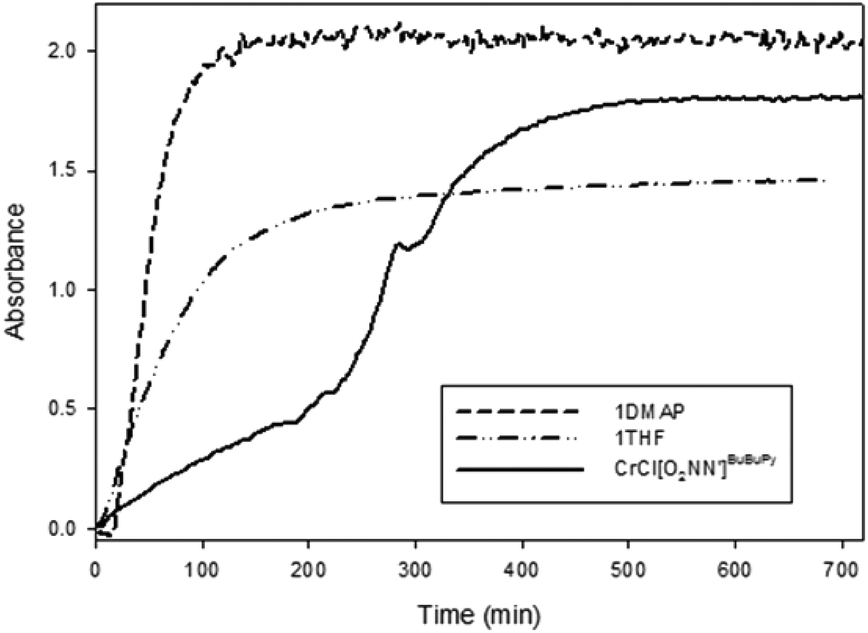 | ||
Fig. 3 First 12 h of the reaction profiles showing the growth of the absorbance of the polycarbonate carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band at 1750 cm−1 catalyzed by 1·DMAP (dashed line), 1·THF (dashed-dotted line) and CrCl[O2NN′]BuBuPy (solid line). Reaction conditions: 40 bar CO2, 60 °C, [Cr]:[CHO]:[DMAP] = 1:500:1. O band at 1750 cm−1 catalyzed by 1·DMAP (dashed line), 1·THF (dashed-dotted line) and CrCl[O2NN′]BuBuPy (solid line). Reaction conditions: 40 bar CO2, 60 °C, [Cr]:[CHO]:[DMAP] = 1:500:1. | ||
The fastest initial reaction rate was exhibited by 1·DMAP followed by 1·THF in the presence of equimolar DMAP. It is also worth noting that signal saturation (reaching a plateau in the absorbance of the polycarbonate ν(CO)) was also achieved within the shortest time by 1·DMAP (∼2 h), whereas CrCl[O2NN′]BuBuPy reached signal saturation after 8 h. The reaction catalyzed by CrCl[O2NN′]BuBuPy exhibited two stages during the first 6 h. The first stage occurs over 4 h and represents the slowest propagation among the three complexes, while the second stage is faster and lasts for ∼150 min before signal saturation occurs. In both stages, propagation was slower than for the 1·DMAP and the 1·THF/DMAP catalyzed reactions. Relative reaction rates were calculated based on the propagation in the first hour of the reaction after stabilization of the reaction conditions (∼20 min) directly from the change of the carbonyl signal intensity as the slope of the plots (Fig. 4). The rates of propagation obey the order of 1·DMAP > 1·THF/DMAP > CrCl[O2NN′]BuBuPy/DMAP and are represented in Table 3.
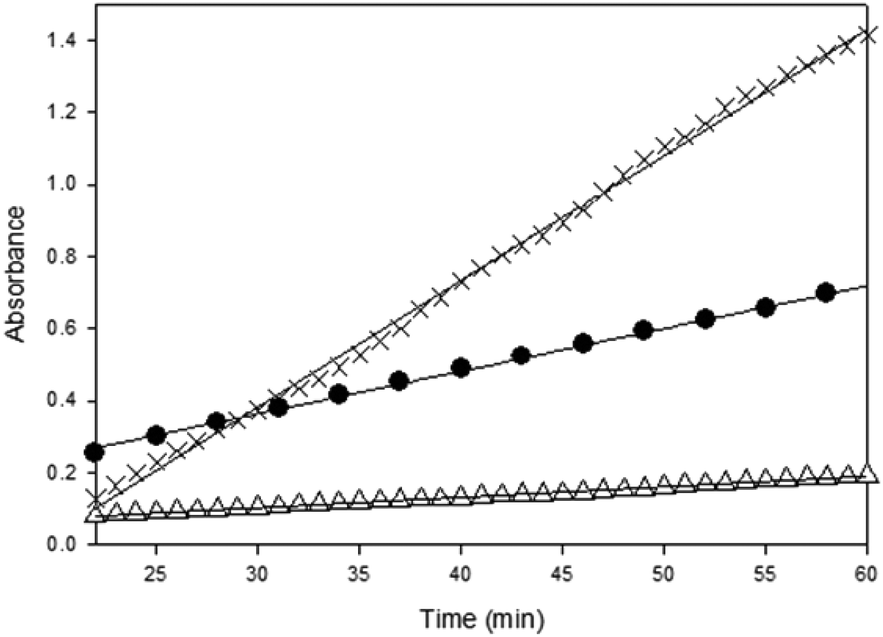 | ||
| Fig. 4 Initial rates of reaction profiles during the first hour based on the absorbance of the ν(CO) of the polycarbonates. 1·DMAP (×), 1·THF (●), CrCl[O2NN′]BuBuPy (△). Lines represent best fits of a linear model to the observed data (see Table 3). | ||
O) of the growing polycarbonate chains and R2 values of the linear regressions
| Complex | Relative reaction rate robs (×10−2 min−1) | Relative reaction rate robs (×10−4 s−1) | R 2 |
|---|---|---|---|
| 1·DMAP | 3.5 ± 0.026 | 5.8 ± 0.043 | 0.9980 |
| 1·THF | 1.2 ± 0.020 | 2.0 ± 0.033 | 0.9978 |
| CrCl[O2NN′]BuBuPy | 0.29 ± 0.0032 | 0.48 ± 0.0053 | 0.9960 |
The first 20 min of the reaction exhibit short initiation periods of approximately 8 min for 1·THF/DMAP and approximately 17 min for 1·DMAP (Fig. S11†). These initiation times are in accordance with the initiation periods observed for copolymerization by Cr(III)-salen complexes.34 The longer initiation with 1·DMAP is possibly due to the relatively strong coordination of DMAP, which results in a more stable complex compared to 1·THF/DMAP. The procedure for use of the 1·THF/DMAP catalyst system involves the addition of DMAP to a solution of 1·THF in CHO where the labile THF could rapidly give rise to an open coordination site, therefore CHO coordination to the chromium center of 1 may occur prior to DMAP binding. DMAP coordination to our chromium(III)-complexes was studied in detail in our group previously and the binding of two equivalents of DMAP was observed by MALDI-TOF MS even under equimolar concentrations of chromium complex 1 and DMAP.52 By comparison, CrCl[O2NN′]BuBuPy is THF-free, but exists as a dimer in the solid state and in non-coordinating solvents.49 The very short initiation time of approximately 5 min may arise from dissociation of the dimeric complex to monomeric species. This may also serve to explain the two stages of the reaction giving different rates or propagation. Dissociation of the dimer into monomers leads to five-coordinate Cr(III) sites that catalyze copolymerization according to the rate observed in the first stage (as modeled in Fig. 4). A faster rate ensues during the second stage, which may be due to DMAP coordination and chloride dissociation. Initiation of epoxide ring opening by chloride nucleophiles is believed to dominate the reaction based on end-group analysis of the polymer by MALDI-TOF MS, where no DMAP-containing end-groups were observed.49,52 Even for the faster second stage, the rate proved to be slower (robs = 0.75 × 10−2 min−1, Fig. S12†) than the rates with para-methoxy-containing 1·DMAP and 1·THF (3.5 × 10−2 min−1 and 1.2 × 10−2 min−1 respectively). Rieger and co-workers observed two different rates of propagation after initiation with a dinuclear Cr(III)-salphen type complex.59 The first stage was attributed to a heterogeneous phase due to the insolubility of the flexibly linked dinuclear complex, which was followed by a homogeneous stage after dissolution of the complex. Our complex, CrCl[O2NN′]BuBuPy proved to be highly soluble in CHO, so this is unlikely the cause for the two different rates in our case.
Polymer end group analysis on polymers produced by 1·THF and 1·DMAP by MALDI-TOF mass spectrometry
MALDI-TOF mass spectrometry is valuable for end-group analysis and the mass spectra of the polymers obtained show multiple end-group series, where repeating units of m/z 142 are observed corresponding to the expected cyclohexane carbonate motif. The variety of end groups is a result of different initiation possibilities demonstrated by these catalyst systems, i.e. the chromium chloride-containing catalyst and the additional DMAP, chloride or azide nucleophile. In the next section, first, the end groups of the polymers produced with 1·DMAP alone and with 1·THF/DMAP will be discussed. Then, the end group analysis of the polymers produced by 1·DMAP and added PPNX salts will follow revealing more information on the initiation of the copolymerization.When 1·DMAP was used according to the conditions in Table 2, entry 7, the MALDI-TOF MS spectrum of the polymer produced shows two major sets of chains (Fig. 5). One series possesses chloride and a hydroxyl end groups, whereas the other series possesses two hydroxyl group ends, which implies that there is either adventitious water present and/or cyclohexene-1,2-diol is produced during polymerization causing rapid chain transfers.60 No DMAP end-groups were observed, showing a probable chloride dissociation and initiation, while DMAP stays coordinated.
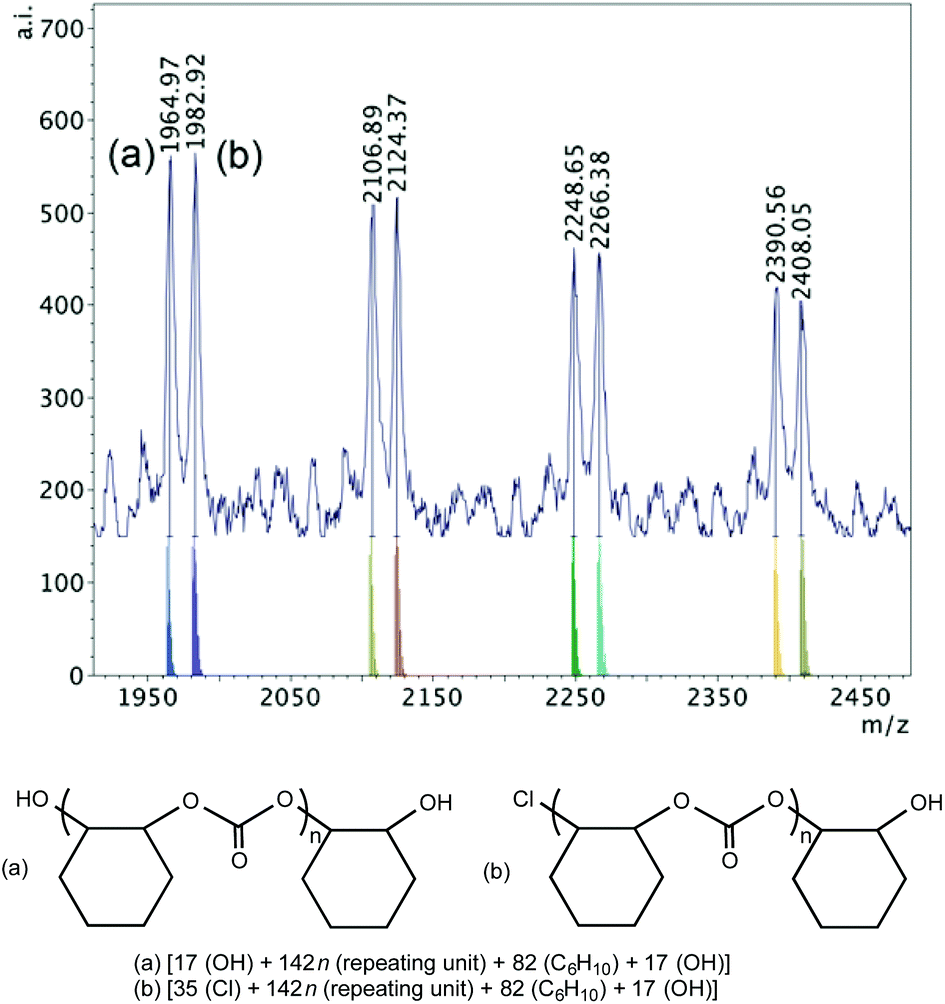 | ||
| Fig. 5 MALDI-TOF mass spectrum (m/z 1950–2450, n = 13–17) produced by 1·DMAP according to Table 2, entry 7 with calculated masses of fragments shown beneath the observed spectrum and the proposed structures of polymers (a) and (b). | ||
The higher mass region of the MALDI-TOF mass spectrum of the polymer produced by 1·THF according to Table 2, entry 3 is shown in Fig. 6. The polymer consists of only one main chain in the higher mass region (m/z 7800–8900, inset of Fig. 6B), which is also chloride initiated and hydroxyl group terminated with a K+ ion in the chain, also corresponding to chloride initiation again.
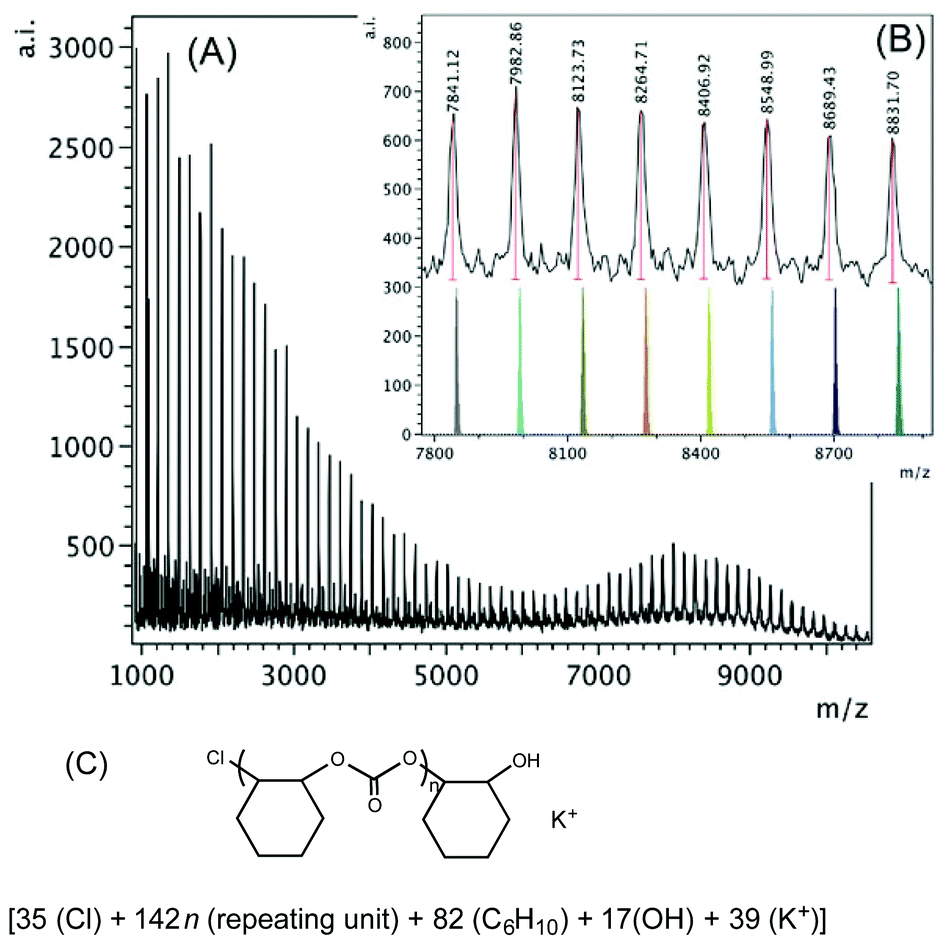 | ||
| Fig. 6 (A) MALDI-TOF mass spectrum produced by 1·THF according to Table 2, entry 3. (B) Higher mass region (m/z 7800–8900, n = 54–61) of the spectrum with calculated masses of fragments shown beneath the observed spectrum. (C) Proposed structure of the high mass polymer. | ||
Inspection of the low molecular weight region of the MALDI-TOF mass spectrum between m/z 1900 to 2700, however, reveals the presence of both DMAP and chloride initiated polymer chains (Fig. S16†). Altogether, four different species were detected. Series (a) with the most intense peaks corresponds to a polymer with DMAP and chloride end groups with one ether linkage in the polymer structure (the ether linkage could be located anywhere in the chain). The presence of DMAP in one end of the polymer end groups indicates that DMAP can also initiate ring opening of the epoxide. Series (b) – the lowest intensity peaks that disappear above m/z 2300 – possess DMAP and chloride termini. Series (c) with two hydroxyl group ends is probably the result of chain transfer reactions caused by trace water contamination.60 Series (d) is represented by chloride and hydroxyl end group-containing polymer, where a sodium ion is also present.
When 1·THF is used with PPNCl, chloride initiation is anticipated as both the complex and the co-catalyst contain chloride groups capable of ring-opening. As shown in Fig. 7, chloride groups are observed in both of the two sets of polymers detected in the higher mass region. Series (a) and (b) are both hydroxyl group terminated and contain sodium ions. The difference between the two is the presence of an ether linkage incorporated in polymer chain (a). The presence of the chloride end groups in both cases, again, is the indication of initiation by chlorides, which can come from the complex or the added PPNCl nucleophile. In the lower mass region (Fig. S17†) three sets of polymer chains are observed; series (a) is dihydroxylated, series (b) contains chloride and hydroxyl termini, and series (c) contains chloride end groups. Series (c) could be the result of intermolecular chain transfer of two chains initiated by chloride.50
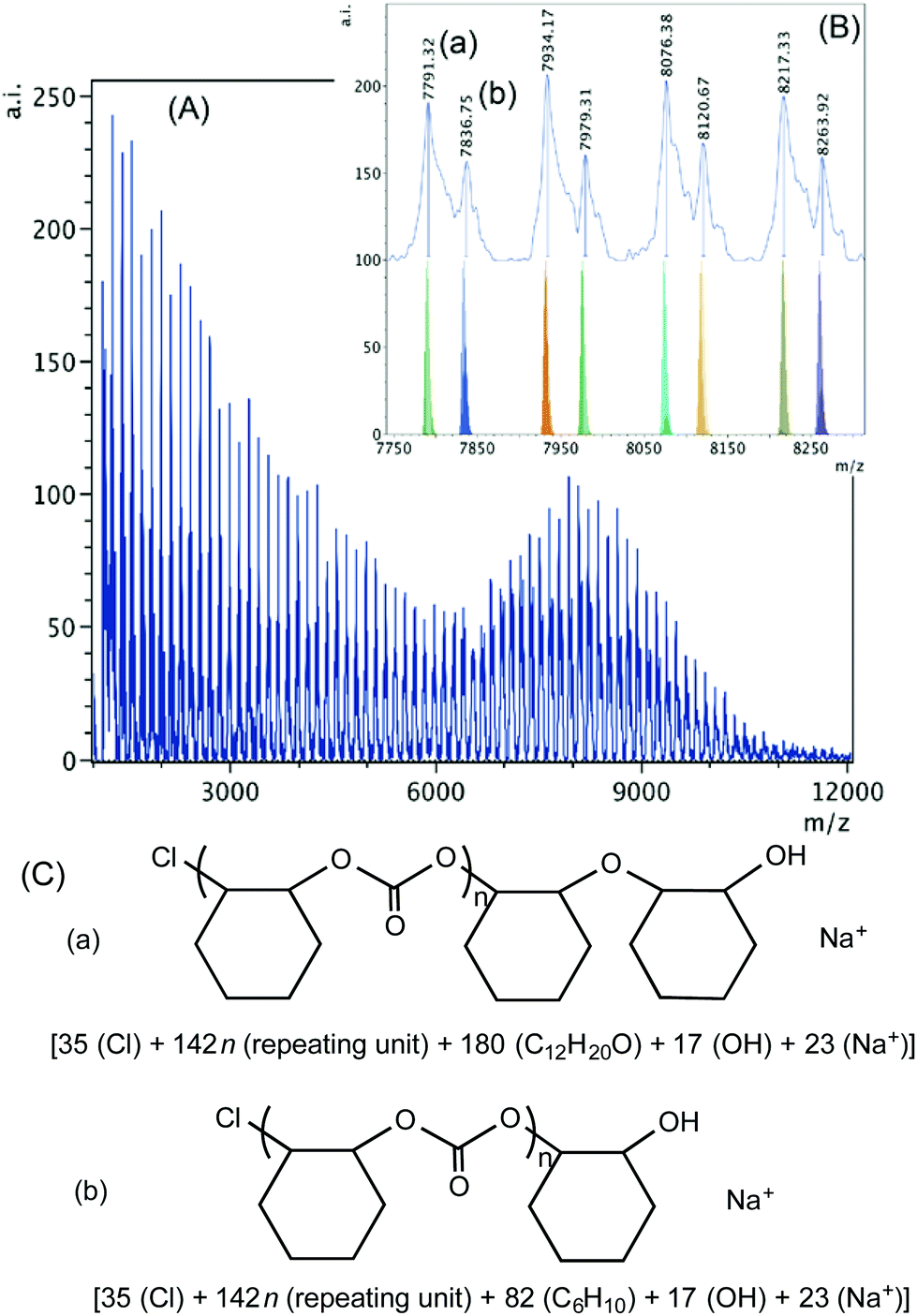 | ||
| Fig. 7 (A) MALDI-TOF mass spectrum produced by 1·THF according to Table 2, entry 5. (B) Higher mass region (m/z 7750–8300, n = 53–56) of the spectrum with calculated masses of fragments shown beneath the observed spectrum. (C) Proposed structures of the high mass range polymers (a) and (b). | ||
Interestingly, when 1·DMAP is used together with PPNCl, only DMAP initiated polymer chains are observed in both the upper and the lower mass region (Fig. 8A). This suggests that the coordinated DMAP is displaced by the anionic chloride and can attack a coordinated cyclohexene oxide. The polymer chain illustrated in the high mass region (Fig. 8B) is present over a wide mass range from 2000–8000 m/z shows initiation by a DMAP molecule and termination by hydrolysis of the metal alkoxide. In the lower mass region (Fig. S18†) we can only see DMAP initiated polymer chains. In series (a) we observe a series with an ether linkage incorporated in the chain terminated by a hydroxyl group, plus a sodium ion. Series (b) consists of a hydroxyl end group and series (c) of a chloride and DMAP termini with a Na+ ion. Most likely series (c) represents an intermolecular chain transfer between a DMAP and a chloride initiated polymer chain. This observation is different from the 1·DMAP-catalyzed reaction without any PPNX salt (Table 2, entry 7), where there were no DMAP initiated polymer chains found. The difference may be attributed to the elevated amount of chloride ions. When PPNCl is added, there are twice as many chloride ions present coming from both the complex and the PPN+ salt compared to where there is no PPNCl added. Consequently, the competition for an open coordination site is higher, which can easily result in DMAP dissociation and incorporation of the polymer chain end.
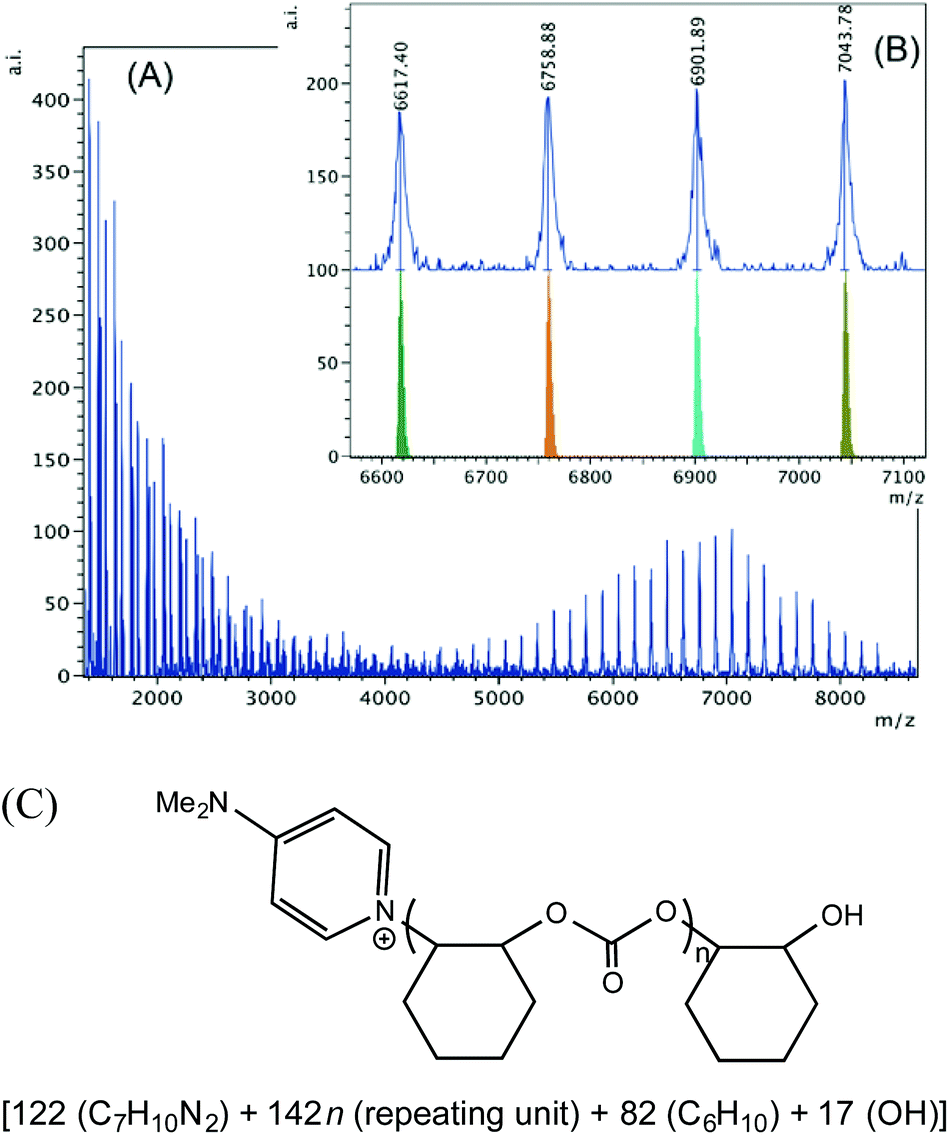 | ||
| Fig. 8 (A) MALDI-TOF mass spectrum of polycarbonate produced by 1·DMAP according to Table 2, entry 10. (B) Higher mass region (m/z 6600–7100, n = 45–48) of the spectrum with calculated masses of fragments shown beneath the observed spectrum. (C) Proposed structure of the high mass range polymer. | ||
The spectrum of the polymer obtained when 1·THF was used in the presence of PPNN3 shows high intensity peaks in the high mass region (Fig. 9). Two sets of polymers can be observed in this region between m/z 7400–8000. Based on the calculation shown in Fig. 9(C), series (a) shows an ether linkage in the polymer chain that was initiated by a nucleophilic attack of an azide group and was terminated by a chloride group. Series (b) is again most likely the result of an intermolecular chain transfer between two polymer chains that were both initiated by azide groups. In the lower mass region, however, azide group-initiated polymer chains are not observed (Fig. S19†). Series (a) with chloride and hydroxyl end groups and series (b) with two hydroxyl termini represent polymers where chain termination and chain transfer could be caused by adventitious water resulting in lower molecular weight polymer chains.
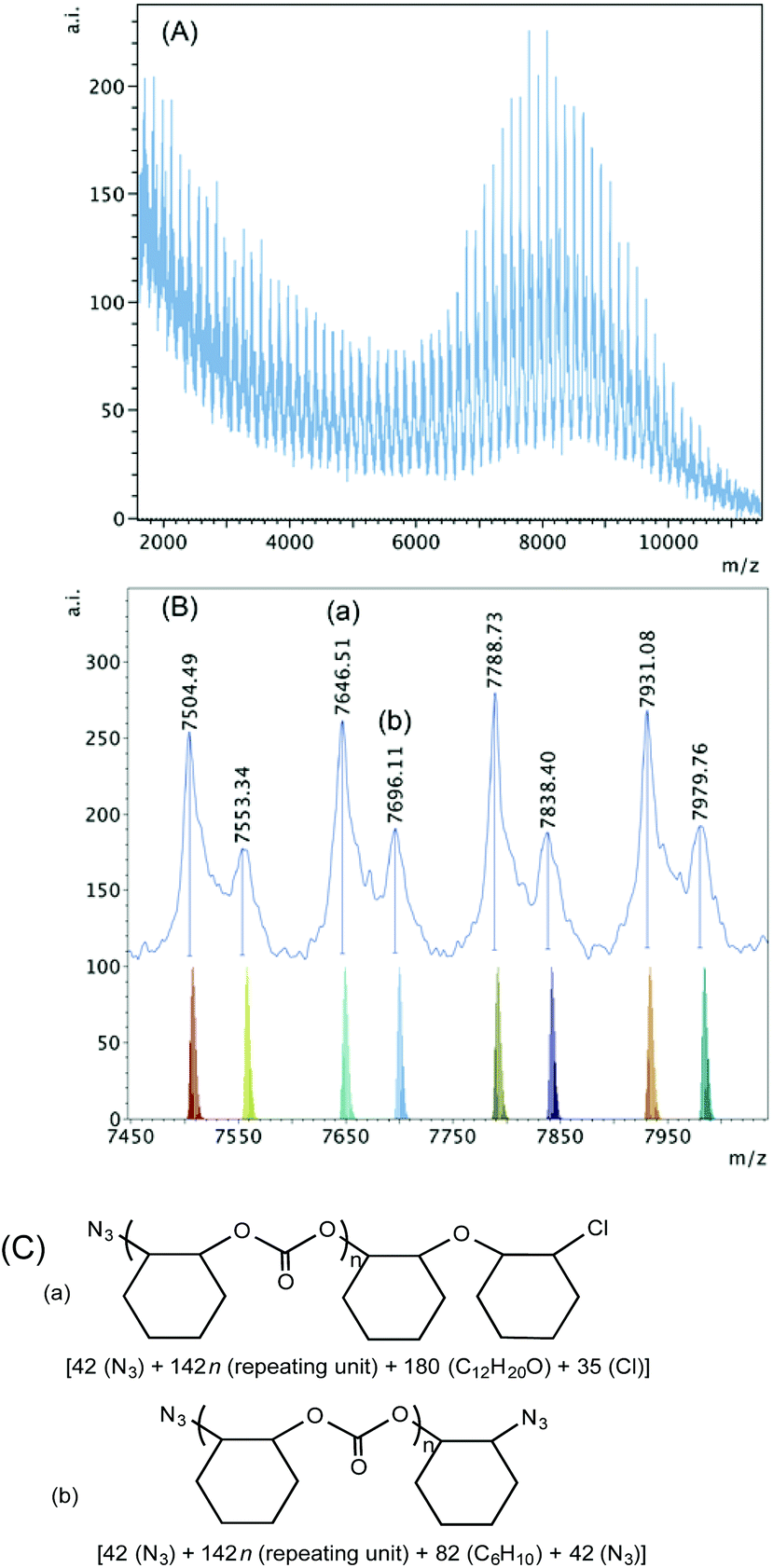 | ||
| Fig. 9 (A) MALDI-TOF mass spectrum produced by 1·THF according to Table 2, entry 6. (B) Higher mass region (m/z 7450–8000, n = 51–54) of the spectrum with calculated masses of fragments shown beneath the observed spectrum. (C) Proposed structures of the high mass range polymers (a) and (b). | ||
Azide initiated polymer chains were also observed when 1·DMAP was used together with PPNN3 (Fig. S20† and Fig. 10). In the high mass region (Fig. S20†) of the spectrum there is only one significant high intensity set of peaks observed, which corresponds to an azide and chloride terminated polymer cationized by Na+. In the lower mass region two main sets of polymers can be discerned from the spectrum (Fig. 10): series (a) possesses azide and chloride groups plus Na+, and series (b) consists of a DMAP and azide terminated polymer, again, probably due to intermolecular chain transfer.
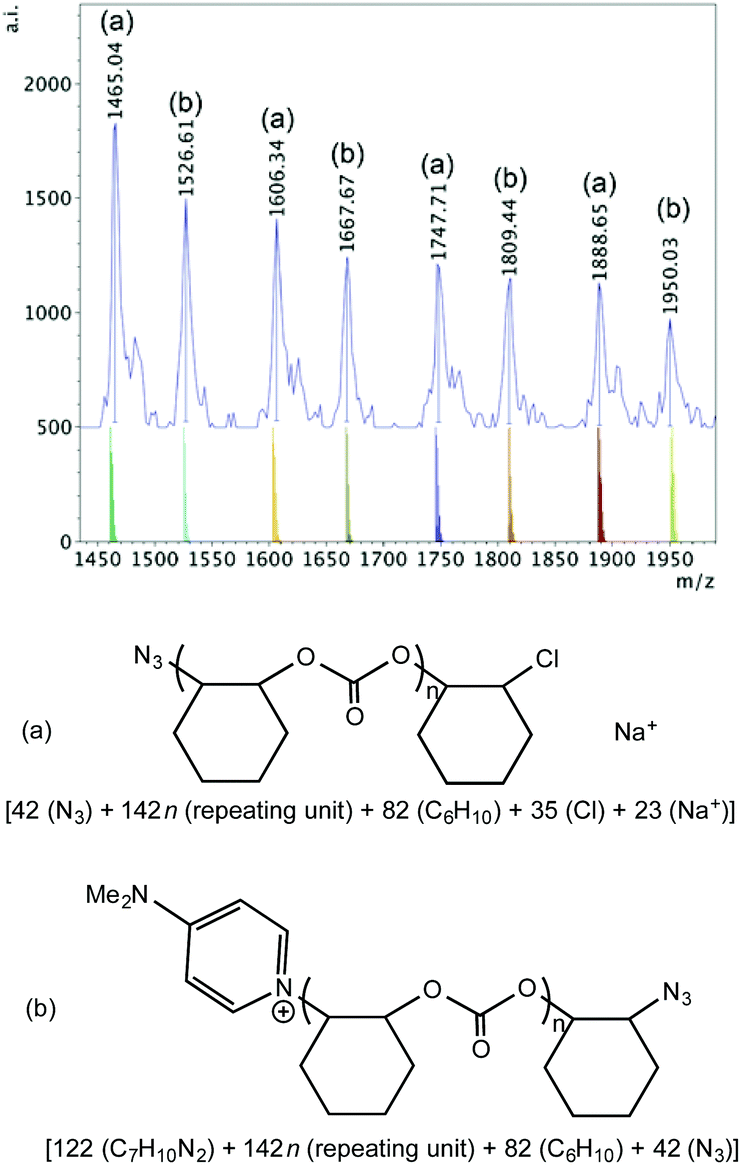 | ||
| Fig. 10 Lower mass region (m/z 1400–2000, n = 9–12) of the MALDI-TOF mass spectrum produced by 1·DMAP according to Table 2, entry 11, with calculated masses of fragments shown beneath the observed spectrum and the proposed structures of polymers (a) and (b). | ||
Mechanistic considerations
The presence of the chloride end group indicates chloride initiation, which may occur via an intermolecular or an intramolecular pathway.49 Displacement of the chloride by an epoxide has been shown to be viable based on the bond strengths between chromium and epoxide or chloride with salen complexes calculated in an elegant recent computational study.61 In this manner one possible pathway can be the replacement of the chloride with an epoxide, which points toward the intermolecular ring opening by the chloride. After the chloride loss, a cationic complex is produced and provides the platform for copolymerization. This possibility of the cationic route is depicted in Scheme 3A. Of course, it cannot be excluded that under polymerization conditions the nucleophilic co-catalyst (DMAP) dissociates from the central metal, e.g. in the case of the 1·DMAP/PPNCl system and ring opens a then coordinated epoxide (Scheme 3B). In this case, the chlorides of PPNCl compete with the DMAP for coordination to the metal center leading to a chromium dichloride intermediate.26 Displacement of one of the chlorides produces a vacant site for epoxide binding and the free DMAP can now serve as nucleophile for epoxide ring-opening. Initiation by a chloride, however, is also possible based on the end group analysis of the 1·DMAP/PPNCl system, where both DMAP and chloride termini were observed in the lower mass region of the MALDI-TOF mass spectrum likely caused by chain transfer. Several different initiation possibilities were extensively elaborated upon and compiled in reviews.7,62,63 Based on our observations, we propose that it is the monometallic intermolecular initiation pathway that most likely occurs with these chromium(III)-bis(phenolate) complexes (Scheme 3).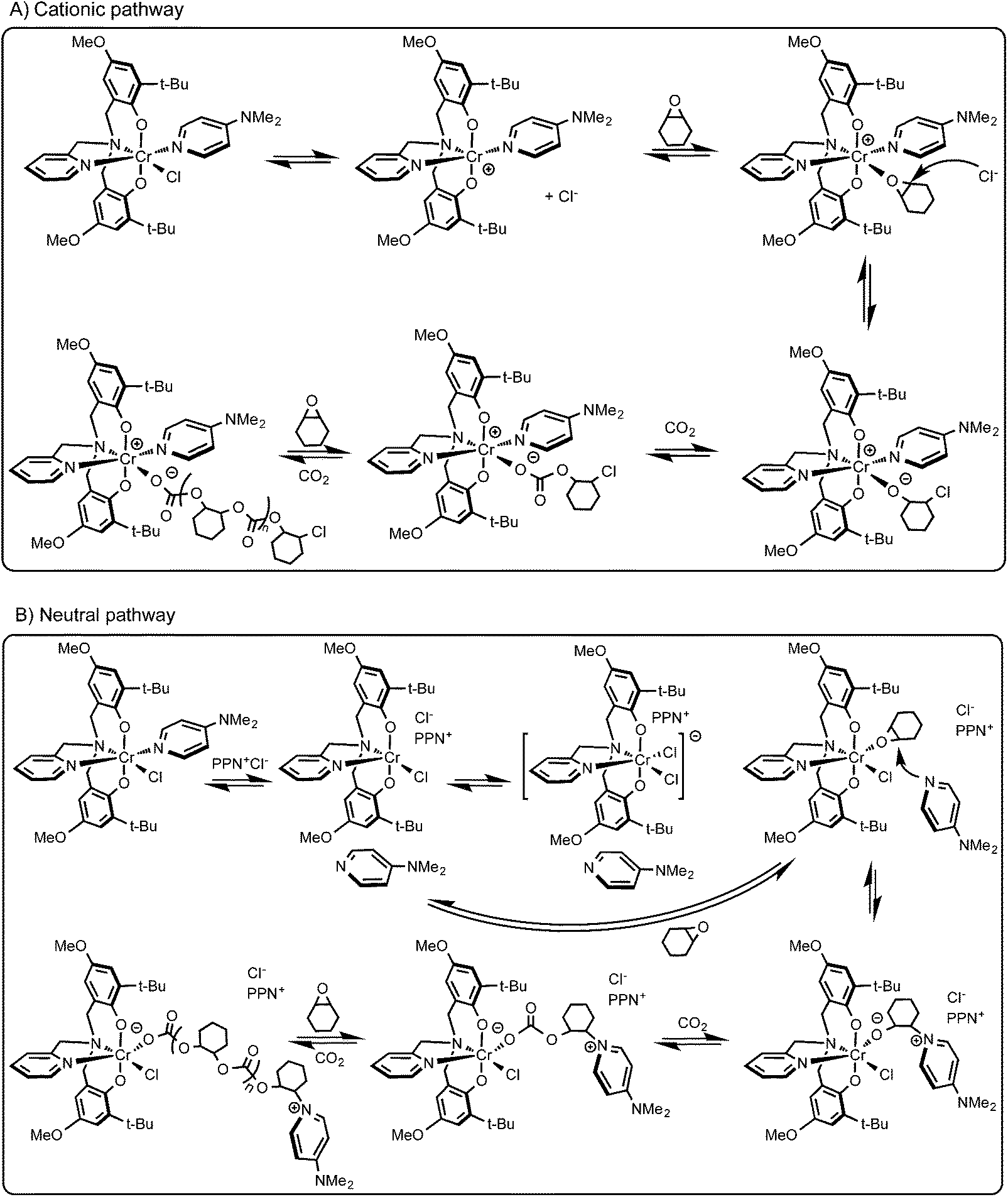 | ||
| Scheme 3 (A) Proposed initiation pathway for the copolymerization of cyclohexene oxide and carbon dioxide with 1·DMAP and (B) with 1·DMAP/PPNCl. | ||
Conclusions
Chromium(III) complexes 1·THF and 1·DMAP show improved catalytic activity over our previously reported complex, CrCl[O2NN′]BuBuPy, in the copolymerization of cyclohexene oxide and CO2. End group analysis of the polymers obtained by 1·THF and 1·DMAP indicates that ring opening is most probably initiated by the anion of the co-catalyst except when 1·DMAP is used with PPNCl, in which case DMAP dissociation is observed, leading to its role as the epoxide ring-opening nucleophile as shown by polymer end group analysis by MALDI-TOF MS. Based on this end group analysis, we propose that CO2/epoxide copolymerization catalyzed by chromium (III) amine-bis(phenolate) complexes is initiated by epoxide ring opening by an external nucleophile, that is, an intermolecular pathway.Acknowledgements
Financial support was provided by the Canada Foundation for Innovation (CFI), Newfoundland and Labrador Research Development Corporation, the Natural Sciences and Engineering Research Council (NSERC) of Canada and Memorial University of Newfoundland.References
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- A. M. Nelson and T. E. Long, Polym. Int., 2012, 61, 1485–1491 CrossRef CAS PubMed.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC.
- W. B. Kim, U. A. Joshi and J. S. Lee, Ind. Eng. Chem. Res., 2004, 43, 1897–1914 CrossRef CAS.
- D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS PubMed.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- M. Tolinski, Plastics and Sustainability: Towards a Peaceful Coexistence Between Bio-Based and Fossil Fuel-Based Plastics, Wiley-Scrivener, Hoboken, NJ, USA, 2011 Search PubMed.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- K. Nakano, T. Hiyama and K. Nozaki, Chem. Commun., 2005, 1871–1873 RSC.
- H. Sugimoto and A. Ogawa, React. Funct. Polym., 2007, 67, 1277–1283 CrossRef CAS PubMed.
- N. Ikpo, L. N. Saunders, J. L. Walsh, J. M. B. Smith, L. N. Dawe and F. M. Kerton, Eur. J. Inorg. Chem., 2011, 5347–5359 CrossRef CAS PubMed.
- D. J. Darensbourg and D. R. Billodeaux, Inorg. Chem., 2005, 44, 1433–1442 CrossRef CAS PubMed.
- X. B. Lu, L. Shi, Y. M. Wang, R. Zhang, Y. J. Zhang, X. J. Peng, Z. C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664–1674 CrossRef CAS PubMed.
- G. P. Wu, S. H. Wei, W. M. Ren, X. B. Lu, T. Q. Xu and D. J. Darensbourg, J. Am. Chem. Soc., 2011, 133, 15191–15199 CrossRef CAS PubMed.
- X. B. Lu, W. M. Ren and G. P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed.
- J. Yoo, S. J. Na, H. C. Park, A. Cyriac and B. Y. Lee, Dalton Trans., 2010, 39, 2622–2630 RSC.
- K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC.
- C. E. Anderson, S. I. Vagin, W. Xia, H. Jin and B. Rieger, Macromolecules, 2012, 45, 6840–6849 CrossRef CAS.
- L. N. Saunders, N. Ikpo, C. F. Petten, U. K. Das, L. N. Dawe, C. M. Kozak and F. M. Kerton, Catal. Commun., 2012, 18, 165–167 CrossRef CAS PubMed.
- M. Reiter, P. T. Altenbuchner, S. Kissling, E. Herdtweck and B. Rieger, Eur. J. Inorg. Chem., 2015, 2015, 1766–1774 CrossRef CAS PubMed.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083–3090 RSC.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972–6980 CrossRef CAS.
- D. J. Darensbourg and A. I. Moncada, Inorg. Chem., 2008, 47, 10000–10008 CrossRef CAS PubMed.
- P. Chen, M. H. Chisholm, J. C. Gallucci, X. Zhang and Z. Zhou, Inorg. Chem., 2005, 44, 2588–2595 CrossRef CAS PubMed.
- R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498–11499 CrossRef CAS.
- S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS.
- D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335–6342 CrossRef CAS PubMed.
- D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS PubMed.
- R. Eberhardt, M. Allmendinger and B. Rieger, Macromol. Rapid Commun., 2003, 24, 194–196 CrossRef CAS PubMed.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers, C. C. Fang, D. R. Billodeaux and J. H. Reibenspies, Inorg. Chem., 2004, 43, 6024–6034 CrossRef CAS PubMed.
- D. J. Darensbourg and R. M. Mackiewicz, J. Am. Chem. Soc., 2005, 127, 14026–14038 CrossRef CAS PubMed.
- D. J. Darensbourg, R. M. Mackiewicz and D. R. Billodeaux, Organometallics, 2005, 24, 144–148 CrossRef CAS.
- B. Li, R. Zhang and X. B. Lu, Macromolecules, 2007, 40, 2303–2307 CrossRef CAS.
- X. Q. Xu, C. M. Wang, H. R. Li, Y. Wang, W. L. Sun and Z. Q. Shen, Polymer, 2007, 48, 3921–3924 CrossRef CAS PubMed.
- B. Li, G. P. Wu, W. M. Ren, Y. M. Wang, D. Y. Rao and X. B. Lu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6102–6113 CrossRef CAS PubMed.
- D. J. Darensbourg and S. B. Fitch, Inorg. Chem., 2009, 48, 8668–8677 CrossRef CAS PubMed.
- D. J. Darensbourg, M. Ulusoy, O. Karroonnirum, R. R. Poland, J. H. Reibenspies and B. Çetinkaya, Macromolecules, 2009, 42, 6992–6998 CrossRef CAS.
- D. J. Darensbourg, R. R. Poland and A. L. Strickland, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 127–133 CrossRef CAS PubMed.
- L. P. Guo, C. M. Wang, W. J. Zhao, H. R. Li, W. L. Sun and Z. Q. Shen, Dalton Trans., 2009, 5406–5410 RSC.
- D. Adhikari, S. T. Nguyen and M.-H. Baik, Chem. Commun., 2014, 50, 2676–2678 RSC.
- D. Y. Rao, B. Li, R. Zhang, H. Wang and X. B. Lu, Inorg. Chem., 2009, 48, 2830–2836 CrossRef CAS PubMed.
- Y. L. Xiao, Z. Wang and K. L. Ding, Macromolecules, 2006, 39, 128–137 CrossRef CAS.
- G. A. Luinstra, G. R. Haas, F. Molnar, V. Bernhart, R. Eberhardt and B. Rieger, Chem. – Eur. J., 2005, 11, 6298–6314 CrossRef CAS PubMed.
- M. H. Chisholm and Z. P. Zhou, J. Am. Chem. Soc., 2004, 126, 11030–11039 CrossRef CAS PubMed.
- M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 CrossRef CAS PubMed.
- R. K. Dean, L. N. Dawe and C. M. Kozak, Inorg. Chem., 2012, 51, 9095–9103 CrossRef CAS PubMed.
- R. K. Dean, K. Devaine-Pressing, L. N. Dawe and C. M. Kozak, Dalton Trans., 2013, 42, 9233–9244 RSC.
- H. Chen, L. N. Dawe and C. M. Kozak, Catal. Sci. Technol., 2014, 4, 1547–1555 CAS.
- C. M. Kozak, A. M. Woods, C. S. Bottaro, K. Devaine-Pressing and K. Ni, Faraday Discuss., 2015 Search PubMed , In press.
- S. Menage, G. Gellon, J.-L. Pierre, D. Zurita and E. Saint-Aman, Bull. Soc. Chim. Fr., 1997, 134, 785–791 CAS.
- K. Devaine-Pressing, J. H. Lehr, M. E. Pratt, L. N. Dawe, A. A. Sarjeant and C. M. Kozak, Dalton Trans., 2015, 44, 12365–12375 RSC.
- S. Heidari, E. Safaei, A. Wojtczak, P. Cotic and A. Kozakiewicz, Polyhedron, 2013, 55, 109–116 CrossRef CAS PubMed.
- L. M. Alcazar-Roman, B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2003, 3082–3087 RSC.
- R. K. Dean, S. L. Granville, L. N. Dawe, A. Decken, K. M. Hattenhauer and C. M. Kozak, Dalton Trans., 2010, 39, 548–559 RSC.
- D. J. Darensbourg, A. I. Moncada, W. Choi and J. H. Reibenspies, J. Am. Chem. Soc., 2008, 130, 6523–6533 CrossRef CAS PubMed.
- S. Klaus, S. I. Vagin, M. W. Lehenmeier, P. Deglmann, A. K. Brym and B. Rieger, Macromolecules, 2011, 44, 9508–9516 CrossRef CAS.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- D. J. Darensbourg and A. D. Yeung, Polym. Chem., 2015, 6, 1103–1117 RSC.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 CAS.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, MALDI-TOF MS, UV-Vis and IR spectra of 1·THF, 2·THF and 1·DMAP, representative 1H and 13C NMR and MALDI-TOF mass spectra of PCHC, and crystallographic information files (CIF) for 1·THF, 2·THF and 1·DMAP. CCDC 1408672–1408674. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5py01028d |
| ‡ Current address: Department of Chemistry and Biochemistry, Wilfrid Laurier University, 75 University Ave. W. Waterloo, Ontario, N2L 3C5, Canada. |
| This journal is © The Royal Society of Chemistry 2015 |