
Multicomponent isocyanide-based synthesis of reactive styrenic and (meth)acrylic monomers and their RAFT (co)polymerization†
Sonja
Schmidt
,
Miriam
Koldevitz
,
Janina-Miriam
Noy
and
Peter J.
Roth
*
Centre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, University of New South Wales, UNSW, Sydney, NSW 2052, Australia. E-mail: P.Roth@unsw.edu.au
First published on 26th September 2014
Abstract
The multicomponent Passerini reaction of aldehydes, carboxylic acids, and isocyanides is used to produce a series of novel reactive (meth)acrylic and styrenic monomers carrying pendant double bond, (trimethylsilyl protected) triple bond, diene, acetate, or pentafluorophenyl functionality. Dichloromethane and water were compared as solvents in the synthesis of 15 different monomers, with water resulting in significantly higher, up to quantitative, isolated yields with minimal purification. Characterization by 1H, 13C, and 19F NMR spectroscopy, FT-IR spectroscopy and mass spectrometry confirmed the synthesis and high purity of the functional α-acyloxycarboxamide products. The monomers are shown to be well suited for the RAFT-synthesis of well-defined homopolymers, statistical copolymers with methyl methacrylate, poly(ethylene glycol) methyl ether methacrylate, and styrene, statistical copolymers produced from two different Passerini-derived monomers, and AB diblock copolymers. SEC-measured polydispersities were generally low, ĐM ≤ 1.29, and 1H NMR spectroscopy confirmed copolymer molar compositions in good agreement with comonomer feed ratios. We expect this synthetic strategy to provide access to a wide range of novel multifunctional materials and demonstrate preliminary postpolymerization modification of a polystyrene derivative by cleavage of its pendent acetate groups and coupling of the dye Methyl Red to the resulting alcohol groups.
Introduction
The chemical modification of repeat units of pre-made polymers has developed into a major synthetic tool in the polymer chemistry arena.1 Commonly termed postpolymerization modification, this method allows for introduction of appropriate functionality into reactive precursors furnishing the resulting materials with tailored properties including biocompatibility,2 or responsiveness to external stimuli.3 The horizon of postpolymerization modification has significantly expanded with the advent of reversible deactivation radical polymerization (RDRP) techniques and their high tolerance toward functional groups. Consequently, major research efforts have focused on vinyl-type monomers and polymers equipped with reactive groups that undergo robust, efficient and orthogonal modification reactions. Activated ester–amine substitution,4 cycloaddition reactions including azide–alkyne addition and Diels–Alder reactions, and radical and nucleophilic thiol–ene reactions5 represent the main cornerstones of such efficient, “click”-type chemistries.6–8 Reactive monomers that undergo such reactions—pentafluorophenyl (meth)acrylate, (trimethylsilyl protected) propargyl (meth)acrylate, furfuryl methacrylate or allyl methacrylate, to name several common, commercially available, examples—are typically monofunctional species prepared by reaction of an appropriately functional alcohol with an activated (meth)acrylic acid derivative.Multicomponent reactions (MCRs) are convergent reactions in which more than two reactants combine to form a single product. Producing multifunctional products often with excellent atom economy, many of these reactions play key roles in the synthesis of libraries of drugs or other biologically relevant species.9 Recently, MCRs have been receiving increasing attention from the polymer chemistry community10,11 as a means to produce monomers,12–14 as a postpolymerization modification strategy,12,15–17 including modification of polymer end groups,16,18,19 and for step growth polymerization of difunctional reactants.20–22 For example, Meier's group presented the synthesis of novel functional monomers for olefin metathesis polymerization from renewable resources12 and acrylic monomers for free radical polymerization14via a Passerini three-component reaction of an aldehyde, carboxylic acid, and isocyanide. Kakuchi and Theato15 reported the copper catalyzed multicomponent reaction between a terminal alkyne, sulfonyl azide, and an amine23 to post-modify an alkyne-functional copolymer in near-quantitative conversion. Tao, Wei, and co-workers16 found the Biginelli reaction of a dione, aldehyde, and urea to proceed with high efficiency, compatibility and bio-orthogonality during the synthesis of functional polymers, for example in the postpolymerization modification of a dione-functional polymethacrylate with urea and benzaldehyde.
Herein, we exploit the Passerini reaction to produce novel acrylic, methacrylic, and styrenic monomers equipped with reactive groups suitable for postpolymerization modification. In this strategy, some of the most commonly used reactive groups, viz. double bond, (protected) triple bond, acetate-protected alcohol, diene, and pentafluorophenyl functionality, are installed into monomers through functional aldehydes or carboxylic acids. The isocyanide component, on the other hand, produces a side chain N-functional amide providing another handle for possible derivatization. Passerini reactions were performed in dichloromethane or water with reactions in the latter, green, solvent proceeding to quantitative conversion for most functional monomers. We further demonstrate the success of RAFT polymerization in producing a range of novel well-defined reactive (co)polymers from the portfolio of reactive monomers and present preliminary results for postpolymerization modification.
Experimental section
Materials
All reagents were purchased from Sigma-Aldrich and were used as received unless stated otherwise. Azobis(isobutyronitrile) (AIBN) was recrystallized from methanol and stored at −24 °C. The comonomers methyl methacrylate (MMA) and poly(ethylene glycol) methyl ether methacrylate (PEGMA, monomer molecular weight 360 g mol−1) were passed through basic Al2O3 to remove inhibitors before polymerization. The chain transfer agents 4-cyano-4-[(phenylcarbonothioyl)thio]pentanoic acid (CTA1)24 and benzyl propyl trithiocarbonate (CTA2)25 were prepared as described elsewhere.Instrumentation
NMR spectroscopic measurements were performed on a Bruker Avance 300 MHz instrument. The internal solvent signal δ(CDCl3) = 7.26 ppm was used as reference.Size exclusion chromatography (SEC) was performed on a Shimadzu system equipped with four 300 × 7.8 mm2 linear phenogel columns (105, 104, 103 and 500 Å) operating at a flow rate of 1 mL min−1 using dimethylacetamide as eluent. The system was calibrated with a series of narrow molar mass distribution polystyrene standards with molar masses ranging from 0.58–1820 kg mol−1.
Fourier transform infrared (FT-IR) spectroscopy was performed on a Bruker IFS 66/S instrument under attenuated total reflectance (ATR) and data was analyzed on OPUS software version 4.0.
Electrospray ionization (ESI) mass spectrometry was performed on a Scientific LTQ Orbitrap XL mass spectrometer operating in positive ion mode with a spray voltage of 1.2 kV, a capillary voltage of 45 V, a capillary temperature of 200 °C, and a tube lens voltage of 120 V.
General procedure for monomer synthesis
Reactions in dichloromethane (DCM): (Meth)acrylic acid (1, 1 mmol) and aldehyde/ketone (2, 1 mmol) were dissolved in DCM (300 μL). Subsequently, isocyanide (3, 1 mmol) was added under stirring. The mixture was stirred for 24 h at room temperature and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using n-hexane–ethyl acetate (ratio given in Table 1 for each monomer). Yields are given in Table 1.| Monomer name, abbreviation, reactivity | Monomer structure | 1 | 2 | 3 | R f in hexane–EtOAc (ratio) | Conditions | Isolated yield (%) |
|---|---|---|---|---|---|---|---|
| N-tert-Butyl-2-(meth)acryloyloxy-2-methylbutanamide, tBu-(M)A-MBu, test reaction |
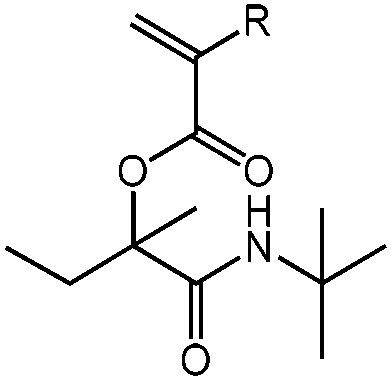
|
1a | 2a | 3a | 0.90 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
R = H, DCM | 42 |
| R = H, H2O | 56 | ||||||
| 1b | 2a | 3a | 0.65 (9:1) |
R = CH3, DCM | 34 | ||
| R = CH3, H2O | 100 | ||||||
| N-tert-Butyl-2-(meth)acryloyloxy-2-(2-furyl)acetamide, tBu-(M)A-Fur, Diels–Alder cycloaddition |
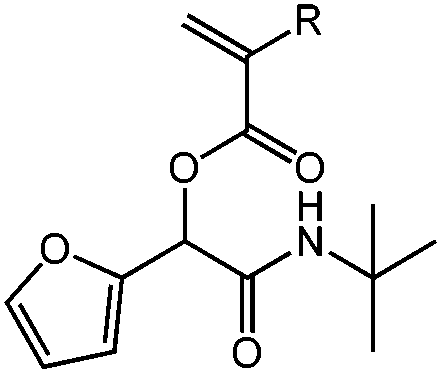
|
1a | 2b | 3a | 0.88 (2:1) |
R = H, DCM | 42 |
| R = H, H2O | 84 | ||||||
| 1b | 2b | 3a | 0.82 (2:1) |
R = CH3, DCM | 80 | ||
| R = CH3, H2O | 100 | ||||||
| N-Cyclohexyl-2-acryloyloxy-2-(2-furyl)acetamide, cHex-A-Fur, Diels–Alder cycloaddition |
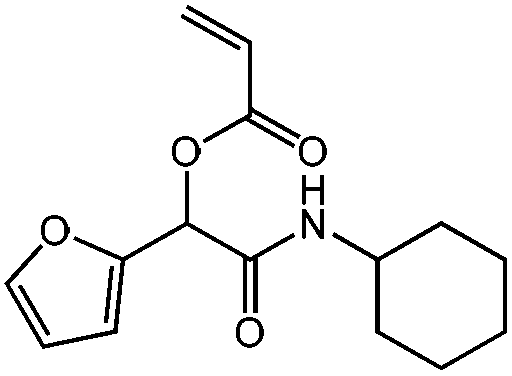
|
1a | 2b | 3b | 0.48 (2:1) |
H2O | 71 |
| N-tert-Butyl-2-(meth)acryloyloxypent-3-enamide, tBu-(M)A-Pentene, radical/electrophilic addition |
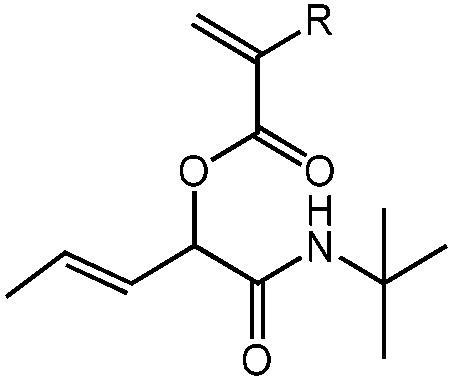
|
1a | 2c | 3a | 0.58 (2:1) |
R = H, DCM | 56 |
| R = H, H2O | 100 | ||||||
| 1b | 2c | 3a | 0.70 (2:1) |
R = CH3, DCM | 86 | ||
| R = CH3, H2O | 95 | ||||||
| N-Cyclohexyl-2-acryloyloxypent-3-enamide, cHex-A-Pentene, radical/electrophilic addition |
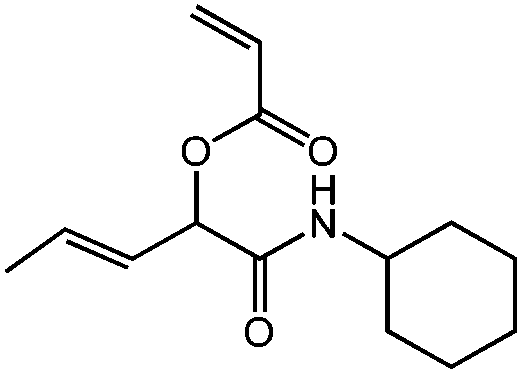
|
1a | 2c | 3b | 0.46 (2:1) |
H2O | 71 |
| N-tert-Butyl-2-(meth)acryloyloxy-2-(4-trimethylsilylethynylphenyl)acetamide, tBu-(M)A-TMSyne, CuAAC, thiol–yne, Glaser coupling (after trimethylsilyl deprotection) |

|
1a | 2d | 3a | 0.79 (2:1) |
R = H, DCM | 91 |
| R = H, H2O | 91 | ||||||
| 1b | 2d | 3a | 0.70 (2:1) |
R = CH3, DCM | 91 | ||
| R = CH3, H2O | 99 | ||||||
| N-tert-Butyl-2-(meth)acryloyloxy-2-(pentafluorophenyl)acetamide, tBu-(M)A-PFP, nucleophilic aromatic substitution |
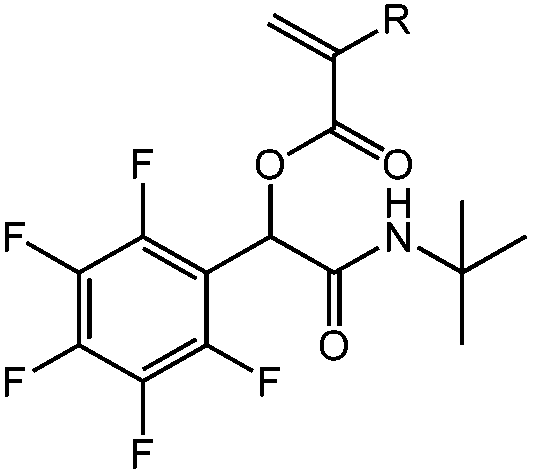
|
1a | 2e | 3a | 0.81 (2:1) |
R = H, DCM | 90 |
| R = H, H2O | 91 | ||||||
| 1b | 2e | 3a | 0.45 (4:1) |
R = CH3, DCM | 91 | ||
| R = CH3, H2O | 97 | ||||||
| N-Cyclohexyl-2-acryloyloxy-2-(pentafluorophenyl)acetamide, cHex-A-PFP, nucleophilic aromatic substitution |
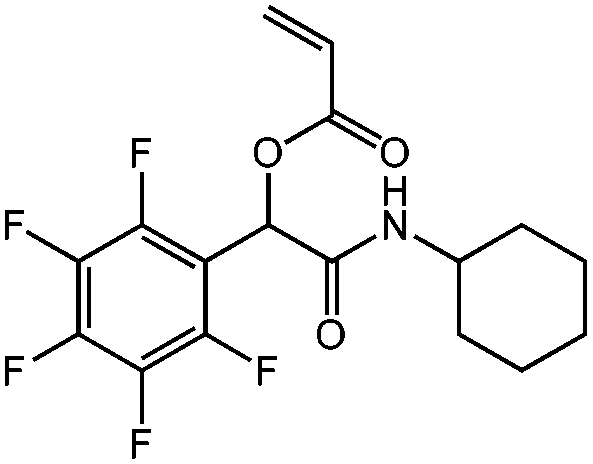
|
1a | 2e | 3b | 0.71 (3:2) |
H2O | 97 |
| N-tert-Butyl-2-acetoyloxy-2-(4-vinylphenyl)acetamide, tBu-Ac-Sty, coupling with activated carbonyls (after acetate deprotection) |
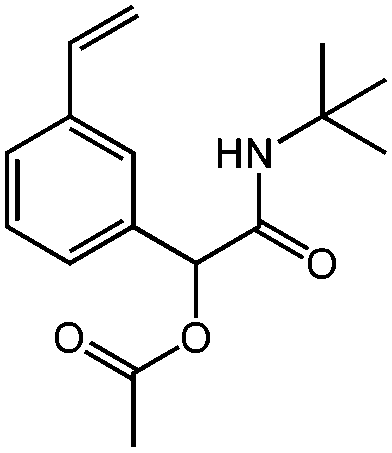
|
1c | 2f | 3a | 0.62 (2:1) |
H2O | 100 |
| N-tert-Butyl-2-(pent-4-ynoyloxy)-2-(4-vinylphenyl)acetamide, tBu-Pentyne-Sty, CuAAC, thiol–yne, Glaser coupling |
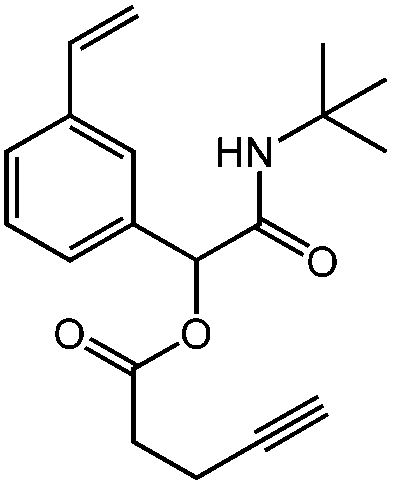
|
1d | 2f | 3a | 0.83 (2:1) |
DCM | 78 |
| H2O | 92 | ||||||
Reactions in water: (Meth)acrylic acid (1, 1 mmol) and aldehyde/ketone (2, 1 mmol) were added to water (300 μL) producing a heterogeneous mixture. Subsequently, isocyanide (3, 1 mmol) was added under stirring. The mixture was stirred for 24 h at room temperature. Most monomers precipitated as a white solid which was filtered and washed with water yielding pure product. tBu-A-MBu and tBu-Ac-Sty were isolated by extraction with ethyl acetate followed by solvent evaporation. tBu-MA-MBu was isolated by evaporating the water. cHex-A-Fur was isolated by filtration followed by column chromatography. tBu-MA-Pentene and cHex-A-PFP were isolated by evaporating water followed by column chromatography. Yields are given in Table 1.
tBu-A-MBu
1H NMR (CDCl3, 300 MHz) δ/ppm = 6.35 (dd, J = 17.3, 1.4 Hz, 1 H, HHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 6.10 (dd, J = 17.3, 10.3 Hz, 1 H, HHCCH–), 6.01 (s, 1 H, NH), 5.84 (dd, J = 10.3, 1.4 Hz, 1 H, HHCCH–), 2.08 (ddt, J = 38.0, 14.2, 7.3 Hz, 2 H, –CH2CH3), 1.63 (s, 3 H, –CH3), 1.34 (s, 9 H, –C(CH3)3), 0.80 (t, J = 7.3 Hz, 3 H, –CH2CH3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 171.4 (–CONH–), 164.0 (–COO–), 131.1 (H2CCH–), 129.0 (H2CCH–), 85.8 (–COO–C(Me,Et)–), 51.2 (–C(CH3)3), 29.9 (–CH2CH3), 28.7 (–C(CH3)3), 22.2 (–CH3), 8.1 (–CH2CH3). FT-IR (ATR) ν/cm−1 = 3338 (m, N–H,), 2973, 2942 (w-m, C–H, alkyl, CCH2 stretch), 1724 (s, CO, ester stretch), 1654 (s, CO, amide stretch), 1141 (s, C–N stretch).
CH–), 6.10 (dd, J = 17.3, 10.3 Hz, 1 H, HHCCH–), 6.01 (s, 1 H, NH), 5.84 (dd, J = 10.3, 1.4 Hz, 1 H, HHCCH–), 2.08 (ddt, J = 38.0, 14.2, 7.3 Hz, 2 H, –CH2CH3), 1.63 (s, 3 H, –CH3), 1.34 (s, 9 H, –C(CH3)3), 0.80 (t, J = 7.3 Hz, 3 H, –CH2CH3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 171.4 (–CONH–), 164.0 (–COO–), 131.1 (H2CCH–), 129.0 (H2CCH–), 85.8 (–COO–C(Me,Et)–), 51.2 (–C(CH3)3), 29.9 (–CH2CH3), 28.7 (–C(CH3)3), 22.2 (–CH3), 8.1 (–CH2CH3). FT-IR (ATR) ν/cm−1 = 3338 (m, N–H,), 2973, 2942 (w-m, C–H, alkyl, CCH2 stretch), 1724 (s, CO, ester stretch), 1654 (s, CO, amide stretch), 1141 (s, C–N stretch).
tBu-MA-MBu
1H NMR (CDCl3, 300 MHz) δ/ppm = 6.10 (s, 1 H, NH), 6.04 (qd, J = 1.0, 0.5 Hz, 1 H, HHCC(CH3)–), 5.59 (p, J = 1.6 Hz, 1 H, HHCC(CH3)–), 2.12 (m, 2 H, –CH2CH3), 1.96 (s, 3 H, H2CC(CH3)–), 1.65 (s, 3 H, –CH3), 1.36 (s, 9 H, –C(CH3)3), 0.81 (t, J = 7.5 Hz, 3 H, –CH2CH3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 171.6 (–CONH–), 165.2 (–COO–), 136.9 (H2CC(CH3)–), 125.7 (H2CC(CH3)–), 86.1 (–COO–C(Me,Et)–), 51.2 (–C(CH3)3), 29.7 (–CH2CH3), 28.8 (–C(CH3)3), 22.4 (–CH3), 18.59 (H2CC(CH3)–), 8.2 (–CH2CH3). FT-IR (ATR) ν/cm−1 = 3346 (w, N–H stretch), 2967 (w, C–H, alkyl CCH2, stretch), 1722 (s, CO, ester stretch), 1673 (s, CO, amide stretch), 1506 (s, C–H bend), 1135 (s, C–N stretch).
tBu-A-Fur
1H NMR (CDCl3, 300 MHz) δ/ppm = 7.41 (dd, J = 1.9, 0.8 Hz, 1 H, –C(–O)CH–CHCH(–O)) 6.58–6.45 (m, 2 H, HHCCH–, –C(–O)CH–CHCH(–O)), 6.36 (ddd, J = 3.4, 1.9, 0.4 Hz, 1 H, –C(–O)CH–CHCH(–O)), 6.25–6.12 (m, 2 H, HHCCH–, –COO–CH(Fur)–), 5.97 (bs, 1 H, NH), 5.93 (dd, J = 10.4, 1.3 Hz, 1 H, HHCCH–), 1.38 (s, 9 H, –C(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 165.0(–CONH–), 164.3 (–COO–), 148.5 (–C(–O)CH–CHCH(–O)), 143.6 (–C(–O)CH–CHCH(–O)), 132.8, 127.5 (H2CCH–), 111.3, 110.8 (–C(–O)CH–CHCH(–O)), 69.1 (–COO–CH(Fur)–), 51.9 (–C(CH3)3), 28.7 (–C(CH3)3). FT-IR (ATR) ν/cm−1 = 3303 (w, N–H stretch), 2975, 2925 (w, C–H, CCH2 stretch), 1716 (s, CO, ester stretch), 1670 (s, CO, amide stretch), 1554 (s, CO stretch), 1187 (s, C–N stretch), 736 (s, C–H, Furan, stretch).
tBu-MA-Fur
1H NMR (CDCl3, 300 MHz) δ/ppm = 7.41 (dd, J = 1.9, 1.0 Hz, 1 H, –C(–O)CH–CHCH(–O)), 6.49 (d, J = 3.3 Hz, 1 H, –C(–O)CH–CHCH(–O)), 6.36 (d, J = 3.3 Hz, 1 H, –C(–O)CH–CHCH(–O)), 6.19 (1 H, HHCC(CH3)–), 6.13 (s, 1 H, –COO–CH(Fur)–), 5.98 (s, 1 H, NH), 5.67(1 H, HHCC(CH3)–), 1.98 (dd, J = 1.5, 0.8 Hz, 3 H, H2CC(CH3)–), 1.38 (d, J = 0.8 Hz, 9 H, –C(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 165.5 (–CONH–), 165.1 (–COO–), 148.6 (–C(–O)=CH–CHCH(–O)), 143.6 (–C(–O)=CH–CHCH(–O)), 135.6 (H2CC(CH3)–), 127.1 (H2CC(CH3)–), 111.2, 110.7 (–C(–O)CH–CHCH(–O)), 69.2 (–COO–CH(Fur)–), 51.8 (–C(CH3)3), 28.8 (–C(CH3)3), 18.4 (H2CC(CH3)–). FT-IR (ATR) ν/cm−1 = 3280 (w-m, N–H stretch), 3081, 2973 (w, C–H, alkyl, CCH2, stretch), 1712 (s, CO, ester, stretch), 1662 (s, CO, amide, stretch), 1556 (s, CC, stretch), 1149 (s, C–N, stretch), 746 (s, C–H, Furan, stretch). MS (ESI) m/z (%) = 553 (100) [2M + Na]+, 288 (55) [M + Na]+.
cHex-A-Fur
1H NMR (CDCl3, 300 MHz) δ/ppm = 7.40 (dd, J = 1.9, 0.8 Hz, 1 H, –C(–O)CH–CHCH(–O)), 6.55– 6.46 (m, 2 H, HHCCH–, –C(–O)=CH–CHCH(–O)), 6.36 (ddd, J = 3.3, 1.9, 0.3 Hz, 1 H, HHCCH–), 6.26–6.14 (m, 2 H, –COO–CH(Fur)–, –C(–O)CH–CHCH(–O)), 6.10–6.01 (m, 1 H, NH), 5.93 (dd, J = 10.4, 1.3 Hz, 1 H, HHCCH–), 3.84 (m, 1 H, –NH–CH<), 2.03–1.08 (m, 10 H, cHex). 13C NMR (CDCl3, 75 MHz) δ/ppm = 165.0 (–CONH–), 164.3 (–COO–), 148.3 (–C(–O)CH–CHCH(–O)), 143.7 (–C(–O)CH–CHCH(–O)), 132.9, (H2CCH–), 127.4 (H2CCH–), 111.5, 110.8 (–C(–O)CH–CHCH(–O)), 69.0 (–COO–CH(Fur)–), 48.5 (–NH–CH<), 33.0, 25.6, 24.8 (cHex). FT-IR (ATR) ν/cm−1 = 3309 (w, N–H, stretch), 2938, 2857 (w, C–H, alkyl, CCH2, stretch), 1724 (s, CO, ester, stretch), 1664 (s, CO, amide, stretch), 1536 (s, CC, stretch), 1251 (s, C–O, ester, stretch), 1047 (s, C–N, stretch), 754 (s, C–H, Furan, stretch).
tBu-A-Pentene
1H NMR (CDCl3, 300 MHz) δ/ppm = 6.47 (dd, J = 17.3, 1.4 Hz, 1 H, HHCCH–), 6.18 (dd, J = 17.3, 10.4 Hz, 1 H, HHCCH–), 5.96–5.76 (m, 3 H, NH, HHCCH–, –CHCH–CH3), 5.56 (qq, 1 H, –CHCH–CH3), 5.49 (dt, 1 h, –COO–CH(propene)–), 1.73 (ddd, J = 6.5, 1.6, 0.8 Hz, 3 H, –CHCH–CH3), 1.34 (s, 9 H, –C(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 167.6 (–CONH–), 164.5 (–COO–), 132.6 (–CHCH–CH3), 132.2 (H2CCH–), 127.9 (H2CCH–), 124.9 (–CHCH–CH3), 74.9 (–COO–CH(propene)–), 51.5 (–C(CH3)3), 28.8 (–C(CH3)3), 18.0 (–CHCH–CH3). FT-IR (ATR) ν/cm−1 = 3311 (w-m, N–H, stretch), 2977, 2927 (w, C–H, alkyl, CCH2, stretch), 1722 (m-s, CO, ester, stretch), 1656 (s, CO, amide, stretch), 1550 (m–s, CC, stretch), 1195 (s, C–N, stretch), 962 (s, C–H, alkene, stretch).
tBu-MA-Pentene
1H NMR (CDCl3, 300 MHz) δ/ppm = 6.17 (dq, J = 1.9, 1.1 Hz, 1 H, HHCC(CH3)–) 5.95–5.77 (m, 2 H, NH, –CHCH–CH3), 5.64 (dq, J = 1.9, 1.1 Hz, 1 H, HHCC(CH3)–), 5.58 (qq, 1 H, –CHCH–CH3), 5.48 (m, 1 H, –COO–CH(propene)–), 1.98 (dd, J = 1.6, 1.0 Hz, 3 H, H2CC(CH3)–), 1.73 (ddd, J = 6.5, 1.6, 0.9 Hz, 3 H, –CHCH–CH3), 1.34 (s, 9 H, –C(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 167.8 (–CONH–), 165.6 (–COO–), 136.0 (H2CC(CH3)–), 132.3 (H2CC(CH3)–), 126.6, 125.0 (–CHCH–CH3), 75.0 (–COO–CH(propene)–), 51.4 (–C(CH3)3), 28.8 (–C(CH3)3), 18.5, 18.0 (H2CC(CH3)–, –CHCH–CH3). FT-IR (ATR) ν/cm−1 = 3282 (w-m, N–H, stretch), 3077, 2975 (w, C–H, alkyl, CCH2, stretch), 1714 (s, CO, ester, stretch), 1656 (s, CO, amide, stretch), 1558 (s, CC, stretch), 1157 (s, C–N, stretch), 948 (s, C–H, alkene, stretch). MS (ESI) m/z (%) = 262 (100) [M + Na]+.
cHex-A-Pentene
1H NMR (CDCl3, 300 MHz) δ/ppm = 6.47 (dd, J = 17.3, 1.4 Hz, 1 H, HHCCH–), 6.18 (dd, J = 17.3, 10.4 Hz, 1 H, HHCCH–), 5.96–5.83 (m, 2 H, HHCCH–, –CHCH–CH3), 5.63–5.52 (m, 2 H, –COO–CH(propene)–, –CHCH–CH3), 3.76 (m, 1 H, –NH–CH<), 1.97–1.03 (m, 10 H, cHex). 13C NMR (CDCl3, 75 MHz) δ/ppm = 167.5 (–CONH–), 164.5 (–COO–), 132.6 (–CHCH–CH3), 132.2 (H2CCH–), 127.8 (H2CCH–), 124.8 (–CHCH–CH3), 74.7 (–COO–CH(propene)–), 48.2 (–NH–CH<), 33.0 (–CHCH–CH3), 24.8, 24.8, 18.0 (cHex), FT-IR (ATR) ν/cm−1 = 3272 (w, N–H, stretch), 2937, 2854 (w, C–H, alkyl, CCH2, stretch), 1729 (s, CO, ester, stretch), 1650 (s, CO, amide, stretch), 1176 (s, C–N, stretch), 964 (s, C–H, alkene, stretch).
tBu-A-TMSyne
1H NMR (CDCl3, 300 MHz) δ/ppm = 7.50–7.34 (m, 4 H, Ar–H), 6.50 (dd, J = 17.3, 1.3 Hz, 1 H, HHCCH–), 6.22 (dd, J = 17.3, 10.4 Hz, 1 H, HHCCH–), 6.00 (s, 1 H, –COO–CH(Ar)–), 5.98–5.85 (m, 2 H, HHCCH–, NH), 1.33 (s, 9 H, –C(CH3)3), 0.23 (s, 9 H, –Si(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 166.9 (–CONH–), 164.4 (–COO–), 136.1 (CH–C (ipso)), 132.7 (H2CCH–), 132.4 (meta CH), 127.6 (H2CCH–), 127.3 (ortho CH), 123.9 (C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– (para)), 104.6, 95.3, (–CC–), 75.4 (–COO–CH(Ar)–), 51.7 (–C(CH3)3), 28.8 (–C(CH3)3), 0.0 (–Si(CH3)3). FT-IR (ATR) ν/cm−1 = 3286 (w, N–H, stretch), 2969 (w, C–H, alkyl, CCH2, stretch), 2356 (m, –CC–, stretch), 1725 (m–s, CO, ester, stretch), 1654 (s, CO, amide, stretch), 1250 (m–s, Si–CH3, stretch), 1174 (s, C–N, stretch), 836 (s, C–H, alkene, stretch).
C– (para)), 104.6, 95.3, (–CC–), 75.4 (–COO–CH(Ar)–), 51.7 (–C(CH3)3), 28.8 (–C(CH3)3), 0.0 (–Si(CH3)3). FT-IR (ATR) ν/cm−1 = 3286 (w, N–H, stretch), 2969 (w, C–H, alkyl, CCH2, stretch), 2356 (m, –CC–, stretch), 1725 (m–s, CO, ester, stretch), 1654 (s, CO, amide, stretch), 1250 (m–s, Si–CH3, stretch), 1174 (s, C–N, stretch), 836 (s, C–H, alkene, stretch).
tBu-MA-TMSyne
1H NMR (CDCl3, 300 MHz) δ/ppm = 7.50–7.33 (m, 4 H, Ar–H), 6.20 (p, J = 1.0 Hz, 1 H, HHCC(CH3)–), 5.99 (s, 1 H, –COO–CH(Ar)–), 5.90 (s, 1 H, NH), 5.68 (p, J = 1.5 Hz, 1 H, HHCC(CH3)–), 2.00 (dd, J = 1.5, 1.0 Hz, 3 H, H2CC(CH3)–), 1.33 (s, 9 H, –C(CH3)3), 0.23 (s, 9 H, –Si(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 167.1 (–CONH–), 165.5 (–COO–), 136.3 (H2CC(CH3)–), 135.7 (CH–C (ipso)), 132.4 (meta CH), 127.2 (ortho CH), 127.0 (H2CC(CH3)–), 123.9 (C–CC– (para)), 104.6, 95.3 (–CC–), 75.5 (–COO–CH(Ar)–), 51.7 (–C(CH3)), 28.8 (–C(CH3)3), 18.4 (H2CC(CH3)–), 0.0 (–Si(CH3)3). FT-IR (ATR) ν/cm−1 = 3305 (w, N–H, stretch), 3081, 2973 (w, C–H, alkyl, CCH2, stretch), 2356, 2159 (m, –CC–, stretch), 1718 (m–s, CO, ester, stretch), 1652 (s, CO, amide, stretch), 1250 (m, Si–CH3, stretch), 1153 (s, C–N, stretch), 836 (s, C–H, alkene, stretch). MS (ESI) m/z (%) = 765 (100) [2M + Na]+, 394 (88) [M + Na]+.
tBu-A-PFP
1H NMR (CDCl3, 300 MHz) δ/ppm = 6.52 (dd, J = 17.2, 1.2 Hz, 1 H, HHCCH–), 6.40 (s, 1 H, –COO–CH(PFP)–), 6.20 (dd, J = 17.2, 10.4 Hz, 2 H, NH, HHCCH–), 6.00 (dd, J = 10.4, 1.2 Hz, 1 H, HHCCH–), 1.40 (s, 9 H, –C(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 164.9 (–CONH–), 163.7 (–COO–), 133.9 (H2CCH), 126.8 (H2CCH–), 65.4 (–C(CH3)3), 52.2 (–COO–CH(PFP)–), 28.6 (–C(CH3)3). 19F NMR (CDCl3, 282 MHz) δ/ppm = −140.9 (2 F, ortho), −152.4 (1 F, para), −161.5 (2 F, meta). FT-IR (ATR) ν/cm−1 = 3276 (w, N–H, stretch), 2981 (w, C–H, alkyl, CCH2, stretch), 1735 (m–s, CO, ester, stretch), 1652 (s, CO, amide, stretch), 1521, 1502 (s, CC, stretch), 1251 (s, C–N, stretch), 1000 (s, C–F, stretch), 804 (s, C–H, alkene, stretch).
tBu-MA-PFP
1H NMR (CDCl3, 300 MHz) δ/ppm = 6.38 (s, 1 H, –COO–CH(PFP)–), 6.26 (s, 1 H, NH), 6.19 (d, J = 1.0 Hz, 1 H, HHCC(CH3)–), 5.76–5.71 (d, J = 1.0 Hz, 1 H, HHCC(CH3)–), 1.98 (dd, J = 1.6, 1.0 Hz, 3 H, H2CC(CH3)–), 1.40 (s, 9 H, –C(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 165.1 (–CONH–), 164.8 (–COO–), 135.1 (H2CC(CH3)–), 128.0 (H2CC(CH3)–), 65.6 (–C(CH3)3), 52.1 (–COO–CH(PFP)–), 28.6 (–C(CH3)3), 18.3 (H2CC(CH3)–). 19F NMR (CDCl3, 282 MHz) δ/ppm = −140.9 (2 F, ortho), −152.4, (1 F, para), −161.5 (2 F, meta). FT-IR (ATR) ν/cm−1 = 3332 (w, N–H, stretch), 2973 (w, C–H, alkyl, CCH2, stretch), 1725 (m–s, CO, ester, stretch), 1670 (s, CO, amide, stretch), 1506 (s, CC, stretch), 1135 (s, C–N, stretch), 1000 (s, C–F, stretch). MS (ESI) m/z (%) = 388 (100) [M + Na]+.
cHex-A-PFP
1H NMR (CDCl3, 300 MHz) δ/ppm = 6.51 (dd, 1 H, HHCCH–), 6.45 (s, 1 H, –COO–CH(PFP)–), 6.34 (1 H, NH), 6.20 (q, 1 H, HHCCH–), 5.99 (dd, 1 H, HHCCH–), 3.82 (m, 1 H, –NH–CH<), 1.97–1.12 (m, 10 H, cHex). 13C NMR (CDCl3, 75 MHz) δ/ppm = 164.7 (–CONH–), 163.6 (–COO–), 133.74 (H2CCH–), 126.6 (H2CCH–), 65.1 (–COO–CH(PFP)–), 48.6 (–NH–CH<), 32.7, 25.4, 24.7 (cHex). 19F NMR (CDCl3, 282 MHz) δ/ppm = −140.7 (ortho), −152.0 (para), −161.5 (meta). FT-IR (ATR) ν/cm−1 = 3307 (w, N-H, stretch), 2930, 2860 (w, C-H alkyl, CCH2, stretch), 1737 (m–s, CO, ester, stretch), 1654 (s, CO, amide, stretch), 1506 (s, CC, stretch), 1128 (s, C–N, stretch), 1000 (s, C–F, stretch).
tBu-Ac-Sty
1H NMR (CDCl3, 300 MHz) δ/ppm = 7.47–7.27 (m, 4 H, Ar–H), 6.71 (dd, J = 17.6, 10.9 Hz, 1 H, H2CCH–), 5.94 (s, 1 H, –COO–CH(Ar)–), 5.87 (s, 1 H, NH), 5.82, 5.70 (2 d, 2 H, H2CCH–), 2.18 (s, 3 H, CH3COO–), 1.36 (s, 9 H, –C(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 169.3, 167.4 (–CONH–, –COO–), 138.3 (H2CCH–C (ipso)), 136.5 (C–CH(OAc)–), 136.4 (H2CCH–), 129.1, 126.9, 126.8, 125.7 (Ar-C), 114.8 (H2CCH–), 75.7 (–COO–CH(Ar)–), 51.7 (–C(CH3)3), 28.8 (–C(CH3)3), 21.2 (CH3COO–). FT-IR (ATR) ν/cm−1 = 3300 (w, N–H, stretch), 2780 (w, C–H, alkyl, CCH2, stretch), 1731 (m–s, CO, ester, stretch), 1658 (s, CO, amide, stretch), 1224 (s, C–N, stretch). MS (ESI) m/z (%) = 573 (100) [2M + Na]+, 298 (65) [M + Na]+.
tBu-Pentyne-Sty
1H NMR (CDCl3, 300 MHz) δ/ppm = 7.47–7.27 (m, 4 H, Ar–H), 6.70 (dd, 1 H, HHCCH–), 5.99 (s, 1 H, –COO–CH(Ar)–), 5.75 (dd, J = 17.6, 0.9 Hz, 1 H, HHCCH–), 5.27 (dd, J = 10.9, 0.9 Hz, 1 H, HHCCH–), 2.73–2.50 (m, 4 H, HCCCH2CH2–), 1.99 (t, J = 2.6 Hz, 1 H, HCCCH2CH2–), 1.36 (s, 9 H, –C(CH3)3). 13C NMR (CDCl3, 75 MHz) δ/ppm = 170.1 (–CONH–), 167.2 (–COO–), 138.2 (H2CCH–C (ipso)), 136.5 (H2CCH–), 136.2 (Ar C–CH(CONHR)–), 129.1, 126.9, 126.8, 125.6 (Ar-C), 114.8 (H2CCH–), 82.4 (HCCCH2CH2–), 76.0 (–COO–CH(Ar)–), 69.6 (HCCCH2CH2–), 51.8 (–C(CH3)3), 33.5 (HCCCH2CH2–), 28.8 (–C(CH3)3), 14.4 (HCCCH2CH2–). FT-IR (ATR) ν/cm−1 = 315 (w, C–H, alkyne, stretch), 3278 (w, N–H, stretch), 2965, 2923 (w, C–H, alkyl, CCH2, stretch), 1731 (m–s, CO, ester, stretch), 1650 (s, CO, amide, stretch), 1550 (s, CC, arom., stretch), 1359 (s, CH2, deform), 1224 (C–O, ester), 1153 (s, C–N, stretch), 636 (s, C–H, deform).
General procedure of (co)polymer synthesis
In a typical experiment, multifunctional monomer (5 mmol, 100 eq.) or a mixture of monomers of predetermined molar composition (5 mmol in total, 100 eq.), chain transfer agent (CTA1 or CTA2 as given in Table 2, 0.05 mmol, 1 eq.), AIBN (0.8 mg, 0.005 mmol, 0.1 eq.), anisole (5 mL) and a magnetic stir bar were combined in a flask which was sealed with a rubber septum. The mixture was purged with nitrogen for 20 min and placed into an oil bath preheated to 70 °C. Polymerization was continued overnight and stopped by quenching to room temperature. Monomer conversions and theoretical molecular weights, Mtheor.n, were determined by a 1H NMR spectroscopic measurement of a reaction sample (100 μL) diluted with CDCl3 (550 μL) by comparison of polymeric signals with the vinyl signals of residual monomers. Polymers were isolated by precipitation into diethyl ether–hexane. p(tBu-MA-TMSyne), p(tBu-MA-Fur), and p(cHex-A-PFP) were purified by dialysis in methanol (regenerated cellulose membranes, MW cut-off 3500 g mol−1). SEC data is summarized in Table 2.| Entry | Abbreviationa | Monomer feed (molar ratio in %) | CTA | Conversion b (%) |
M
theor.nb (kg mol−1) |
M
SECnc (kg mol−1) |
Đ
SECMc |
|---|---|---|---|---|---|---|---|
| a Molar composition measured by 1H NMR spectroscopy. b Global monomer conversion and theoretical molar mass determined by NMR spectroscopy before purification by quantification of residual monomer vinyl signals. c Determined by size exclusion chromatography in DMAc. | |||||||
| 1 | p(tBu-MA-Fur) | tBu-MA-Fur (100) | CTA1 | 57 | 15.3 | 7.6 | 1.25 |
| 2 | p(tBu-MA-Fur0.58-co-MMA0.42) |
tBu-MA-Fur (60):MMA (40) |
CTA1 | 89 | 18.5 | 7.6 | 1.16 |
| 3 | p(tBu-MA-Pentene0.37-co-MMA0.63) |
tBu_MA-Pentene (45):MMA (55) |
CTA1 | 81 | 12.6 | 9.8 | 1.11 |
| 4 | p(tBu-MA-Pentene0.34-co-PEGMA0.66) |
tBu_MA-Pentene (35):PEGMA (65) |
CTA1 | 73 | 21.0 | 22.6 | 1.47 |
| 5 | p(tBu-MA-TMSyne) | tBu-MA-TMSyne (100) | CTA1 | 72 | 27.0 | 21.9 | 1.29 |
| 6 | p(tBu-MA-TMSyne0.50-co-MMA0.50) |
tBu-MA-TMSyne (55):MMA (45) |
CTA1 | 87 | 16.1 | 15.7 | 1.18 |
| 7 | p(tBu-MA-PFP) | tBu-MA-PFP (100) | CTA1 | 87 | 32.0 | 15.9 | 1.26 |
| 8 | p(tBu-MA-PFP0.51-co-MMA0.49) |
tBu-MA-PFP (50):MMA (50) |
CTA1 | 95 | 22.5 | 20.7 | 1.14 |
| 9 | p(tBu-MA-PFP0.31-co-PEGMA0.69) |
tBu-MA-PFP (30):PEGMA (70) |
CTA1 | 81 | 26.2 | 35.2 | 1.39 |
| 10 | p(cHex-A-PFP) | cHex-A-PFP (100) | CTA2 | 77 | 14.9 | 5.5 | 1.18 |
| 11 | p(tBu-MA-PFP0.69-co-tBu-MA-Fur0.31) |
tBu-MA-PFP (70):tBu-MA-Fur (30) |
CTA1 | 84 | 22.6 | 16.8 | 1.25 |
| 12 | p(tBu-Ac-Sty) | tBu-Ac-Sty (100) | CTA1 | 76 | 21.2 | 16.7 | 1.08 |
| 13 | p(tBu-Ac-Sty0.56-co-Sty0.44) |
tBu-Ac-Sty (50):Sty (50) |
CTA1 | 28 | 7.9 | 10.4 | 1.05 |
| 14 | p[(tBu-MA-TMSyne)0.43-block-MMA0.57] | MMA (100) | p(tBu-MA-TMSyne) (entry 5) | 50 | 36.6 | 30.7 | 1.34 |
| 15 | p[(tBu-MA-TMSyne0.23-co-MMA0.23)-block-MMA0.54] | MMA (100) | p(tBu-MA-TMSyne0.50-co-MMA0.50) (entry 6) | 69 | 26.3 | 34.1 | 1.25 |
Chain extension experiments
Homopolymer p(tBu-MA-TMSyne) with an NMR-determined degree of polymerization, DPNMR, of 72 (entry 5 in Table 2) (50 mg, approx. 1.85 μmol RAFT end groups, 1 eq.), MMA (35 mg, 351.5 μmol, 190 eq.), AIBN stock solution (containing 0.06 mg AIBN, 0.2 eq.) and anisole (1 mL) were mixed, purged with nitrogen for 30 min, and allowed to polymerize in a preheated oil bath (70 °C) for 15 h. After cooling to room temperature, a 1H NMR spectrum of a reaction sample was acquired. Comparison of the signals at δ = 3.78 ppm (s, residual monomer –OCH3), 3.63 (bs, polymer –OCH3), and 0.29 (bs, –Si(CH3)3) revealed a monomer conversion of 50%, resulting in copolymer p[(tBu-MA-TMSyne)0.43-block-MMA0.57] with a DPNMR of the pMMA block of 96, and an Mtheor.n of 36.6 kg mol−1. The copolymer was precipitated into methanol. MSECn = 30.7 kg mol−1, ĐM = 1.34.In an analogous procedure, copolymer p(tBu-MA-TMSyne0.50-co-MMA0.50) with a DPNMR of 87 (entry 6 in Table 2) (100 mg, 6.21 μmol RAFT end groups, 1 eq.) was chain extended using MMA (92 mg, 919 μmol, 148 eq.), AIBN stock solution (containing 0.2 mg AIBN, 0.2 eq.) and anisole (1 mL). Monomer conversion determined by 1H NMR spectroscopy (taking into account the MMA repeat units of the macro-RAFT agent) was 69% resulting in an Mtheor.n of 26.3 kg mol−1 for the resulting p[(tBu-MA-TMSyne0.23-co-MMA0.23)-block-MMA0.54] and a DPNMR of the extended pMMA block of 102. The (A-co-B)-b-B copolymer was precipitated into diethyl ether–hexane 1:4. MSECn = 34.1 kg mol−1, ĐM = 1.25.
Postpolymerization modification of p(tBu-Ac-Sty)
p(tBu-Ac-Sty) (107.2 mg, 0.39 mmol repeat units) was treated with sodium methoxide solution (25% in methanol) in dry methanol (10 mL) with stirring at room temperature overnight. The product was isolated by dialysis in methanol yielding 87.6 mg (96%) of white powder. 1H NMR (CDCl3, 300 MHz) δ/ppm = 6.97, 6.43 (m, 4 H, Ar-H), 4.61 (bs, 1 H, Ar–CH(OH)CONH–), 2.63–0.74 (m, 3 H, backbone), 1.31 (bs, 9 H, tBu). Deacetylated polymer (40 mg, 0.171 mmol repeat units), dimethylaminopyridine (DMAP, 1.9 mg, 0.0016 mmol), Methyl Red (21 mg, 0.078 mmol), and dichloromethane (2 mL) were mixed in a nitrogen atmosphere. Dicyclohexyl carbodiimide (DCC, 19.3 mg, 0.094 mmol) was added and the mixture was stirred overnight at room temperature. Precipitated urea was removed by filtration and the dye-labelled polymer was isolated by several precipitations into hexane. Successful removal of Methyl Red reagent was confirmed by TLC. 1H NMR (CDCl3, 300 MHz) δ/ppm = 8.25–7.56 (m, Methyl Red Ar-H), 7.01, 6.41 (m, Ar-H), 4.64 (bs, 1 H, Ar–CH(OH)CONH–), 3.06 (bs, –N(CH3)2), 2.27–0.74 (m, backbone), 1.32 (bs, 9 H, tBu).Results and discussion
Monomer synthesis
As shown in Scheme 1, the Passerini reaction occurs between a carboxylic acid, an isocyanide and an aldehyde or ketone and produces an α-acyloxycarboxamide as the single product. In order to exploit this reaction to synthesize reactive (meth)acrylate monomers we chose as reagents acrylic and methacrylic acid, 1a and 1b, to provide the desired polymerizable functionalities, tert-butyl and cyclohexyl isocyanide, 3a and 3b, as commercially available isocyanide reagents, and a selection of functional aldehydes to introduce reactive groups. In addition to the ketone butanone 2a used for initial test reactions, the selection included furfural 2b (carrying a diene known to undergo Diels–Alder cycloaddition of dienophiles),8 crotonaldehyde 2c (featuring a double bond susceptible to electrophilic and radical addition reactions),5,7,8 4-[(trimethylsilyl)ethynyl]benzaldehyde 2d (containing a protected triple bond exploitable for azide–alkyne cycloadditions and thiol–yne reactions),6,7 and 2,3,4,5,6-pentafluorobenzaldehyde 2e (bearing a perfluorinated aromatic amenable to nucleophilic aromatic substitution).8 Functional styrenic monomers were prepared using 3-vinyl-benzaldehyde 2f to provide the eponymous monomeric handle and carboxylic acids carrying reactive groups. Acetic acid 1c provided an acetate-protected (and somewhat less reactive) alcohol group while 4-pentynoic acid 1d contained a triple bond. To the best of our knowledge, this is the first published report on styrenic monomers prepared through an isocyanide-based MCR. Table 1 provides a summary of synthesized monomers and their reactivity. Monomer abbreviations are based on the full chemical name and reflect the respective synthetic reagents in the order isocyanide–carboxylic acid–aldehyde/ketone.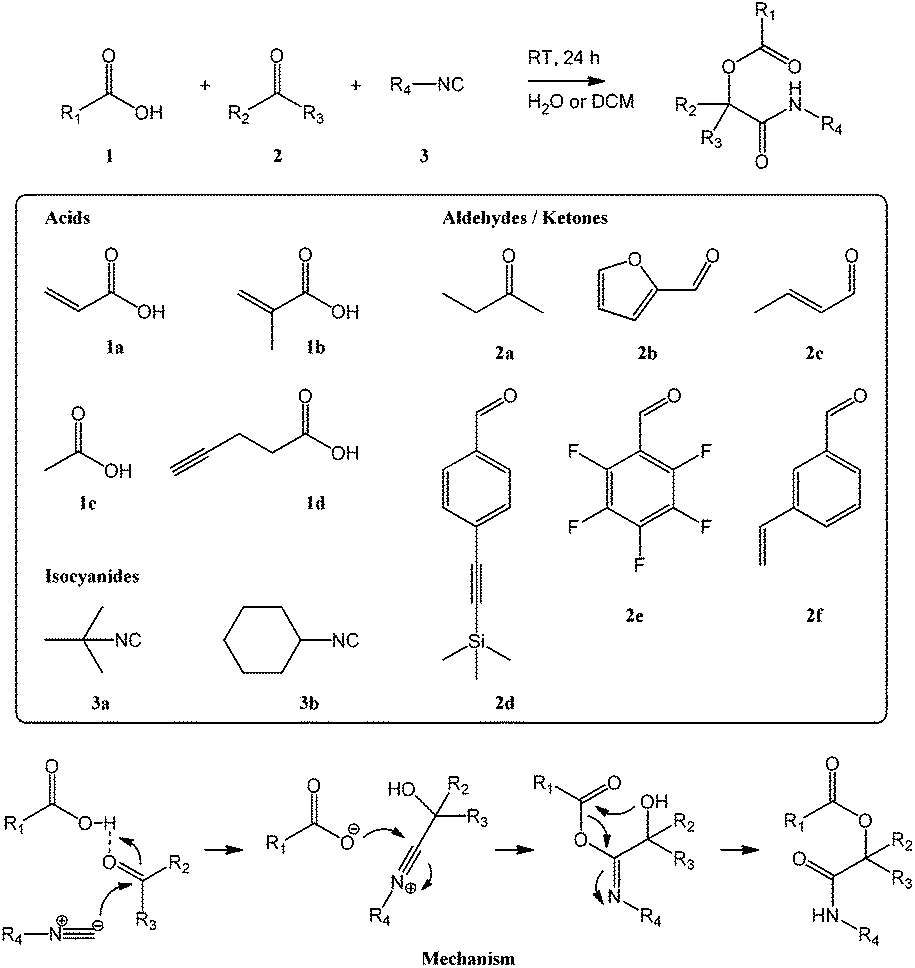 | ||
| Scheme 1 Reaction scheme and mechanism of the Passerini reaction with structures of employed carboxylic acids 1, keto components 2, and isocyanides 3. | ||
Reactions were carried out at high equimolar concentrations (3.33 M) of all reagents in dichloromethane or water with stirring at room temperature overnight. When using water as solvent, most monomers precipitated as a white solid and could be isolated by washing with water and drying resulting in very high to quantitative isolated yields. Most reactions done in dichloromethane, on the other hand, did not proceed to completion and required purification by column chromatography. Isolated yields, solvent mixtures used for column chromatography, and retention factors for thin layer chromatography (TLC) are summarized in Table 1. Water has previously been shown to accelerate isocyanide-based MCRs,26 presumably because reactions occur in a highly concentrated immiscible organic phase, and our results on Passerini synthesis of reactive monomers corroborate this finding. Products were characterized by 1H, 13C, and, for pentafluorophenyl-functional species, 19F NMR spectroscopy, FT-IR spectroscopy, and mass spectrometry. All measurements conformed to the expected structures and confirmed high purity; please see the ESI† for details.
RAFT (Co)polymer synthesis
With a series of novel reactive (meth)acrylic and styrenic monomers in hand, we next explored their suitability for the production of well-defined homo- and copolymers. We chose RAFT polymerization mediated by a dithioester, CTA1, or trithiocarbonate, CTA2 (structures given in Scheme 2) as a versatile, well-documented27 polymerization method. (Co)polymerizations were carried out in anisole overnight at 70 °C using a concentration ratio [monomers]:[CTA]:[AIBN] of 100:1:0.1 with methyl methacrylate (MMA), poly(ethylene glycol) methyl ether methacrylate (PEGMA), and styrene (Sty) as common comonomers. (Co)polymer products were isolated by precipitation into diethyl ether–hexane or by dialysis in methanol. After drying, the resulting materials were characterized by 1H NMR spectroscopy, SEC, and FT-IR spectroscopy. Table 2 provides a summary of homo- and copolymers with monomer feed ratios and molar compositions determined by 1H NMR spectroscopy for copolymers, conversions, apparent molar masses, MSECn, and polydispersities, ĐSECM, measured by SEC, and theoretical molar masses, Mtheor.n, calculated from conversions and compositions.
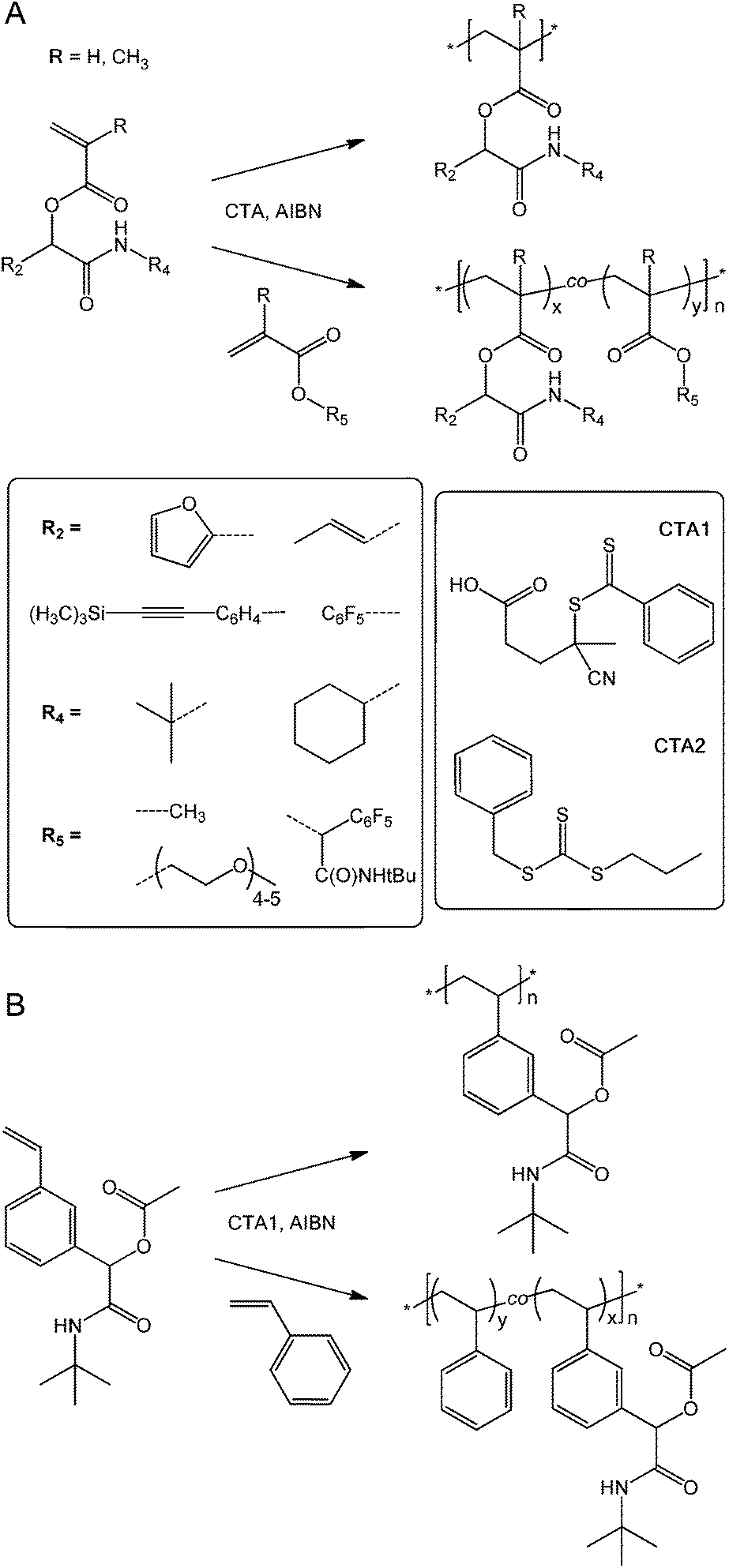 | ||
| Scheme 2 (Meth)acrylic (A) and styrenic (B) (co)polymer synthesis with structures of (co)monomers and chain transfer agents. | ||
Overall, the reactive, Passerini-produced monomers proved well-amenable to RAFT (co)polymerization and produced a series of well-defined homo- and copolymers. In all cases, the comonomer composition found by 1H NMR spectroscopy was very similar to the comonomer feed ratio suggesting high control over copolymer composition. Despite typically high conversions up to 95%, SEC-determined polydispersities ranged between 1.05 and 1.29 suggesting well controlled radical (co)polymerization, with the exception of two copolymers, both including PEGMA as comonomer, which had polydispersities of 1.47 (entry 4 in Table 2) and 1.39 (entry 9), respectively. We assumed that these broader size distributions stem from crosslinker impurities within the PEG-based comonomer, since the respective reactive monomers, tBu-MA-Pentene and tBu-MA-PFP, produced well defined copolymers with MMA (ĐSECM = 1.11 and 1.14, entries 3 and 8, respectively). SEC curves were typically nearly symmetrical and monomodal, see Fig. 1 and the ESI.† The low polydispersity ĐSECM = 1.11 measured for p(tBu-MA-Pentene0.37-co-MMA0.63) containing the double bond functional Passerini-monomer also suggested absence of radical side reactions of the double bond functionality resulting in crosslinking reactions. The styrenic monomer tBu-Ac-Sty was homopolymerized (Table 2, entry 12, MSECn = 16.7 kg mol−1, ĐSECM = 1.08) and copolymerized in equimolar feed with styrene (Table 2, entry 13, MSECn = 10.4 kg mol−1, ĐSECM = 1.05), both experiments resulting in products with very narrow molecular weight distributions, see curves a and b in Fig. 1. We also prepared a copolymer of two different Passerini-prepared monomers, tBu-MA-PFP and tBu-MA-Fur, employed in a 70:30 molar ratio, resulting in p(tBu-MA-PFP0.69-co-tBu-MA-Fur0.31) (Table 2, entry 11) with a measured nearly identical molar composition, a MSECn of 16.8 kg mol−1, and a low ĐSECM of 1.25, see trace c in Fig. 1, highlighting successful preparation of a well-defined dual-functional product.
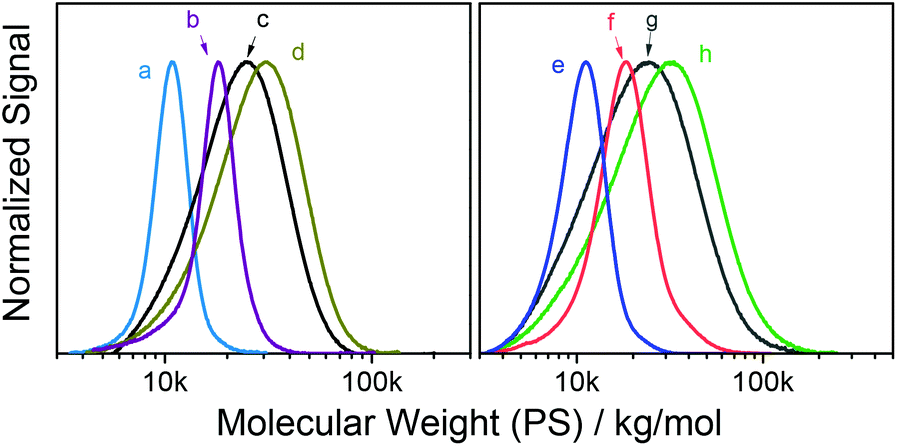 | ||
| Fig. 1 SEC traces of reactive homo- and copolymers; (a) p(tBu-Ac-Sty0.56-co-Sty0.44) (Table 2, entry 13), (b) p(tBu-Ac-Sty) (entry 12), (c) p(tBu-MA-PFP0.69-co-tBu-MA-Fur0.31) (entry 11), (d) p(tBu-MA-TMSyne) (entry 5), (e) p(tBu-MA-Pentene0.37-co-MMA0.63) (entry 3), (f) p(tBu-MA-Fur0.58-co-MMA0.42) (entry 2), (g) p(tBu-MA-Fur) (entry 1), and (h) p(tBu-MA-TMSyne0.50-co-MMA0.50) (entry 6). | ||
Copolymer p(tBu-MA-Pentene0.37-co-MMA0.63) (Table 2, entry 3) is used as a representative example to demonstrate the integrity of the side chain functionality after polymerization by NMR spectroscopy. 1H NMR spectra of monomer tBu-MA-Pentene and its copolymer with MMA are shown in Fig. 2. The upper, copolymer, spectrum in Fig. 2A clearly shows the broad signals of the pendent double bond, marked b’ and e’ at 5.90 and 5.46 ppm, and the signal of the allylic C–H group, marked f’ at 5.32 ppm, with integrals conforming to quantitative presence of the side chain double bond. 1H NMR spectra of a styrenic example are shown in Fig. 2B. The bottom spectrum of monomer tBu-Ac-Sty shows the typical complex pattern of a meta-substituted benzene derivative at 7.48–7.30 ppm, the common set of three dd resonances for the vinyl group and three singlets originating from the benzylic/α-amide/α-acetoxy proton (5.95 ppm, 1 H), the acetate group (2.18 ppm, 3 H) and the tert-butyl amide component (1.35 ppm, 9 H). The upper spectrum in Fig. 2B shows the broad signals expected for homopolymer p(tBu-Ac-Sty), with a slight upfield shift of the aromatic protons compared to those of the monomer due to the less electronegative effect of the aliphatic backbone compared to the vinyl group. Integration of signals (taking into account an overlapping resonance of the amide NH protons) conformed to the expected structure of a polystyrene derivative carrying an acetate-protected alcohol side group.
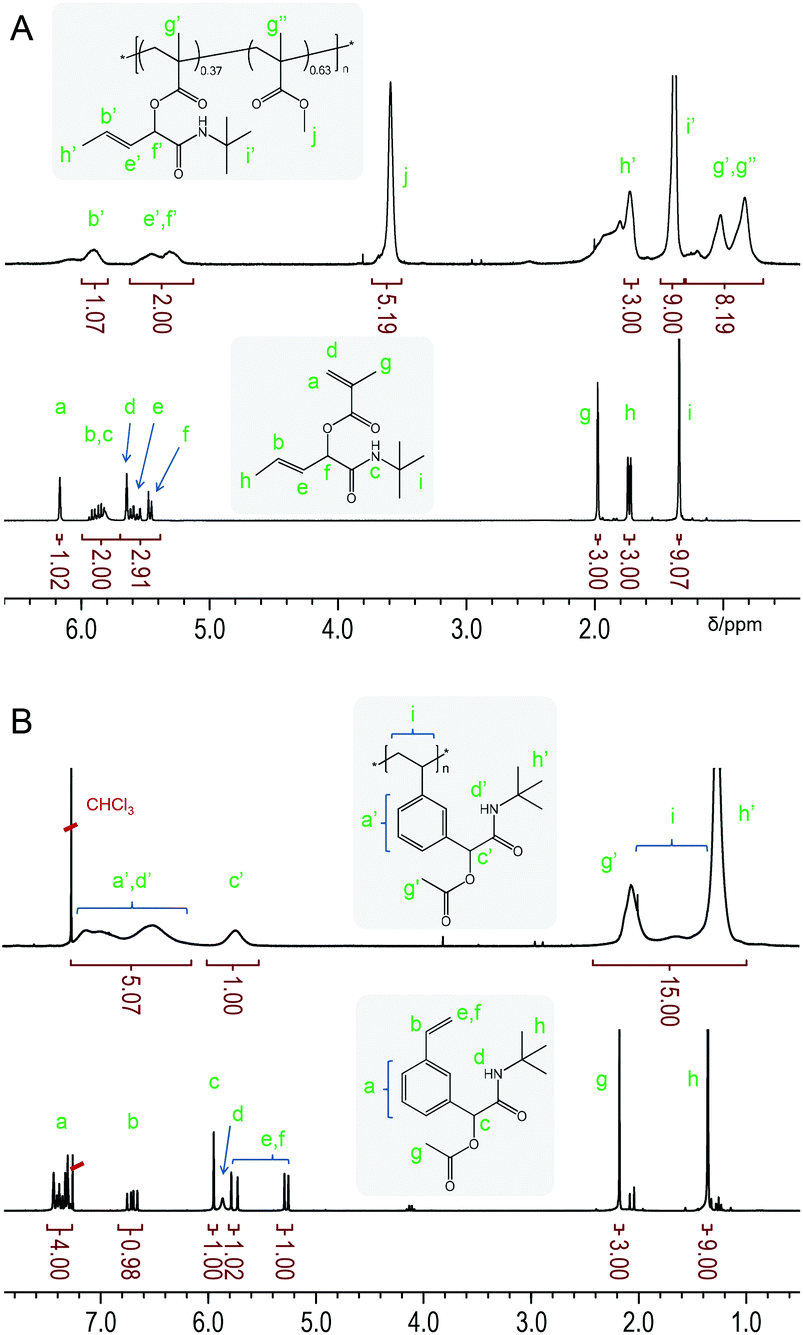 | ||
| Fig. 2 Representative 1H NMR spectra of a methacrylic monomer and its copolymer with MMA (A) and of a styrenic monomer with its homopolymer (B). | ||
FT-IR spectra of a Passerini-made monomer and its homopolymer are shown in Fig. 3 (curves a and b) for the example of tBu-Ac-Sty. The spectra are very similar and show the characteristic bands of ester CO stretching (ν = 1740 cm−1), amide CO stretching (ν = 1660 cm−1), methyl C–H rocking (ν = 1360 cm−1), ester C–O stretching and amide C–N stretching (overlapping at ν = 1220 cm−1) vibrations in agreement with the N-tert-butyl-2-acetoyloxy-acetamide structure. The spectrum of the monomer additionally exhibits signals attributed to vibrations of the vinyl group such as alkene C–H stretching (ν = 3090 cm−1) and alkene CC bending (ν = 910 cm−1) which are absent in the spectrum of the polymer.
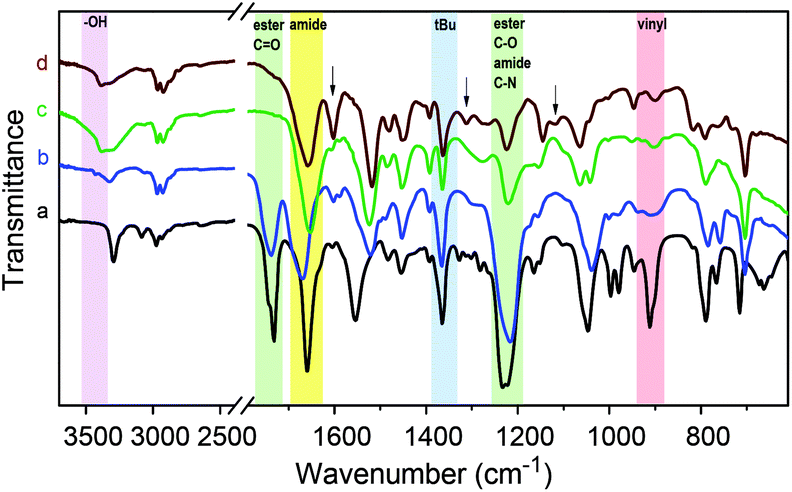 | ||
| Fig. 3 Sections of FT-IR spectra of (a) monomer tBu-Ac-Sty, (b) homopolymer p(tBu-Ac-Sty), (c) hydroxy-functional polystyrene derivative p(tBu-OH-Sty) after acetate deprotection, and (d) after dicyclohexylcarbodiimide (DCC) coupling with Methyl Red. Characteristic bands of O–H stretching (violet), ester CO stretching and ester C–O stretching (the latter overlapping with C–N stretching) (green), amide CO stretching (yellow), methyl C–H rocking (blue), and vinyl CC bending (red) vibrations are marked. Characteristic resonances of the attached dye molecule are highlighted by arrows in the top spectrum. | ||
After analysis by SEC, NMR spectroscopy, and FT-IR spectroscopy indicated successful synthesis of well-defined reactive (co)polymers, we additionally confirmed high RAFT end group fidelity through chain extension experiments. Homopolymer p(tBu-MA-TMSyne) (Table 2, entry 5) and copolymer p(tBu-MA-TMSyne0.50-co-MMA0.50) (Table 2, entry 6) were used as macro-RAFT agents in the polymerization of a block of MMA. The resulting species p[(tBu-MA-TMSyne)0.43-block-MMA0.57] (Table 2, entry 14) and p[(tBu-MA-TMSyne0.23-co-MMA0.23)-block-MMA0.54] (Table 2, entry 15) (indices referring to NMR-determined molar compositions) were further characterized by SEC (traces shown in the ESI†, results summarized in Table 2). Molecular weight distributions of the A-b-B and (A-co-B)-b-B copolymer species were shifted entirely toward higher apparent molecular weights compared to their respective macro-RAFT agents, with only slightly increased dispersities. This indicated that the majority of RAFT-made reactive (co)polymer chains carried RAFT agent end groups that allowed for re-initiation and extension with a second block. These experiments suggest that the set of Passerini-made monomers lends itself well to the construction of a wide range of complex architectures available through RDRP techniques.
A further expedient feature of these reactive building blocks was the solubility of the derived (co)polymers in organic solvents; details are compiled in the ESI†. All tested (co)polymers were soluble in anisole, chloroform, ethyl acetate, tetrahydrofuran, dimethylformamide, dimethylacetamide, dimethylsulfoxide, acetonitrile, and, unlike many polystyrene and poly(meth)acrylate derivatives, ethanol and methanol. All (co)polymers were found to be insoluble in diethyl ether and hexane. While the PEGMA-containing copolymers were soluble in cold water, all other (co)polymers were insoluble in water. Good solubility in polar organic solvents is presumably conferred by the N-functional amide side chain groups originating from the isocyanide reagents which are likely to undergo hydrogen bonding with polar solvents. An example that highlights the influence of the amide groups on polymer solubility is the comparison of p(tBu-MA-Fur) with literature-known poly(furfuryl methacrylate) in which the tert-butyl carbamoyl group is formally replaced by a hydrogen. While poly(furfuryl methacrylate) is insoluble in methanol,28 we found its N-tert-butyl amide-functional sister polymer to be soluble in methanol; dialysis in this solvent being, in fact, the preferred means of purification.
Postpolymerization modification
Given the successful preparation of 15 different homo- and copolymers with, generally, low polydispersities, the portfolio of Passerini-prepared monomers offers great potential for manifold postpolymerization modifications. As mentioned in the introduction, alkynes, double bonds, dienes, and pentafluorophenyl groups have been exploited for the efficient modification of a wide range of diversely structured polymeric substrates and can be expected to provide simple synthetic access to a multitude of novel materials derived from the series of multifunctional (co)polymers presented herein. As a preliminary example of a postpolymerization modification of a Passerini monomer-derived polymer, we chose a homopolymer carrying the, arguably, least reactive functional side group, an acetate-protected hydroxy group. Homopolymer p(tBu-Ac-Sty) was treated with sodium methoxide in anhydrous methanol, after which complete disappearance of the acetate –CH3 resonance at δ = 2.18 ppm and the shift of the benzylic proton from δ = 5.72 ppm (>CH–OAc) to δ = 4.61 ppm (>CH–OH) in a 1H NMR spectrum confirmed complete deprotection. An FT-IR spectrum, plotted in Fig. 3 (curve c) showed the appearance of a broad O–H stretching band at ν = 3390 cm−1, the complete disappearance of the ester CO stretching band at ν = 1740 cm−1, and, in the absence of ester C–O stretching vibration, a reduction of the absorbance at ν = 1220 cm−1, while the characteristic amide and alkane backbone resonances remained unchanged, likewise suggesting selective cleavage of the ester group. In order to demonstrate further chemical modification of the resulting hydroxy-functional p(tBu-OH-Sty), the pendent alcohol group was partially modified with the carboxylic acid functional dye Methyl Red by means of a dicyclohexylcarbodiimide coupling, see Fig. 4. A shortage of 45 mol% of Methyl Red reagent was employed using dichloromethane as solvent. After purification by repeated precipitation into hexane, the absence of residual unconjugated dye was confirmed by TLC and the resulting polymer was obtained as a dark brown solid. Integration of the broad signal at δ = 3.06 ppm in an 1H NMR spectrum attributed to the dimethylamino fragment of attached Methyl Red dye suggested a conversion of 67% of dye reagent resulting in a copolymer dye-labelled in approx. 30 mol% of its repeat units. Successful dye conjugation was also apparent from a weak band at ν = 1735 cm−1, (visible as a shoulder on the adjacent amide CO stretching band) in an FT-IR spectrum, shown in Fig. 3 (curve d), which was attributed to the CO stretching vibration of the ester linking the Methyl Red to the polymer. Characteristic absorbance bands of the dye were visible and are marked with arrows in Fig. 3. The modified copolymer dissolved in ethanol containing a drop of aqueous HCl with distinct red color, visually confirming successful sequential postpolymerization modification, see photograph in Fig. 4.
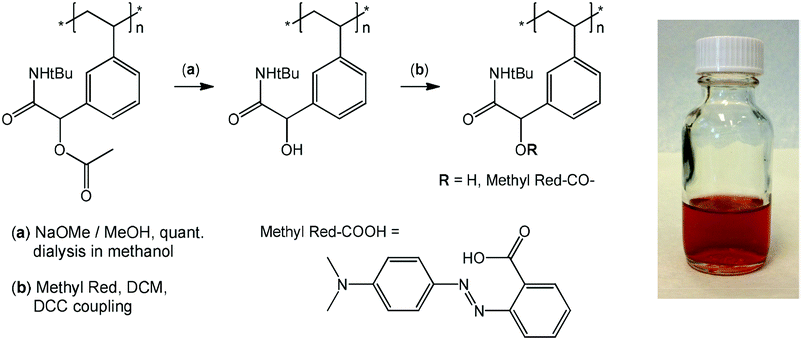 | ||
| Fig. 4 Example of postpolymerization modification of p(tBu-Ac-Sty) by alcohol deprotection and coupling with the carboxylic acid functional dye Methyl Red with photograph of purified polymer (0.5 g L−1) in acidified ethanol. | ||
Conclusions
We investigated dichloromethane and water as solvents for the multicomponent Passerini reaction using functional aldehydes and carboxylic acids to generate novel (meth)acrylic and styrenic monomers carrying common reactive groups amenable to efficient postpolymerization modification. Producing up to quantitative yields after simple purification, water proved to be the more expedient solvent choice. RAFT (co)polymerization successfully provided a series of well-defined homo- and copolymers equipped with pendent double bond, protected triple bond, diene, pentafluorophenyl, and acetate functionality (or a combination thereof) with generally low polydispersities and high control over molar composition and end group fidelity. In addition to polymerizable and reactive groups, the Passerini-prepared series of monomers contained a further functional amide group derived from the isocyanide component offering good (co)polymer solubility in polar organic solvents and providing potential for further functionalization and fine-tuning of the obtainable polymeric materials. Together with the well-documented reactivity of the pendent functional groups which we demonstrated exemplarily by the dye-functionalization of a hydroxy-functional polystyrene derivative, these novel monomers and (co)polymers offer large potential for postpolymerization modification (as well as the generation of libraries of multifunctional monomeric building blocks) and the synthesis of novel multifunctional materials with tailored properties.Acknowledgements
The authors acknowledge Mr E. Alipour for synthetic assistance. P.J.R. acknowledges the Australian Research Council (ARC) for funding through a Discovery Early Career Researcher Award (DE120101547).References
- K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Polym. Chem., 2013, 51, 1–28 CrossRef.
- G. B. H. Chua, P. J. Roth, H. T. T. Duong, T. P. Davis and A. B. Lowe, Macromolecules, 2012, 45, 1362–1374 CrossRef CAS.
- P. J. Roth, Macromol. Chem. Phys., 2014, 215, 825–838 CrossRef CAS.
- P. Theato, J. Polym. Sci., Polym. Chem., 2008, 46, 6677–6687 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176–191 RSC.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46–48 CrossRef CAS PubMed.
- J. G. Rudick, J. Polym. Sci., Polym. Chem., 2013, 51, 3985–3991 CrossRef CAS.
- O. Kreye, T. Tóth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790–1792 CrossRef CAS PubMed.
- O. Kreye, O. Türünç, A. Sehlinger, J. Rackwitz and M. A. R. Meier, Chem. – Eur. J., 2012, 18, 5767–5776 CrossRef CAS PubMed.
- A. Sehlinger, O. Kreye and M. A. R. Meier, Macromolecules, 2013, 46, 6031–6037 CrossRef CAS.
- R. Kakuchi and P. Theato, ACS Macro Lett., 2013, 2, 419–422 CrossRef CAS.
- C. Zhu, B. Yang, Y. Zhao, C. Fu, L. Tao and Y. Wei, Polym. Chem., 2013, 4, 5395–5400 RSC.
- A. E. J. de Nooy, D. Capitani, G. Masci and V. Crescenzi, Biomacromolecules, 2000, 1, 259–267 CrossRef CAS.
- L. Li, X.-W. Kan, X.-X. Deng, C.-C. Song, F.-S. Du and Z.-C. Li, J. Polym. Sci., Polym. Chem., 2013, 51, 865–873 CrossRef CAS.
- B. Yang, Y. Zhao, C. Fu, C. Zhu, Y. Zhang, S. Wang, Y. Wei and L. Tao, Polym. Chem., 2014, 5, 2704–2708 RSC.
- X.-X. Deng, L. Li, Z.-L. Li, A. Lv, F.-S. Du and Z.-C. Li, ACS Macro Lett., 2012, 1, 1300–1303 CrossRef CAS.
- I.-H. Lee, H. Kim and T.-L. Choi, J. Am. Chem. Soc., 2013, 135, 3760–3763 CrossRef CAS PubMed.
- L. Li, A. Lv, X.-X. Deng, F.-S. Du and Z.-C. Li, Chem. Commun., 2013, 49, 8549–8551 RSC.
- S. H. Kim, S. H. Park, J. H. Choi and S. Chang, Chem. – Asian J., 2011, 6, 2618–2634 CrossRef CAS PubMed.
- P. J. Roth, M. Collin and C. Boyer, Soft Matter, 2013, 9, 1825–1834 RSC.
- J. Y. Quek, P. J. Roth, R. A. Evans, T. P. Davis and A. B. Lowe, J. Polym. Sci., Polym. Chem., 2013, 51, 394–404 CrossRef CAS.
- M. C. Pirrung and K. D. Sarma, J. Am. Chem. Soc., 2003, 126, 444–445 CrossRef PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- J. He, Y. Zhang and E. Y. X. Chen, J. Polym. Sci., Polym. Chem., 2013, 51, 2793–2803 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 19F NMR spectra of all monomers, additional SEC traces of (co)polymers and for chain extension experiments, table summarizing (co)polymer solubility. See DOI: 10.1039/c4py01147c |
| This journal is © The Royal Society of Chemistry 2015 |