
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyamide 6/silica hybrid materials by a coupled polymerization reaction†
L.
Kaßner
a,
K.
Nagel
a,
R.-E.
Grützner
b,
M.
Korb‡
c,
T.
Rüffer‡
c,
H.
Lang‡
c and
S.
Spange
*a
aPolymer Chemistry, Technische Universität Chemnitz, 09107 Chemnitz, Germany. E-mail: stefan.spange@chemie.tu-chemnitz.de
bBASF SE, Carl-Bosch-Str. 38, 67063 Ludwigshafen am Rhein, Germany
cInorganic Chemistry, Technische Universität Chemnitz, 09107 Chemnitz, Germany
First published on 10th July 2015
Abstract
Polyamide 6/SiO2 hybrid materials were produced by a coupled polymerization reaction of three monomeric components namely 1,1′,1′′,1′′′-silanetetrayltetrakis-(azepan-2-one) (Si(ε-CL)4), 6-aminocaproic acid (ε-ACA) and ε-caprolactam (ε-CL) within one process. Si(ε-CL)4 together with ε-ACA has been found suitable as a precursor monomer for the silica and PA6 components. The accurate adjustment of the molar ratio of both components, as well as the combination of the overall process for producing the polyamide 6/SiO2 hybrid material with the hydrolytic ring opening polymerization of ε-caprolactam is of great importance to achieve homogeneous products with a low extractable content. Water in comparison with ε-ACA has been found unsuitable as an oxygen source to produce uniformly distributed silica. The procedure was carried out in a commercial laboratory autoclave at 8 bar initial pressure. The molecular structure and morphology of the hybrid materials have been investigated by solid state 29Si and 13C NMR spectroscopy, DSC and FTIR spectroscopy and electron microscopy measurements.
Introduction
Polyamide 6 (PA6) is one of the most important engineering plastics, due to the combination of high mechanical and thermal stability, chemical resistance and processability.1 Its physical properties can be further improved by combination with other components, e.g. layered silicates,2–4 glass or carbon fibres5–8 and metal oxides,9–12 to fabricate hybrid materials or composites. These materials are suitable for several applications, especially in the automobile industry, for example as the inlet for fuel systems, wheel trims and engine covers.13 PA6/silica hybrid materials are of special interest because incorporation of SiO2 into PA6 improves mechanical properties such as hardness and elastic modulus.14,15 Furthermore, silica is non-toxic and colorless and nanoparticles as well as hybrid materials could be obtained by the sol–gel process under mild reaction conditions.Strategies for producing PA6/SiO2 hybrid materials can be classified into different categories. In the simplest way, both components, the preformed SiO2 and PA6, are mixed together by extrusion, melting or by another appropriate procedure.16–21 More elegant ways use the in situ formation of the SiO2 component, i.e. by sol–gel processing22 or the in situ polymerization of ε-caprolactam in the presence of preformed silica particles.15,23–26 The grafting of PA6 on surface functionalized SiO2 particles is also a suitable route to fabricate polymer/SiO2 hybrid materials.27,28 As an additional way, the simultaneous formation of both the polymer components within one procedure is known.14,29 So far, there has been no report on the simultaneous synthesis of PA6 and SiO2 from a combined monomer within one coupled polymerization reaction.
In the literature, the terms composite and hybrid material have been used in different ways for these types of materials.16 Composite materials are mixtures of both components on a length scale of 100 nm to 10 m, whereas inorganic/organic hybrid materials are combined at the molecular level up to several nm. The challenge is to create nanostructured hybrid materials, which show a stronger improvement in physical properties than macroscopic mixtures even at low filler contents of a few weight percent.
To achieve a nanostructured hybrid material, the simultaneous formation of both components in vicinity is required.30 Therefore, monomers have been constructed in such a way that two polymers are formed from one single source monomer. This strategy has been established for monomers which contain two different moieties suitable for polymerization, one for chain polymerization and another one, i.e. for sol gel processes.31–34 However, in this case both groups do polymerize independently of each other. For step-growth polymerization processes, the twin polymerization has been established as an elegant route to fabricate nanostructured inorganic/organic hybrid materials. The formation of both polymers occurs mechanistically coupled, which is the reason for the smooth nanostructure formation.30,35,36 Monomers which are used in twin polymerization are called twin monomers.
The objective of this publication is the development of a coupled polymerization procedure for the synthesis of PA6/SiO2 hybrid materials within one process. Therefore, three different reactants, namely 1,1′,1′′,1′′′-silanetetrayltetrakis-(azepan-2-one) (Si(ε-CL)4), 6-aminocaproic acid (ε-ACA) and ε-caprolactam (ε-CL) are used in different ratios to adjust the amount of the formed silica. The combined monomer Si(ε-CL)41 which contains the ε-caprolactam moiety covalently bound via the N-atom to the silicon is used as a precursor for the formation of silica as well as polyamide 6. Basically for the purpose of PA6 production, cyclohexanone oxime (CHO) as an industrial precursor molecule for ε-CL also seems eligible as a component in monomer 2 Si(CHO)4 (Scheme 1). However, in this case the BECKMANN rearrangement of this monomer would be an essential step before polymerization takes place.
 | ||
| Scheme 1 Molecular structures of silicon monomers derived from ε-caprolactam (monomer 1) and cyclohexanone oxime (monomer 2). For synthetic procedure see the Experimental part. | ||
Both types of silicon monomers are investigated for PA6/SiO2 hybrid material synthesis. It must be mentioned that the single polymerization of 1 or 2 is unsuitable to produce PA6/SiO2 as the overall stoichiometry is not complied. Water is essential as a co-component (Scheme 2). Therefore, it must be emphasized that monomers 1 and 2 are not ideal twin monomers but they are related to deficient twin monomers due to the possible mechanistically coupled formation of the inorganic and organic polymers. The polymerization process as shown in Scheme 2 is related to the apparent twin polymerization.37
 | ||
| Scheme 2 Strategy for fabrication of PA6/SiO2 hybrid materials by coupling the reaction of Si(ε-CL)41 with the polycondensation of ε-ACA and the hydrolytic ring opening polymerization of ε-CL. | ||
The challenge of PA6/SiO2 hybrid material synthesis according to Scheme 2 is the improvement of the operative coupling of several reactions. One crucial aspect is how the water equivalent can be realized among the occurring processes. Water can be used directly or from a suitable source like the polycondensation of 6-aminocaproic acid. ε-ACA seems to be eligible, because it polymerizes to PA6 and initiates hydrolytic lactam polymerization. The coupling of the water delivering and the water consuming reaction (eqn (1) and (2) in Scheme 2) to produce PA6/SiO2 has been explored as a function of the ratio of 1 and ε-ACA. As a third reaction component ε-caprolactam has been found suitable for a good homogenization of the reaction melt (according to eqn (3) in Scheme 2). By variation of reactant concentrations different SiO2 amounts are adjustable. The reactions have been carried out using a typical procedure, which is suitable to fabricate PA6 from ε-CL.
Experimental
Materials and methods
ε-Caprolactam (>99%) and silicon tetrachloride (99%) were purchased from Sigma Aldrich. 6-Aminocaproic acid (99%) was purchased from Alfa Aesar. Cyclohexanone oxime (97%) was purchased from Acros. Toluene was dried by standard methods and distilled before use under an argon atmosphere. CDCl3 was dried with molecular sieves 4 Å and stored under argon.Liquid state 1H NMR (250.1 MHz), 13C NMR (62.9 MHz) and 29Si NMR (49.7 MHz) spectra were recorded with a Bruker Avance 250 NMR spectrometer. The residual signal of the solvent CDCl3 was used as an internal standard (δ = 7.26 ppm).
Solid state NMR spectra were recorded using a Bruker Digital Avance 400 spectrometer, equipped with double tuned probes capable of MAS (magic angle spinning). 13C{1H}-CP-MAS NMR spectra were recorded at 100.6 MHz using 3.2 mm standard zirconium oxide rotors spinning at 15 kHz. 29Si{1H}-CP-MAS NMR spectra were recorded at 79.5 MHz with a sample spin rate of 12 kHz. Cross polarization with a contact time of 3 ms was used to enhance sensitivity. The recycle delay was 6 s. The spectra were referenced externally to tetrakis(trimethylsilyl)silane (1H, δ = 0.27 ppm; 13C, δ = 3.55 ppm; 29Si, δ = −9.5 ppm). The spectra were recorded by 1H decoupling using the TPPM pulse sequence.
DSC measurements were performed by using a DSC 1 (Mettler Toledo). All measurements were performed in 40 μL aluminium pans and a N2-flow of 50 mL min−1 in a temperature range of 25–250 °C with a heating rate of 10 K min−1. In cyclic measurements with two heating segments and one cooling segment, the highest, respectively lowest temperature was held for 2 min.
ATR-FTIR spectra were recorded with a Golden Gate ATR accessory from LOT-Oriel GmbH & Co. KG, Darmstadt, using a BioRad FT-IR 165 spectrometer (Bio-Rad Laboratories, Philadelphia, PA, USA).
The molecular weight distribution of PA6 was determined by SEC at BASF SE using an App_P apparatus with SDV as the stationary phase, and the column temperature was 65 °C. 1,1,1,3,3,3-Hexafluoro-2-propanol containing 0.05% of potassium trifluoroacetate was used as the eluent with an elution rate of 1 mL min−1 and the sample concentration was 1.5 mg mL−1. PMMAs of narrow and defined molecular weights were used as calibration standards.
The transmission electron microscopy (TEM) was carried out using the Libra 120 (120 kV) transmission electron microscope of Zeiss by TEM Laboratory, BASF SE. Before the measurements, ultrathin cuts were performed by using a Leica ultramicrotome EM UC7 and a cryogenic chamber Leica EM FC7 (80–120 nm).
Electron microscopy images were taken by using an instrument from type Nova NanoSEM 200 of FEI Company after sputtering with platinum (TU Chemnitz, Laboratory of Solid Surfaces Analysis).
Single crystal X-ray structural analyses were performed with an Oxford Gemini diffractometer with Cu-Kα-radiation (λ = 154.184 pm) at TU Chemnitz, Laboratories of Inorganic Chemistry.
Quantitative elemental analyses of the elements C, H und N were performed with varioMICRO CHNS from Elementar Analysensysteme GmbH (TU Chemnitz, Laboratory of Organic Chemistry).
Thermogravimetric measurements were realized on a Thermogravimetric Analyzer 7 (TGA 7), of Perkin Elmer Company (TU Chemnitz, Laboratory of Physical Chemistry). First the samples were heated from 30 to 700 °C with a heating rate of 20 K min−1 under a constant helium flow. Upon further heating to 900 °C the gas flow switched to air. This temperature was held for another 30 min.
Synthesis of monomers 1 and 2
In a typical procedure, either ε-caprolactam or cyclohexanone oxime (20.0 g, 0.177 mol) was dissolved under stirring in anhydrous toluene (300 mL). Triethylamine (20.2 g, 0.200 mol) was added in slight excess in a single portion. This solution was cooled in a water bath and SiCl4 (7.5 g, 0.044 mol), dissolved in 100 mL anhydrous toluene, was added slowly through a dropping funnel under vigorous stirring. Immediately a white precipitate from triethylammonium chloride was observed. Subsequently, the solution was stirred at room temperature for 16 h. Triethylammonium chloride was separated by filtration. After the removal of the solvent under vacuum, a white to beige solid was obtained.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 922 (Si–N). Found: C, 60.17; H, 9.23; N, 11.64. Calc. C24H40N4O4Si: C, 60.47; H, 8.46; N, 11.75.
N), 1447 (CH2), 940 (Si–O). Found: C, 59.51; H, 8.49; N, 11.53. Calc. C24H40N4O4Si: C, 60.47; H, 8.46; N, 11.75.
O), 922 (Si–N). Found: C, 60.17; H, 9.23; N, 11.64. Calc. C24H40N4O4Si: C, 60.47; H, 8.46; N, 11.75.
N), 1447 (CH2), 940 (Si–O). Found: C, 59.51; H, 8.49; N, 11.53. Calc. C24H40N4O4Si: C, 60.47; H, 8.46; N, 11.75.
Synthesis of hybrid materials
Composites were synthesized using a high pressure lab autoclave of Berghof company. A Teflon beaker was used as an insert. Before heating to a reaction temperature of 230 °C, the reactants ε-ACA, ε-CL and 1 were filled in the autoclave in the specified molar ratios (Table 1) at room temperature and then it was purged with argon up to 8 bar and afterwards relaxed to atmospheric pressure thrice. The reaction took place under 8 bar initial pressure (argon) for at least 210 min, including a ca. 60 min heating phase. During the reaction, pressure increased to approximately 14 bar. 15 min before termination of the reaction time, pressure was released slowly to start the post-condensation phase. After cooling down to ambient temperature, the white to beige-colored monolithic samples were crushed into smaller pieces. To remove residual monomers or oligomers, the hybrid materials were purified by Soxhlet extraction for 48 h with methanol and then dried in a vacuum oven at 40 °C to a constant mass. The reference was synthesized according to the same polymerization procedure except for the addition of 1.| No. | SiO2 amount | Molar ratios of reactants | Extractables (48 h, MeOH) | ||
|---|---|---|---|---|---|
| ε-ACA | Si(ε-CL)41 | ε-CL | |||
| R | Reference (pure PA6) | 1 | 0 | 4.4 | 10.0% |
| P1_1 | 1 wt% SiO2 | 10 | 1 | 38.6 | 13.9% |
| P1_2 | 1 wt% SiO2 | 7.2 | 1 | 41.4 | 18.7% |
| P1_3 | 1 wt% SiO2 | 4 | 1 | 43.5 | 31.9% |
| P2 | 2 wt% SiO2 | 4.3 | 1 | 18 | 13.9% |
| P5_1 | 5 wt% SiO2 | 4 | 1 | 1.2 | 11.5% |
| P5_2 | 5 wt% SiO2 | 2.4 | 1 | 3.7 | 21.0% |
To increase the average molecular weight, a post-condensation reaction can be carried out at 200 °C and 5 mbar for 12 h afterwards.
The hybrid material synthesis procedure is reproducible several times. For further information of the reproducibility by the example of P2, see the ESI† (Fig. S1 for ATR-FTIR spectra, Fig. S2 for DSC traces and Table S1 for the amount of extractables and quantitative elemental analysis).
Results and discussion
Both monomers 1 and 2 are not described accurately in the literature. 1 is only briefly mentioned in a patent.The synthesis of 1 and 2 has been performed by the reaction of SiCl4 with ε-CL or cyclohexanone oxime using an appropriate amine base to bind HCl (see the Experimental part). Both new monomers have been characterized by spectroscopic methods and single X-ray structure analysis. Fig. 1 shows the molecular structures of the synthesized monomers. For bond lengths and angles, as well as data acquisition details see the ESI† (Tables S2–S4). Both molecules have a S4-symmetry with tetrahedral geometry at the silicon atom. Bonding to silicon causes planarization of the nitrogen atoms (sum of angles at the nitrogen atoms ∼359°).38–41 Conspicuous in 1 is the small distance of 2.77 Å between silicon and oxygen in the solid state, which is shorter than the sum of the van der Waals radii of the Si and O atoms (3.62 Å).42 Comparable Si–O and Si–N distances were found by Rong and Wollenweber et al. who discussed comparable structures with tosyl groups as substituents at the nitrogen atoms in terms of a [4 + 4] octacoordination with a tetrahedral SiN4 core and four oxygen atoms in the “outer sphere”, capping the tetrahedral planes.43–45 The chemical shifts of the 29Si NMR signals of monomer 1 in the solid state and the solution state are similar which indicates the same bonding motif (Fig. S3 in the ESI†). The solid state 29Si NMR signal appears at δ = −43.0 ppm, which could be explained by the electron withdrawing effect of the carbonyl groups next to the nitrogen atom. Therefore, no strong effect of an octacoordination could be observed by 29Si NMR spectroscopy. Consequently, it is unclear whether the short Si–O distances are due to an additional stabilization or just steric reasons.
 | ||
| Fig. 1 ORTEP drawing of monomers 1 (left) and 2 (right), with ellipsoids drawn at the 50% probability level. Hydrogens are omitted for clarity. | ||
All attempts to polymerize 2 to any PA6 hybrid materials failed. It remains intractable for BECKMANN rearrangement towards the ε-CL component. Neither acid treatment nor heating in different melt compositions have been successful (Table S5 in the ESI†). Therefore, solely 1 was further investigated for hybrid material synthesis.
In spite of the fact that water is suitable to induce the thermal polymerization of ε-CL, the use of free water as the source for the synthetic procedure was not favorable because of a lower conversion or discoloration of the products (Table S6 in the ESI†). The amino acid ε-ACA as a water source has been found to be the most convenient way.
In preliminary studies, the reaction of 1 with ε-ACA has been studied by DSC measurements to optimize the polymerization temperature for the overall process (Fig. 2). Monomer 1 shows a complex temperature dependence. Endothermic melting of ε-ACA and its polymerization under water release starts at temperatures above 209 °C. The combination of ε-ACA with 1 leads to a decrease of the polymerization temperature below 200 °C. In addition, an increasing ε-ACA amount requires a higher reaction temperature. In spite of the lower polymerization temperature in the mixture of ε-ACA with 1, a processing temperature of 230 °C was chosen for hybrid material synthesis because of the high melting point of PA6 (220 °C).
 | ||
| Fig. 2 DSC curves of mixtures of ε-ACA with 1 in different molar ratios at a heating rate of 10 K min−1. | ||
The overall process for synthesizing PA6/SiO2 hybrid materials has been carried out in a high pressure autoclave suitable for PA6 synthesis (Experimental part). It must be mentioned that the order of reactant addition can be varied. Pre-polymerization of the reactants ε-ACA and ε-CL and the subsequent addition of 1 is possible, but the products show discoloration due to their contact with air while heating. Reaction time and polymerization temperature are important for homogeneity and extractable amounts, so that for a better comparability all experiments are performed under constant reaction conditions in an autoclave. All reactants are inserted at the beginning of the heating period and the reactions are performed under an inert atmosphere at a temperature of 230 °C for 3.5 h including the heating and post-condensation phases. For further information see Table S7 in the ESI†.
The observed extractables of the hybrid materials amount to 10–32% and depend on the molar ratios of reactants (Table 1). Particularly for the samples P1 and P5 a decreasing amount of extractables can be detected with a higher ε-ACA ratio.
The resulting PA6/SiO2 hybrid materials are homogeneous solid materials (Fig. 3). Primary monolithic products were received but also granules can be fabricated. Furthermore, the resulting thermoplastic materials can be extruded to films.
 | ||
| Fig. 3 Images of PA6/SiO2 hybrid materials according to sample P2 (Experimental part, Table 1) as monolith, granules and film. | ||
The TEM images of P2 show SiO2 agglomerates with primary particles of 35–60 nm in size (Fig. 4).
 | ||
| Fig. 4 TEM images of a monolithic sample of P2 after freezing ultrathin cuts in different magnifications. | ||
The homogeneity of samples as well as the agglomeration tendency are affected by the containing SiO2 amount as can be seen from electron microscopy images (Table 2 and Fig. S4–S9 in the ESI†). The higher the SiO2 content which is formed during polymerization, the more agglomeration of inorganic particles can be observed. The resulting SiO2 agglomerates build particles with different shapes, for example up to 30 μm long needle-like (P1_2) or shell-like particles around hybrid materials (P5_1) but often spherical as can be seen from the other examples. With the highest SiO2 content (P5_2) particles larger than 100 μm are obtained. Furthermore, the molar ratios of reactants seem to have an influence on the agglomeration tendency. Hence, for example, in an experiment with constant SiO2 amounts (P1 and P5) an increasing ε-ACA ratio causes a decrease of the SiO2 particle size. The obtained wide particle size distribution could have an influence on mechanical behavior but this is a part of further work.
| Sample (SiO2 amount) | Electron microscopy picture | |
|---|---|---|
| a The obtained element fluorine is due to the mechanical crushing of the composite material after the polymerization process in the used Teflon beaker. | ||
| P1_1 (1 wt%) |

|
|
| Molar ratio | ||
| ε-ACA | Si(ε-CL)4 | |
| 10 | 1 | |
| P1_2 (1 wt%) |

|
|
| Molar ratio | ||
| ε-ACA | Si(ε-CL)4 | |
| 7.2 | 1 | |
| P1_3 (1 wt%) |

|
|
| Molar ratio | ||
| ε-ACA | Si(ε-CL)4 | |
| 4 | 1 | |
| P2 (2 wt%) |

|
|
| Molar ratio | ||
| ε-ACA | Si(ε-CL)4 | |
| 4.3 | 1 | |
| P5_1 (5 wt%) |

|
|
| Molar ratio | ||
| ε-ACA | Si(ε-CL)4 | |
| 4 | 1 | |
| P5_2 (5 wt%) |

|
|
| Molar ratio | ||
| ε-ACA | Si(ε-CL)4 | |
| 2.4 | 1 | |
The solid state 13C and 29Si NMR spectra of the hybrid material P2 evidence the molecular structure which relates to the PA6/SiO2 (Fig. 5). The solid state 13C NMR spectrum is in agreement with the literature data of pure PA6 high in α-crystallinity.46 The solid state 29Si NMR spectrum of the hybrid material shows Q3 and Q4 signals, which indicate Si atoms bound to 3 or 4 other Si atoms over siloxane-bridges. It is not possible to distinguish if the Q3 signal is caused by Si–OH or Si–OC groups. Therefore, bonding of carboxylic acid groups to silicon cannot be excluded.
 | ||
| Fig. 5 Solid state 13C{1H}-CP-MAS (above) and 29Si{1H}-CP-MAS NMR spectra (below) of a 2 wt% SiO2 sample P2. | ||
DSC traces of the samples R, P1_2, P2 and P5_1 show the typical thermal behavior of thermoplastic PA6 with a melting point around 220 °C (Fig. 6).
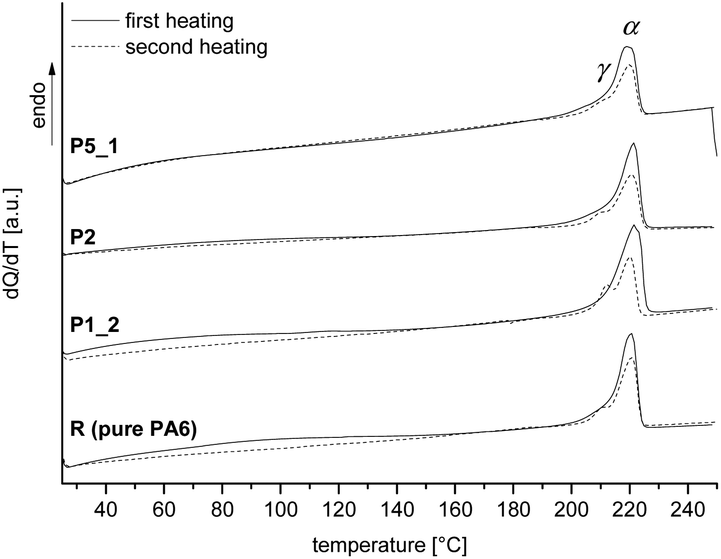 | ||
| Fig. 6 First and second heating of a cyclic DSC measurement of pure polyamide 6 in comparison with hybrid materials with 1, 2 and 5 wt% of SiO2 (extracted samples); signals of α- and γ-crystal modification are marked. | ||
As already described for the solid state 13C NMR spectra, the crystallinity of PA6 is predominated by α-modification. Variation of the SiO2 amount has no influence on the melting or crystallization temperature and the crystal modification. The degree of crystallization with values between 30 and 40% is independent of the SiO2 content and indicates the presence of crystalline and amorphous regions in the organic polymer. For further information see Table S8 (ESI†). Independent of the SiO2 amount, all samples show similar thermal decomposition behavior (thermogravimetric analysis, Fig. S10, ESI†).
In agreement with solid state 13C NMR and 29Si spectra and DSC measurements, ATR-FTIR spectra indicate typical bands for the polyamide 6 like amide I band at 1636 cm−1 and the amide II band at 1536 cm−1 as well as N–H at 3295 cm−1. Furthermore, the typical Si–O stretching vibration at 1073 cm−1 for SiO2 is observed (see Fig. 7). Additionally, the ATR-FTIR spectra give information about the crystallization behavior of PA6. The position of the amide V band at 690 cm−1 and the amide VI band at 580 cm−1 indicate crystallization mainly in α-modification as evidenced from DSC and solid state 13C NMR measurements, too. The amorphous polymer shows broad signals, which can also be observed. Crystallization in the γ-modification would induce bands at 712 cm−1 and 625 cm−1 for amides V and VI.47–49
 | ||
| Fig. 7 ATR-FTIR spectra of hybrid materials with 1, 2 and 5 wt% of SiO2 in comparison with pure polyamide 6 (extracted samples); above: full wavenumber range; below: fingerprint region. | ||
For additional characterization of the obtained hybrid materials, the molar mass of the obtained PA6 and the influence of post-condensation on the molecular weights and the polydispersity index (PDI) were investigated by size exclusion chromatography (SEC, Table 3, Fig. 8 and S11 in the ESI†). All polymers show a monomodal distribution of the molar mass. The SiO2 content seems to have a slight influence on the molecular weight and its distribution. Therefore, Mw as well as Mn and PDI increase with the increasing SiO2 amount that occurs with an increasing ratio of ε-ACA in comparison with the other reactants, namely 1 and ε-caprolactam. Generally, high amino acid concentrations lead to an increasing reaction rate of polycondensation reaction and therefore higher chain lengths and higher conversion. For example, a decrease in the ε-ACA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio for P1_1 to P1_3 causes a decrease in the obtained molar mass due to a reduced conversion of the reactants. A diametrical effect is determined for P5_1 and P5_2. It must be mentioned that for these cases a higher ε-ACA content leads to lower chain lengths due to a decrease of the ε-caprolactam unit amount and a higher concentration of amino or carboxylic acid end groups.
:1 ratio for P1_1 to P1_3 causes a decrease in the obtained molar mass due to a reduced conversion of the reactants. A diametrical effect is determined for P5_1 and P5_2. It must be mentioned that for these cases a higher ε-ACA content leads to lower chain lengths due to a decrease of the ε-caprolactam unit amount and a higher concentration of amino or carboxylic acid end groups.
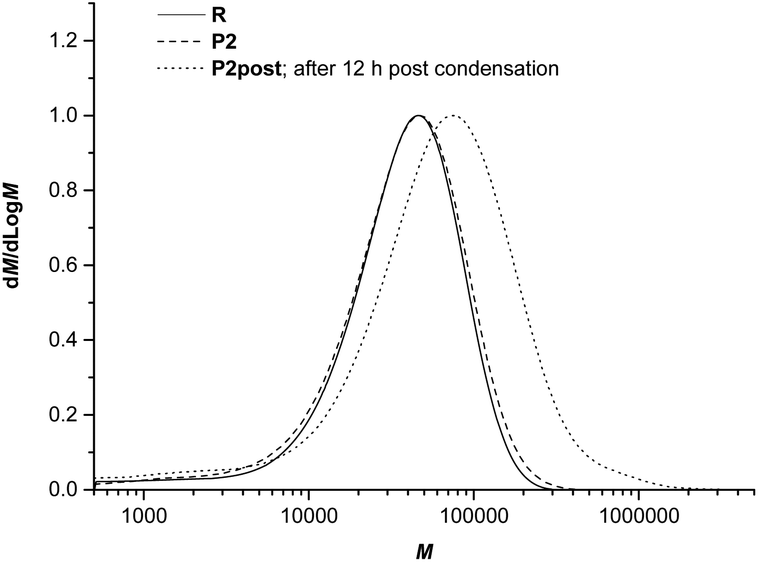 | ||
| Fig. 8 SEC profiles of hybrid material P2 with 2 wt% of SiO2 before and after post-condensation at 200 °C and 5 mbar for 12 h in comparison with PA6 as the reference; extracted samples (48 h, MeOH); normalized to peak height. | ||
| Sample | M w | M n | PDI |
|---|---|---|---|
| Reference (pure PA6) | 48000 |
17500 |
2.7 |
| P1_1 | 54000 |
18900 |
2.9 |
| P1_2 | 46200 |
17500 |
2.6 |
| P1_3 | 35800 |
14400 |
2.5 |
| P2; 2 wt% SiO2 | 49700 |
17400 |
2.8 |
| Before post-condensation | |||
| P2post; 2 wt% SiO2 | 104000 |
19200 |
5.4 |
| After post-condensation | |||
| P5_1 | 62600 |
19900 |
3.1 |
| P5_2 | 80100 |
22900 |
3.5 |
As expected, the post-condensation reaction of P2 at 200 °C leads to a much higher Mw. Additionally, a broader distribution of the molar masses with a higher PDI after post-condensation is observed. This is due to the unreached equilibrium of the post-condensation reaction. Additionally, the water content in the polymer grains decreases from the inner to the outer sphere. Therefore a different molecular weight distribution over the polymer grain is ascertained.50 In some cases, a microscopic nonuniformity of the solid polymers, especially a distribution of crystallite sizes, is discussed as a further reason.51
Kinetic and thermodynamic investigations of the presented polymerization type are still under study.
Conclusions
In this study, polyamide 6/SiO2 hybrid materials were produced by a coupled polymerization reaction of three monomeric components namely 1,1′,1′′,1′′′-silanetetrayltetrakis-(azepan-2-one), 6-aminocaproic acid and ε-caprolactam within one process. The amount of SiO2 is tunable by variation of the reactant stoichiometry, whereas the ratio of 1,1′,1′′,1′′′-silanetetrayltetrakis-(azepan-2-one) to 6-aminocaproic acid is of great importance for high conversion and homogeneity.The investigations have shown that the filler has no significant effect on thermal properties. The average molecular weight slightly rises with the increasing SiO2 content. Furthermore, the latter increased during the post-condensation reaction and, additionally, a broader PDI was determined. Examination of the crystallization behavior of the obtained PA6 by NMR as well as FTIR spectroscopy and DSC show a predominated α-crystallinity and amorphous regions. The unfavorable aspect is the increasing agglomeration tendency of the primary nanoparticles with higher SiO2 amounts. In contrast to conventional in situ procedures or the compounding for the synthesis of PA6/filler hybrid materials, the preceding modification of the filler and negative effects of the modifier, like the thermal degradation,52,53 can be omitted with the described method. Additionally, problems in terms of processing potential toxic nanoparticle powders are avoided.54
Acknowledgements
We thank Fonds der Chemischen Industrie and Deutsche Forschungsgemeinschaft (DFG Sp 392/39-1) for financial support. Furthermore we acknowledge R. John, M. Martin and T. Windberg for their experimental work. We thank Prof. Hietschold and T. Jagemann (TU Chemnitz) for providing the opportunity of measuring electron microscopy images.References
- S. Geier and C. Bonten, Polym. Eng. Sci., 2014, 54, 247–254 CAS.
- Y. Kojima, A. Usuki, M. Kawasumi, A. Okada, Y. Fukushima, T. Kurauchi and O. Kamigaito, J. Mater. Res., 1993, 8, 1185–1189 CrossRef CAS.
- Y. Kojima, A. Usuki, M. Kawasumi, A. Okada, T. Kurauchi and O. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 983–986 CrossRef CAS PubMed.
- K. Masenelli-Varlot, E. Reynaud, G. Vigier and J. Varlet, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 272–283 CrossRef CAS PubMed.
- T. J. Bessell and J. B. Shortall, J. Mater. Sci., 1977, 12, 365–372 CrossRef CAS.
- S.-H. Wu, F.-Y. Wang, C.-C. M. Ma, W.-C. Chang, C.-T. Kuo, H.-C. Kuan and W.-J. Chen, Mater. Lett., 2001, 49, 327–333 CrossRef CAS.
- K. Han, Z. Liu and M. Yu, Macromol. Mater. Eng., 2005, 290, 688–694 CrossRef CAS PubMed.
- A. Güllü, A. Özdemir and E. Özdemir, Mater. Des., 2006, 27, 316–323 CrossRef PubMed.
- B. Ou, Z. Zhou, Q. Liu, B. Liao, Y. Xiao, J. Liu, X. Zhang, D. Li, Q. Xiao and S. Shen, Polym. Compos., 2014, 35, 294–300 CrossRef CAS PubMed.
- H. R. Pant, M. P. Bajgai, K. T. Nam, Y. A. Seo, D. R. Pandeya, S. T. Hong and H. Y. Kim, J. Hazard. Mater., 2011, 185, 124–130 CrossRef CAS PubMed.
- M. Zhu, Q. Xing, H. He, Y. Zhang, Y. Chen, P. Pötschke and H.-J. Adler, Macromol. Symp., 2004, 210, 251–261 CrossRef CAS PubMed.
- A. D. Erem, G. Ozcan and M. Skrifvars, Text. Res. J., 2011, 81, 1638–1646 CrossRef CAS PubMed.
- I. B. Page, Polyamides as Engineering Thermoplastic Materials, iSmithers Rapra Publishing, 2000, vol. 11, p. 13 Search PubMed.
- L. Shen, Q. Du, H. Wang, W. Zhong and Y. Yang, Polym. Int., 2004, 53, 1153–1160 CrossRef CAS PubMed.
- Y. Ou, F. Yang and Z.-Z. Yu, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 789–795 CrossRef CAS.
- M. Nanko, Adv. Technol. Mater. Mater. Proc., 2009, 11, 1–8 CAS.
- M. García, J. Barsema, R. E. Galindo, D. Cangialosi, J. Garcia-Turiel, W. E. van Zyl, H. Verweij and D. H. A. Blank, Polym. Eng. Sci., 2004, 44, 1240–1246 Search PubMed.
- M. M. Hasan, Y. Zhou, H. Mahfuz and S. Jeelani, Mater. Sci. Eng., A, 2006, 429, 181–188 CrossRef PubMed.
- V. N. Dougnac, B. C. Peoples and R. Quijada, Polym. Int., 2011, 60, 1600–1606 CrossRef CAS PubMed.
- P. Theil-Van Nieuwenhuyse, V. Bounor-Legaré, P. Bardollet, P. Cassagnau, A. Michel, L. David, F. Babonneau and G. Camino, Polym. Degrad. Stab., 2013, 98, 2635–2644 CrossRef CAS PubMed.
- W. E. van Zyl, M. García, B. A. G. Schrauwen, B. J. Kooi, J. T. M. De Hosson and H. Verweij, Macromol. Mater. Eng., 2002, 287, 106–110 CrossRef CAS.
- M. García, G. van Vliet, M. G. J. ten Cate, F. Chávez, B. Norder, B. Kooi, W. E. van Zyl, H. Verweij and D. H. A. Blank, Polym. Adv. Technol., 2004, 15, 164–172 CrossRef PubMed.
- F. Yang, Y. Ou and Z. Yu, J. Appl. Polym. Sci., 1998, 69, 355–361 CrossRef CAS.
- Y. Li, J. Yu and Z.-X. Guo, J. Appl. Polym. Sci., 2002, 84, 827–834 CrossRef CAS PubMed.
- L. F. Cai, eXPRESS Polym. Lett., 2010, 4, 397–403 CrossRef CAS.
- G. Rusu and E. Rusu, High Perform. Polym., 2006, 18, 355–375 CrossRef CAS PubMed.
- H. Gu, Y. Guo, S. Y. Wong, Z. Zhang, X. Ni, Z. Zhang, W. Hou, C. He, V. P. W. Shim and X. Li, Microporous Mesoporous Mater., 2013, 170, 226–234 CrossRef CAS PubMed.
- J. Liu, H. Yi, H. Lin, T. Wei and B. Zheng, Polym. Compos., 2014, 35, 435–440 CrossRef CAS PubMed.
- C. Zhao, P. Zhang and S. Lu, J. Mater. Sci., 2007, 42, 9083–9091 CrossRef CAS.
- S. Spange and S. Grund, Adv. Mater., 2009, 21, 2111–2116 CrossRef CAS PubMed.
- M. W. Ellsworth and B. M. Novak, J. Am. Chem. Soc., 1991, 113, 2756–2758 CrossRef CAS.
- B. M. Novak and C. Davies, Macromolecules, 1991, 24, 5481–5483 CrossRef CAS.
- M. W. Ellsworth and B. M. Novak, Chem. Mater., 1993, 5, 839–844 CrossRef CAS.
- B. M. Novak, Adv. Mater., 1993, 5, 422–433 CrossRef CAS PubMed.
- S. Grund, P. Kempe, G. Baumann, A. Seifert and S. Spange, Angew. Chem., Int. Ed., 2007, 119, 636–640 CrossRef PubMed.
- S. Spange, P. Kempe, A. Seifert, A. A. Auer, P. Ecorchard, H. Lang, M. Falke, M. Hietschold, A. Pohlers, W. Hoyer, G. Cox, E. Kockrick and S. Kaskel, Angew. Chem., Int. Ed., 2009, 121, 8403–8408 CrossRef PubMed.
- T. Ebert, A. Seifert and S. Spange, Macromol. Rapid Commun., 2015 DOI:10.1002/marc.201500182.
- K. Hedberg, J. Am. Chem. Soc., 1955, 77, 6491–6492 CrossRef CAS.
- J. He, J. F. Harrod and R. Hynes, Organometallics, 1994, 13, 2496–2499 CrossRef CAS.
- N. W. Mitzel, A. Schier, H. Beruda and H. Schmidbaur, Chem. Ber., 1992, 125, 1053–1059 CrossRef CAS PubMed.
- N. Mitzel, A. Schier and H. Schmidbaur, Chem. Ber., 1992, 125, 2711–2712 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. Wollenweber, K. Reinhart and S.-E. Helen, Z. Naturforsch., 1998, 53 b, 145–148 Search PubMed.
- G. Rong, R. Keese and H. Stoeckli-Evans, Eur. J. Inorg. Chem., 1998, 1998, 1967–1973 CrossRef.
- F. Carre, C. Chuit, R. J. P. Corriu, A. Fanta, A. Mehdi and C. Reye, Organometallics, 1995, 14, 194–198 CrossRef CAS.
- G. R. Hatfield, J. H. Glans and W. B. Hammond, Macromolecules, 1990, 23, 1654–1658 CrossRef CAS.
- A. Miyake, J. Polym. Sci., 1960, 44, 223–232 CrossRef CAS PubMed.
- Nylon plastics handbook, ed. M. I. Kohan, Hanser Publishers, Munich, New York, Cincinnati, 1995 Search PubMed.
- I. Matsubara and J. H. Magill, Polymer, 1966, 7, 199–215 CrossRef CAS.
- K. Meyer, Angew. Makromol. Chem., 1973, 34, 165–167 CrossRef CAS PubMed.
- J. Zimmermann, J. Polym. Sci., Part C: Polym. Lett., 1964, 2, 955–958 CrossRef PubMed.
- W. Xie, Z. Gao, W.-P. Pan, D. Hunter, A. Singh and R. Vaia, Chem. Mater., 2001, 13, 2979–2990 CrossRef CAS.
- T. D. Fornes, P. J. Yoon and D. R. Paul, Polymer, 2003, 44, 7545–7556 CrossRef CAS PubMed.
- D. Napierska, L. C. Thomassen, D. Lison, J. A. Martens and P. H. Hoet, Part. Fibre Toxicol., 2010, 7, 39 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1058156 and 1058182. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5py00815h |
| ‡ Pertaining to single X-ray structure analysis. |
| This journal is © The Royal Society of Chemistry 2015 |