
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Degradable cross-linked polymer vesicles for the efficient delivery of platinum drugs†
Q.
Fu‡
,
J.
Xu‡
,
K.
Ladewig
,
T. M. A.
Henderson
and
G. G.
Qiao
*
Polymer Science Group, Department of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville, VIC 3010, Australia. E-mail: gregghq@unimelb.edu.au; Fax: +61-3-83444153
First published on 16th September 2014
Abstract
Nontoxic and acid-degradable cross-linked polymeric vesicles as efficient nanocarriers for the delivery of platinum drugs are reported. Well-defined linear-brush diblock copolymers were synthesized via a robust ring opening metathesis polymerization technique and self-assembled in a selective solvent to form polymeric vesicles with bilayer structure. Vesicles were subsequently cross-linked using acid-degradable diamino ketal cross-linkers and the anticancer drug cis-platin was conjugated to the vesicles with a high loading content (17.6%) through the formation of stable chelate ring structure. This acid-degradable linker leads to a faster drug release in saline under acidic conditions (pH = 5.5) such as those encountered in the late endosome compared to physiological pH (pH = 7.4). Cancer cells tended to readily internalize the vesicles and dose–response cytotoxicity studies suggested that the drug-loaded cross-linked vesicles were more efficient in delivering the platinum drug into cancer cells compared to internalization of the free drug. Additionally, polymer vesicles prior to platinum conjugation were nontoxic towards cancer cells.
Introduction
cis-Dichlorodiaminoplatinum(II) (cisplatin, CDDP) is one of the most potent chemotherapy drugs, which has been widely used to treat various types of cancers, including sarcomas, lung, ovarian, testicular, lymphomas, and germ cell tumors.1 This platinum complex acts in vivo, by binding to and cross-linking of DNA, which ultimately triggers apoptosis.2,3 However, platinum drugs have well recognized drawbacks, such as inactivity when administered orally, low solubility in water, very short half-lives in the body, and induction of severe side-effects including nausea, vomiting, renal dysfunction, neurotoxicities as well as drug resistance.1,4In recent years, significant effort has been devoted to develop matrix-based systems for the delivery of platinum drugs5–9 since these have the capability to increase drug solubility, protect drugs from undesirable biological binding events, reduce systemic toxicity and localize platinum drugs at tumor sites via the enhanced permeability and retention (EPR) effect.10,11 Stenzel and co-workers12,13 reported the synthesis of polymer micelles for cisplatin delivery and controlled release. More recently, Johnson et al.14 have prepared star-like polymeric nanoparticles carrying multiple therapeutic agents, including doxorubicin, camptothecin, and cisplatin. Although the concept of using matrix systems as platinum carrier has been well established in the laboratory, it is still desirable to design novel more efficient drug delivery systems.
The polymer vesicles, consisting of aqueous interiors entrapped by bilayer lipid-like membranes, are a class of nanoscale self-assemblies from synthetic or natural polymers inspired by complex biological systems such as cells and viral capsids.15–17 In contrast to matrix system, polymer vesicles exhibit versatile transport properties, as hydrophobic drugs can be enclosed in the membrane of the carrier, whereas hydrophilic drugs are encapsulated in their aqueous cavity.5
Vesicles therefore enable the encapsulation of larger amounts of drugs than matrix systems, which means that much smaller amounts of vectors can be administered.18
Recently, we reported a simple and robust approach to fabricate polymer vesicles from linear-brush diblock copolymers.19 These synthetic vesicles provided desirable bidentate functionalities within their bilayer membrane which enable the binding of cisplatin by forming a stable platinum complex via 7-member rings.12,20 We previously showed that this approach afforded a high drug loading content within the vesicles and a controlled drug release profile in physiological media.
Herein, we report the further development of an ameliorated vesicular system containing acid-sensitive cross-linkers, which can stabilize the assembled structure in physiological media and degrade in the endosomal environment of cells for a more efficient cisplatin drug delivery. The polymer vesicles were constructed by self-assembly of well-defined linear-brush polyoxanorbornene-based diblock copolymers in dry acetone, followed by cross-linking between pendant anhydrides and acid-cleavable diamino ketal cross-linkers. Hydrolysis of the anhydrides led to the formation of diacid functionalities, which were subsequently employed to conjugate cisplatin. Vesicles with and without drugs were characterized by dynamic light scattering (DLS), transmission electron microscopy (TEM) and thermogravimetric analysis (TGA). Drug release profiles, cytotoxicity and cellular uptake of drug-loaded vesicles were then investigated in detail.
Experimental
Materials
N-Propargylamine (98%), copper(I) bromide (CuBr, 98%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), 2nd generation Grubbs catalyst (H2IMes)(PCy3)(Cl)2RuCHPh, cis-dichlorodiaminoplatinum(II) (CDDP, 99.9%), 2,2′-(propane-2,2-diyl bis(oxy))diethanamine (PDDE), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (98%, EDC), anhydrous pentane (≥99%), anhydrous pyridine (99.8%) and phosphate-citrate buffer tablet were all purchased from Aldrich and used as received. Phosphate-buffered saline (PBS, Life technologies), paraformaldehyde (Life technologies) N,N-dimethylformamide (DMF, 99.8%, Merck), n-hexane (AR, Chem-Supply) and diethyl ether (AR, Chem-Supply) were also used as received. 4′,6-Diamidino-2-phenylindole (DAPI) and Alexa Fluor® 594 (AF594) were purchased from Life Technologies and used as received. Acetone (AR, Chem-Supply) was distilled from calcium hydride under argon. Tetrahydrofuran (THF, AR, Chem-Supply) was distilled from benzophenone and sodium metal under argon. exo-7-Oxabicyclo(2.2.1)-5-heptene-2,3-dicarboxylic anhydride (oxa-norbornenyl anhydride, ONBAn),21 ω-oxanorbornenyl poly(ethylene oxide) (ONB-PEG, Mn = 2.0 kDa) macromonomer,22 and pyridine modified 2nd generation Grubbs catalyst (H2IMes)(pyr)2(Cl)2RuCHPh23 were synthesized according to literature procedures.Instruments
Gel permeation chromatography (GPC) (THF as eluent) was performed on a Shimadzu liquid chromatography system fitted with a Wyatt DAWN EOS multi-angle laser light scattering (MALLS) detector (690 nm, 30 mW) and a Wyatt OPTILAB DSP interferometric refractometer (690 nm), using three Phenomenex Phenogel columns (500, 104, and 106 Å porosity; 5 μm bead size) operated at 1 mL min−1 with column temperature set at 40 °C. Astra software (Wyatt Technology Corp.) was used to process the data to determine the molecular weights based on the assumption of 100% mass recovery of the polymer where the dn/dc value was unknown. 1H NMR spectroscopic analysis was performed on a Varian Unity Plus 400 MHz spectrometer using the deuterated solvent as reference. Hydrodynamic diameters (Dh) and size distributions of the nanoparticles in aqueous solutions were determined by dynamic light scattering (DLS). DLS measurements were performed using a Malvern high performance particle sizer (HPPS) with a 3.0 mW He–Ne laser operated at 633 nm at an angle of 173° (back scattering) and a constant temperature of 25 ± 0.1 °C. A laboratory-built humidity-controlled vitrification system was used to prepare the samples for cryo-transmission electron microscopy (cryo-TEM). Humidity was kept close to 80% for all experiments, and ambient temperature was 22 °C. 200-mesh copper grids coated with perforated carbon film (Lacey carbon film: ProSciTech, Qld, Australia) were used for all experiments. Grids were first glow discharged in nitrogen to render them hydrophilic. 4 μL aliquots of the sample were pipetted onto each grid prior to plunging. After 30 seconds adsorption time grids were blotted manually using Whatman 541 filter paper, for approximately 2 seconds. Blotting time was optimised for each sample. Grids were then plunged into liquid ethane cooled by liquid nitrogen. Frozen grids were stored in liquid nitrogen until required. The samples were examined using Tecnai 12 Transmission Electron Microscope (FEI, Eindhoven, The Netherlands) at an operating voltage of 120 kV, equipped with a Gatan 626 cryoholder (Gatan, Pleasanton, CA, USA) and an Eagle 4k × 4k CCD camera (FEI, Eindhoven, The Netherlands). At all times low dose procedures were followed, using an electron dose of 8–10 electrons Å−2 for all imaging. Images were recorded using a magnification in the range 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× to 30000×. Thermogravimetric analysis (TGA) was performed on a PerkinElmer Pyris-1 thermogravimetric analyzer, and the samples were heated from 70 to 700 °C at a heating rate of 10 K min−1 under an atmosphere flow (20 mL min−1). UV-vis spectrometry was performed on a Varian Cary 50 Bio UV-Visible spectrophotometer using quartz cuvettes with a 1 cm path length. Confocal microscope was performed on a Leica TCS SP2 confocal microscope.
000× to 30000×. Thermogravimetric analysis (TGA) was performed on a PerkinElmer Pyris-1 thermogravimetric analyzer, and the samples were heated from 70 to 700 °C at a heating rate of 10 K min−1 under an atmosphere flow (20 mL min−1). UV-vis spectrometry was performed on a Varian Cary 50 Bio UV-Visible spectrophotometer using quartz cuvettes with a 1 cm path length. Confocal microscope was performed on a Leica TCS SP2 confocal microscope.
Synthesis of cross-linked polymer vesicles
The experimental details of the synthesis of diblock copolymer, poly(ONBAn-b-ONB-PEG) via the ring-opening metathesis polymerization (ROMP) can be found in our previous publication.19 For the poly(ONBAn35-b-ONB-PEG5), Mn (GPC) = 14.9 kDa, Mw/Mn = 1.21. 1H NMR (400 MHz, CDCl3, 25 °C, δH ppm): 7.80–7.75 (s, 1H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of triazole ring), 6.05 (s, 2H, –CHCH–, trans), 5.75 (s, 2H, –CHCH–, cis), 5.05–4.80 (m, 2H, –CH–O–CH–, cis), 4.60 (s, 2H, –N–CH2–), 4.55–4.40 (m, 4H, –CH–O–CH– trans and –N–CH2CH2O–), 3.75 (s, 2H, –N–CH2CH2O–), 3.60–3.40 (m, 4H, –CH2CH2O–), 3.25–3.05 (m, 5H, –CH–CO and –O–CH3).
C of triazole ring), 6.05 (s, 2H, –CHCH–, trans), 5.75 (s, 2H, –CHCH–, cis), 5.05–4.80 (m, 2H, –CH–O–CH–, cis), 4.60 (s, 2H, –N–CH2–), 4.55–4.40 (m, 4H, –CH–O–CH– trans and –N–CH2CH2O–), 3.75 (s, 2H, –N–CH2CH2O–), 3.60–3.40 (m, 4H, –CH2CH2O–), 3.25–3.05 (m, 5H, –CH–CO and –O–CH3).
The formed linear-brush diblock copolymer (Mn = 14.9 kDa, 100 mg, 6.71 × 10−6 mol) was dissolved in 20 mL distilled THF at a concentration of 5 mg mL−1. The sample was then filtered through a PTFE filter (pore size 0.45 μm). Subsequently, 10 mL distilled acetone was added dropwise into the solution under moderate stirring at room temperature. The mixture was then dialyzed against distilled acetone for 24 h using a dialysis membrane (MWCO 7.0 kDa) to remove THF. The particle size was confirmed by DLS as ∼95 nm.
The nanoparticles with different degrees of cross-linking (DOC, 14 mol%, 28 mol% and 56 mol%) were prepared in parallel according to similar procedures. A typical procedure (DOC = 28 mol%) can be described as followed. A solution of the cross-linker PDDE (2.7 mg, 1.67 × 10−5 mol) was prepared in 100 μL distilled acetone and added slowly to 5 mL vesicle solution (10 mg mL−1) with stirring. The mixture was kept stirring overnight. The final solution was dialyzed against distilled acetone for 2 days to remove all impurities using a dialysis membrane (MWCO 7.0 kDa). Morphological observation of the cross-linked vesicle, XV was performed by DLS and TEM.
50 mg XV were then added into 5 mL 0.1 M NaOH solution. This solution was stirred for 30 min and then dialyzed against distilled water for 2 days to remove NaOH using Spectra/Por (Spectrum Laboratories Inc.) regenerated cellulose dialysis membrane (MWCO 5.0 kDa). After freeze-drying, hydrolysed cross-linked vesicle HXV was obtained as a grey-coloured powder. Morphological observation of the hydrolysed cross-linked vesicles was performed by DLS and cryo-TEM.
Drug loading and release
The conjugation of cis-platin (CDDP) to the polymer vesicles was achieved by following a slightly modified procedure compared to our previous report.19 Thereby incorporation of the CDDP was achieved via the reaction of hydrolysed, cross-linked vesicles (HXV) with a cis-diaminediaqua-platinum(II) complex obtained by hydrolysis of CDDP. Briefly, CDDP (10 mg) was suspended in 10 mL distilled water and mixed with silver nitrate ([AgNO3]/[CDDP] = 1.955) to form the aqueous complex. The solution was stirred in the dark at room temperature for 4 h. As the reaction proceeded a white precipitate of AgCl was observed. The mixture was then centrifuged at 4400 rpm for 1 hour to remove the AgCl precipitate and the supernatant was purified by passing through a 0.22 μm filter. HXV (40 mg, dissolved in 4 mL of DI water) were added to the above CDDP aqueous solution and left to react in a water bath at 37 °C for 12 h with gentle shaking resulting in platinum-loaded polymer nanocarriers. The prepared conjugates were purified by dialysis against distilled water using regenerated cellulose dialysis membrane (MWCO 5.0 kDa), followed by freeze-drying, yielding a light brown powder. The platinum conjugation efficiency to the cross-linked vesicles was determined by TGA as 85.2% by eqn (1).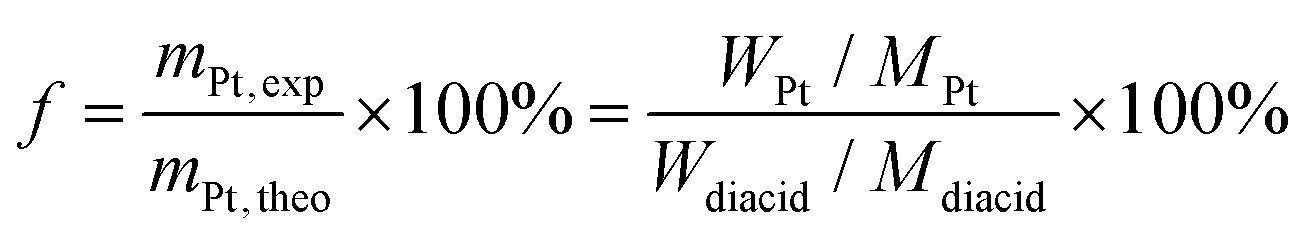 | (1) |
The platinum loading content within the polymer nanoparticles is determined as 17.6% by eqn (2).
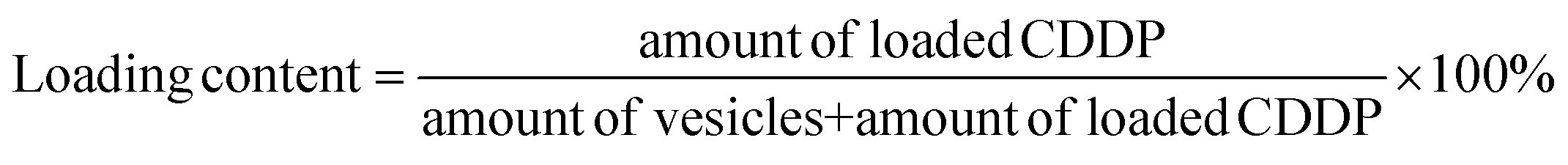 | (2) |
The in vitro release of the CDDP was quantified using a dialysis method. Thereby, the platinum-loaded vesicles (Pt–V, 10 mg) were dissolved in 4 mL saline solution (0.9 wt% of NaCl, 0.15 M). The solution was evenly added to two dialysis tubings (MWCO = 3.5 kDa) and dialyzed against pH buffer solutions (phosphate-citrate buffers, pH = 7.4 and 5.5) (200 mL) at 37 °C, respectively. 0.9 wt% of NaCl was added into these buffer solutions to trigger the platinum drug release. At certain time intervals, the release media was sampled and analysed by means of o-phenylenediamine colorimetric assay (o-PDA) according to previously published methods.20,24 Samples with an unknown Pt content were added to 1 mL of o-PDA solution in N,N′-dimethylformamide (DMF) (2 mg mL−1) and heated for 10 min at 100 °C. The amount of platinum present in the sample was determined by measuring the absorbance at 703 nm using CDDP as a standard curve. The concentration of platinum released from the conjugate was expressed as a ratio of the amount platinum in the releasing solution and that in the initial sample. The percentage of platinum released was calculated by eqn (3):
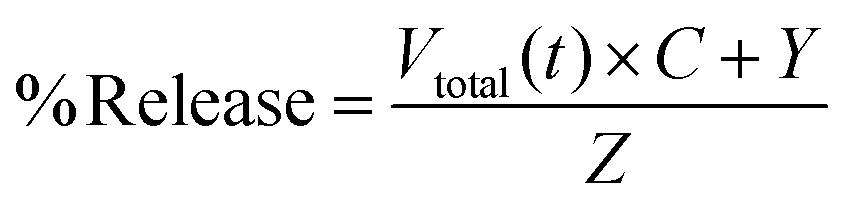 | (3) |
| % CDDP released = Aexp(−t/τ) + y0. | (4) |
| R 2 | y 0 | A | τ | |
|---|---|---|---|---|
| pH 5.5 | 0.986 | 83.2 | −72.3 | 47.8 |
| pH 7.4 | 0.981 | 51.2 | −46.5 | 39.1 |
Cell culture
HeLa cells were maintained in ‘complete’ DMEM medium (supplemented with 10% FBS, 1× GlutaMAX™, and 1× antibiotic–antimycotic) in a humidified atmosphere containing 5% CO2 at 37 °C. Usually, cells were seeded in a T175 flask (ca. 3 × 106 cells mL−1) and passaged twice a week prior to the performance of the subsequent cell viability or cell uptake imaging studies.Cytotoxicity assay
Cytotoxicity of the polymers was assessed using Invitrogen's alamarBlue® cell viability reagent following the manufacturer's instructions. Briefly, a 96 well ‘mixing plate’ with a concentration gradient of the drug-loaded vesicles (2 × 10−6–4 × 10−3 mol Pt L−1, 1 × 10−7–2 × 10−4 mol polymer L−1) and corresponding gradients of free polymer, empty vesicles and degraded vesicles (1 × 10−7–2 × 10−4 mol polymer L−1) as well as free drug and degraded drug-loaded vesicle (2 × 10−6–4 × 10−3 mol Pt L−1) was prepared. In another 96 well plate, ‘the experimental plate’, 80 μL of cell suspension was seeded in each well (ca. 10000 cells per well) except for the ‘medium blanks’ in which 80 μL of medium was added instead. 20 μL of the corresponding well on the ‘mixing plate’ was then added to the cell suspension on the ‘experimental plate’ which was subsequently incubated in a humidified atmosphere containing 5% CO2 at 37 °C. After 48 h, 10 μL of alamarBlue® cell viability reagent was added to each well (except for three wells containing medium only). After 3–3.5 h of incubation under the same growth conditions, the absorbance at 570 and 600 nm of each well was measured by UV-vis spectrophotometer. The absorbances of each well were corrected against the medium-only wells without alamarBlue® reagent and the Ro correction factor for each plate was calculated as follows: Ro = Corrected λ570 of alamarBlue® in media/Corrected λ600 of alamarBlue® in media. Subsequently, the amount of reduced alamarBlue® (AR570) was calculated for each well as follows:| AR570 = Corrected λ570 − (Corrected λ600 × Ro) |
AR570 for each well was then expressed as a percentage of growth control. Note that all experiments were conducted in triplicate on three different occasions and error bars shown represent the standard error of the mean of independent experiments.
Cellular uptake
The hydrolysed, cross-linked vesicle, HXV (2 mg) were dissolved in 1 mL DI water, then 0.34 mg EDCI ([COOH]:[EDC] = 4) and 0.005 mg AF594 were added to the solution. The mixture was then reacted at R.T. for 48 h after which any excess AF594 was removed by dialysis against deionized (DI) water.
On day-1 sterile round glass cover slips (∅ 15 mm) were placed in the wells of a 12 well plate and HeLa cells were seeded in ‘complete DMEM’ medium at a cell density of 200000 cells per well in 0.5 mL seeding volume before the plate was placed in a humidified incubator (95–100% humidity, 5% CO2) overnight. On day 0, the seeding medium was removed from each well and cells were gently washed with 1 mL sterile PBS. Approx. 0.5 mL fresh ‘complete DMEM’ medium and then 100 μL vesicle suspension was added to each well. The plate was returned to the incubator for 12–24 h. On day 1, the medium was removed from each well and cells were gently washed with PBS. Samples were fixed using paraformaldehyde before being mounted onto microscopy slides using DAPI Fluoromount G™ (ProSciTech). Samples were imaged by confocal microscope.
Results and discussion
The linear-brush diblock copolymers were synthesized via ring-opening metathesis polymerization (ROMP) of two (macro)monomers, oxanorbornenyl anhydride (ONBAn) and ω-oxanorbornenyl poly(ethylene glycol) (ONB-PEG, Mn = 2.0 kDa) according to established procedures.19 Copolymers were subsequently self-assembled in dry acetone to afford polymeric vesicles with pendant anhydrides (Scheme 1). Thereafter, the acid-cleavable cross-linker 2,2′-(propane-2,2-diyl bis(oxy))diethanamine (PDDE) was utilized to stabilize the vesicular structure through reaction with the anhydride units in the bilayer membrane. After hydrolysis of any unreacted anhydrides, the platinum drug was loaded into the cross-linked vesicles with high loading content. The cross-linked vesicles are designed to degrade gradually at pH = 5.5, accompanied by the release of cisplatin in saline solution.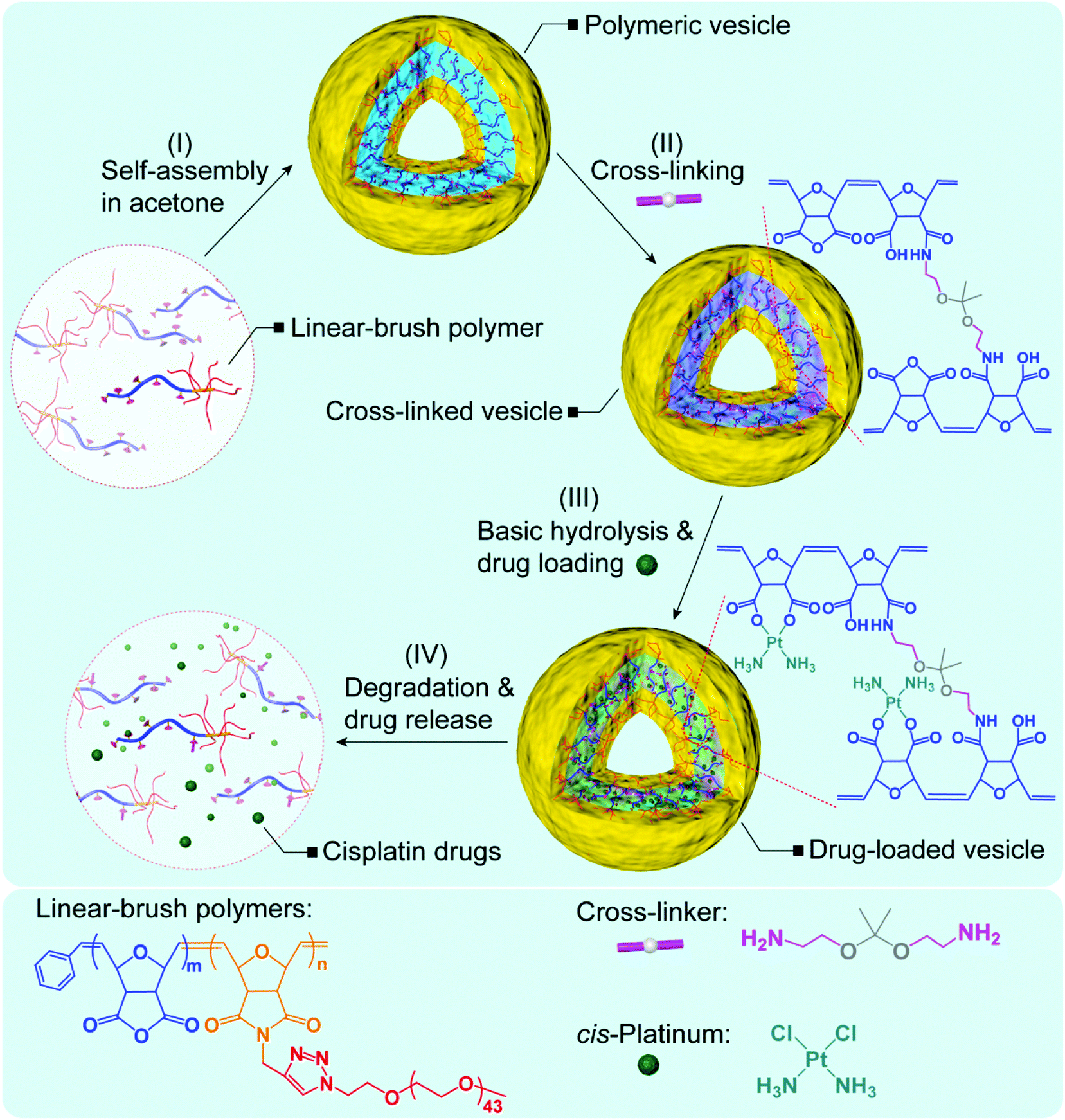 | ||
| Scheme 1 The synthetic route towards nontoxic, acid-degradable polymer vesicles for the delivery of platinum drugs. | ||
Synthesis of cross-linked polymer vesicles
ROMP is a powerful and broadly applicable approach for the preparation of macromolecular materials.25,26 The linear-brush diblock copolymer poly(ONBAn35-b-ONB-PEG5) was prepared via efficient ROMP as follows: ONBAn (35.0 equiv.) was exposed to pyridine modified Grubbs second-generation catalyst (cat., 1.0 equiv.) for 20 min. ONB-PEG (5.0 equiv.) was then added, and the mixture was stirred for 1 h at room temperature. The formed linear-brush copolymer has a molecular weight of 14.9 kDa with a narrow polydispersity of 1.21 (Fig. 1A). The high conversion of ONB-PEG macromonomers (MMs) in ROMP was revealed by 1H NMR spectroscopy. In Fig. 1B, the olefin proton resonances a at δ 6.50 ppm (refer to spectrum 1) totally disappeared after ROMP, which confirms the quantitative consumption of the MMs. The ‘ene’ proton resonance of the linear-brush backbone a′ (trans/cis) at δ 5.4–5.8 ppm (refer to spectrum 2) derived from ring-opening metathesis was observed, which further verified the formation of the linear-brush diblock copolymer.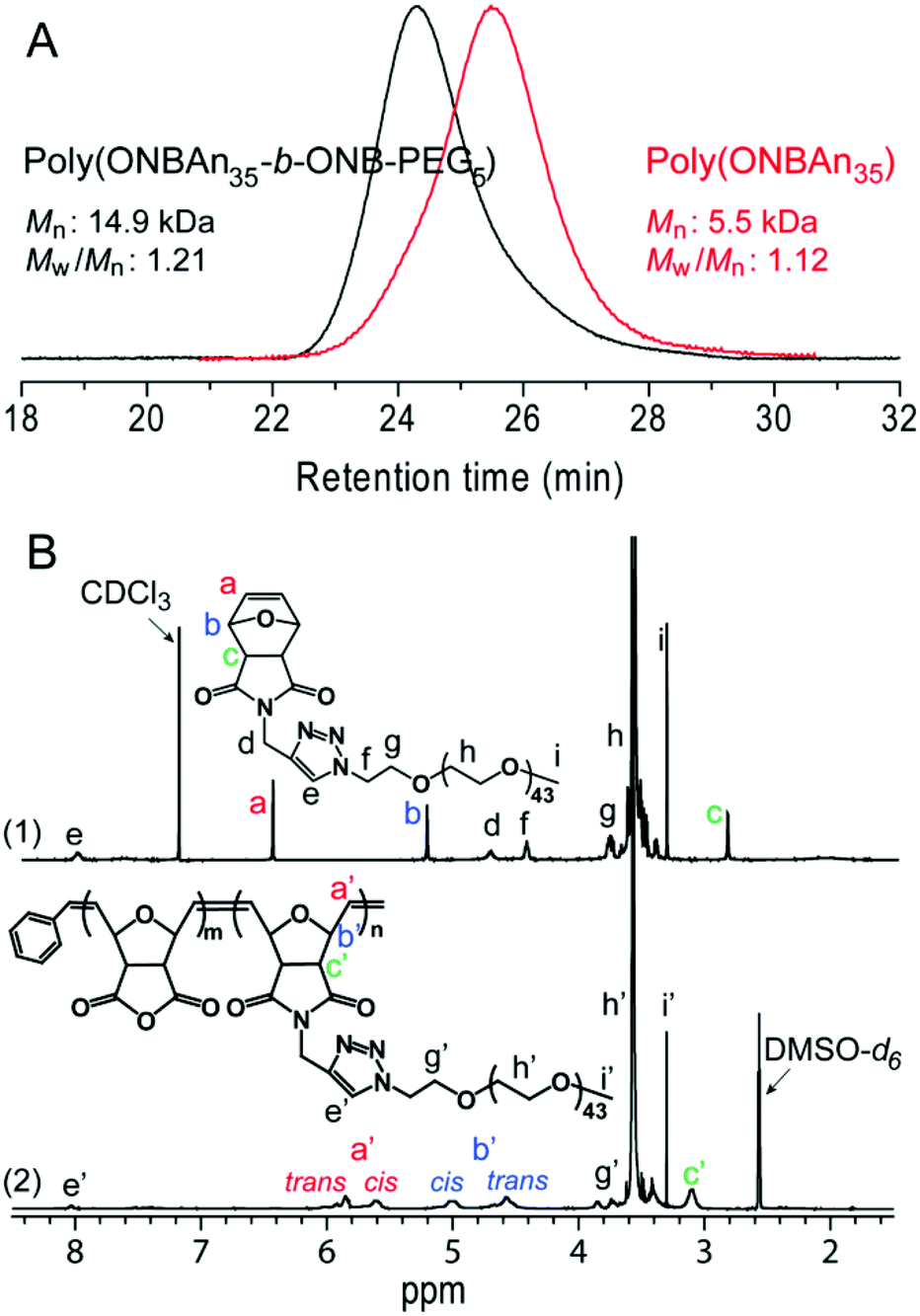 | ||
| Fig. 1 Characterization of linear-brush diblock copolymers poly(ONBAn35-b-ONB-PEG5) (A) GPC measurements and (B) 1H NMR spectroscopic analysis. | ||
The self-assembly of poly(ONBAn35-b-ONB-PEG5) was then performed in dry solvents to protect the anhydride units. The assembled structure formed upon addition of acetone (which is a non-solvent for the poly(ONBAn) block of the diblock copolymer) to the common solvent THF. Firstly, the diblock copolymer was dissolved in dry THF. Subsequently, the slow addition of dry acetone led to the aggregation of the poly(ONBAn) block to form a micellar solution. Residual THF was then removed via dialysis against an excess of dry acetone. TEM analysis indicated the formation of micellar structure (see Fig. S1 in ESI†) and dynamic light scattering (DLS) measurements revealed an intensity-average hydrodynamic diameter, Dh, of ∼95 nm and a polydispersity of 0.03 (see Fig. S2 in ESI†) for these assembles.
The self-assembled structure was then stabilized through partial cross-linking of the ONBAn units (i.e. 28 mol%) using an equivalent amount of the acid-degradable cross-linker PDDE. Utilization of this cross-linker means, that the resulting cross-linked polymer vesicles are biodegradable in an endosomal/lysosomal environment (pH ∼ 5). Given once degraded into the linear-brush diblock copolymers no further degradation into molecular components is possible, the degradation products can nonetheless be cleared from the body since their molecular weights are below the renal clearance threshold (40 kDa for linear PEG).27 The DLS measurement of the cross-linked nanoparticles revealed a Dh of ∼96 nm (see Fig. S2 in ESI†). In addition, nanoparticles with different degrees of cross-linking (DOC, i.e., 14 mol% and 56 mol%) were prepared in parallel according to similar procedures.
Thereafter, hydrolysis of the anhydride functionalities was carried out in 0.1 M NaOH solution for 30 minutes. The ONBAn units were converted to ONB-diacid functionalities according to our previous study.19 After dialysis against deionized (DI) water, the micellar solutions were freeze-dried to give hydrolysed, cross-linked vesicles. Cryo-TEM images without staining showed uniform nanoparticles with an increased Dh of ∼240 nm (Fig. 2A, DOC of 28 mol%) due to increased solubility resulting in swelling of nanoparticles, which agreed with the results from DLS measurements (see Fig. S3 in ESI†). Additionally, the definable bilayer membrane with ∼50 nm thickness was observed from cryo-TEM images (Fig. 2B), clearly indicating a vesicular morphology. Additionally, TEM observation also indicated some asymmetric and imperfect vesicles.
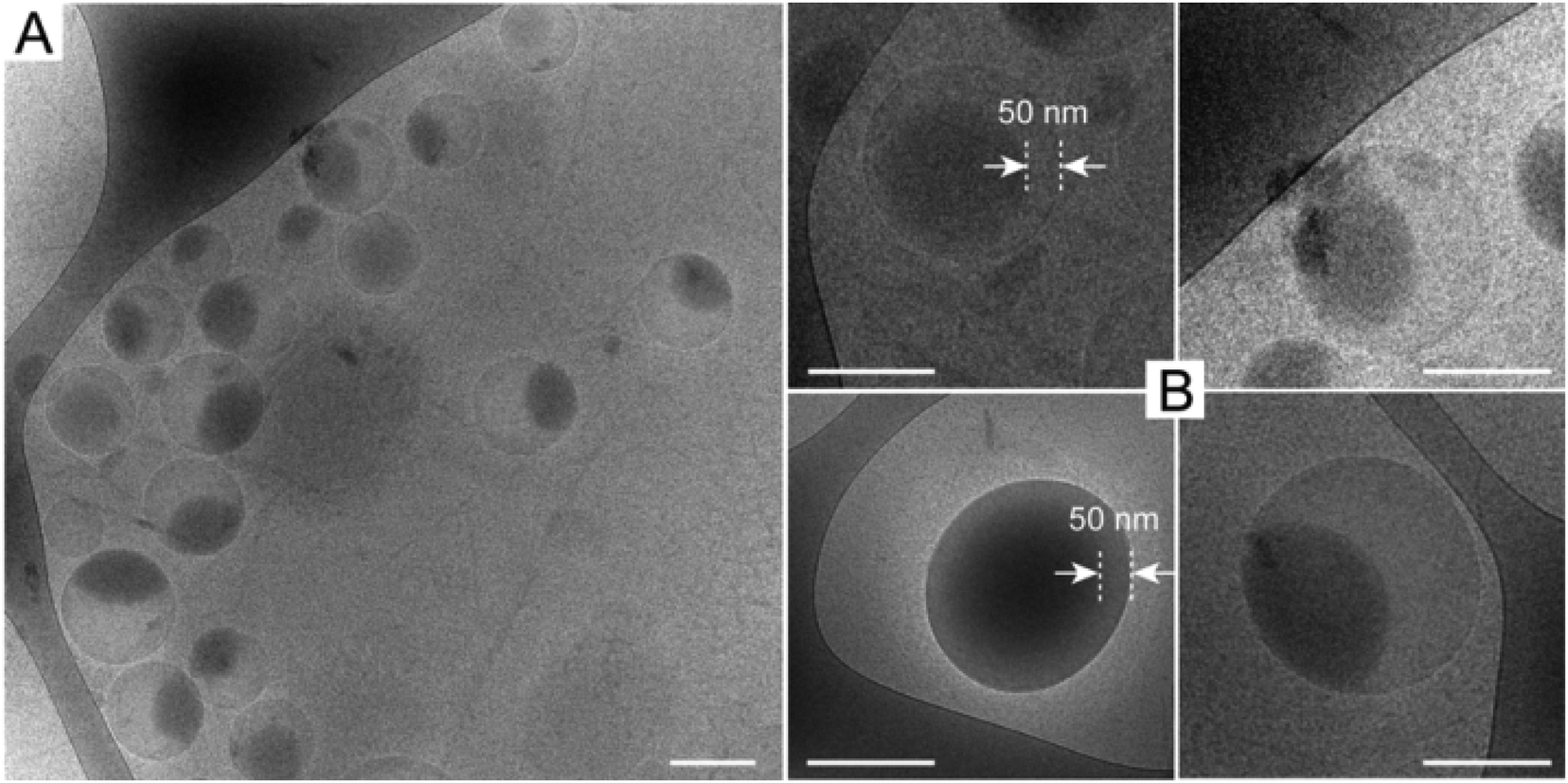 | ||
| Fig. 2 Cryo-TEM images of cross-linked polymeric vesicles without staining. The scale bar represents 200 nm. | ||
Fig. 3A reveals the effect of DOC on vesicle size. At low DOC (∼14 mol%), the hydrolysed particles presented a Dh of 330 nm. In contrast, high DOCs of 28 mol% and 56 mol% resulted in a significantly reduced Dh of 240 nm. DLS studies also confirmed the pH responsiveness of the cross-linked nanoparticles with DOC of 28 mol%. Fig. 3B shows that the particle size did not change as the pH decreased from 13 to 5 with Dh of ∼240 nm, indicating stable structure of the cross-linked vesicles under a range of pH value after cross-linking. However, the particle size abruptly decreased from 240 nm to 170 nm as pH decreased from 5 to 3. This observation cannot be attributed to the disassembly of cross-linked vesicles but is suggestively attributed to inter-polymer complex (IPC) formation between poly(ONB-diacid) block and poly(ONB-PEG) brush, as discovered by us previously.28,29 It should be emphasized that due to the short exposure (<3 min) of cross-linked vesicles to acidic media (pH < 5), no degradation of cross-linked vesicles was observed during DLS measurements.
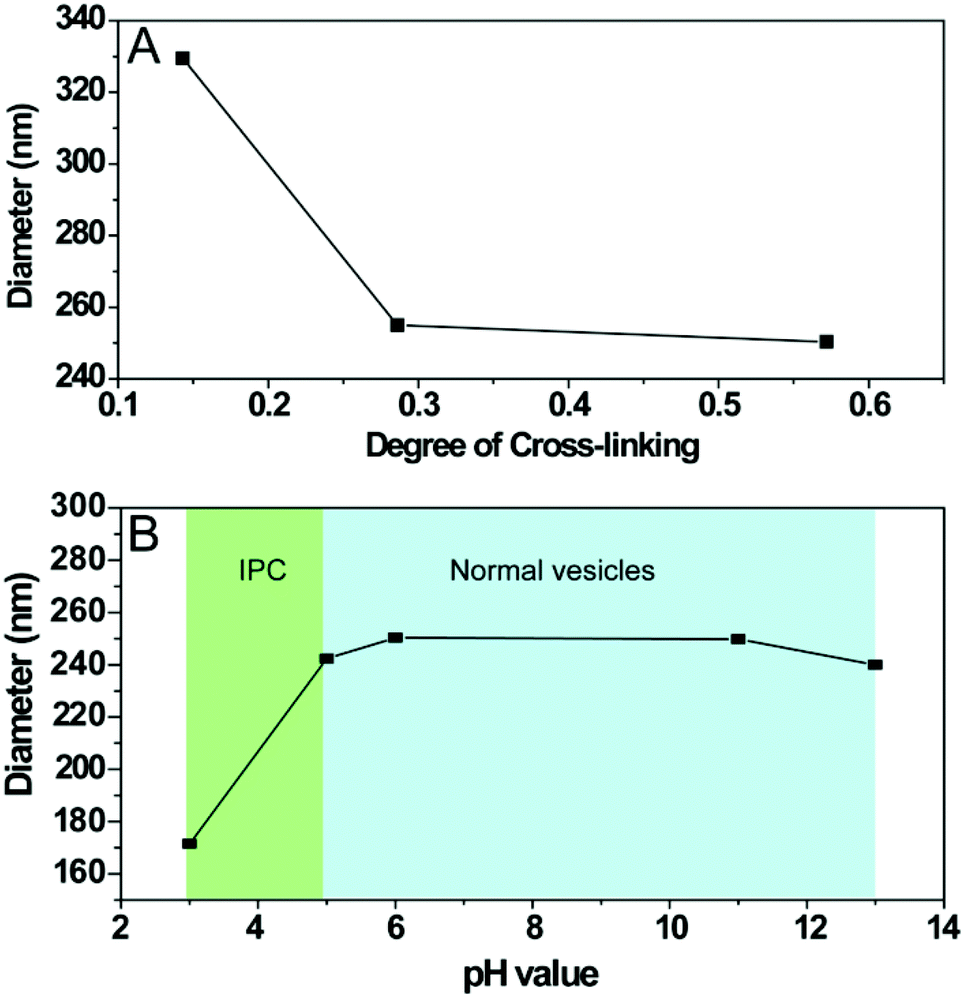 | ||
| Fig. 3 Hydrodynamic diameter of cross-linked polymeric vesicles as measured by DLS as a function of (A) degree of cross-linking (DOC) and (B) pH values. | ||
Drug loading and release
The bidentate functionality (di-acid) is an ideal conjugation site for cisplatin due to its strong tendency to bind cisplatin through the formation of dicarboxylate-platinum complexes.19,30 These metal complexes with a 6–8 member ring structure provide increased stability and high loading contents to the conjugates.30 In the present study, the hydrolysed poly(ONB-diacid) segments of the cross-linked vesicles with DOC of 28 mol% were conjugated to cisplatin according to a previously reported procedure.13 The formed drug-loaded vesicles exhibited a high drug conjugation efficiency of 85.2% as determined by TGA (Fig. 4A). This result indicates that the anhydride groups were converted to bidentate diacid functionalities and most of them have been involved in the complex formation. The drug loading content was calculated to be 17.6 wt% (see ESI†), which is much higher than other complex counterparts in matrix systems reported in the literature (∼10 wt%).4,31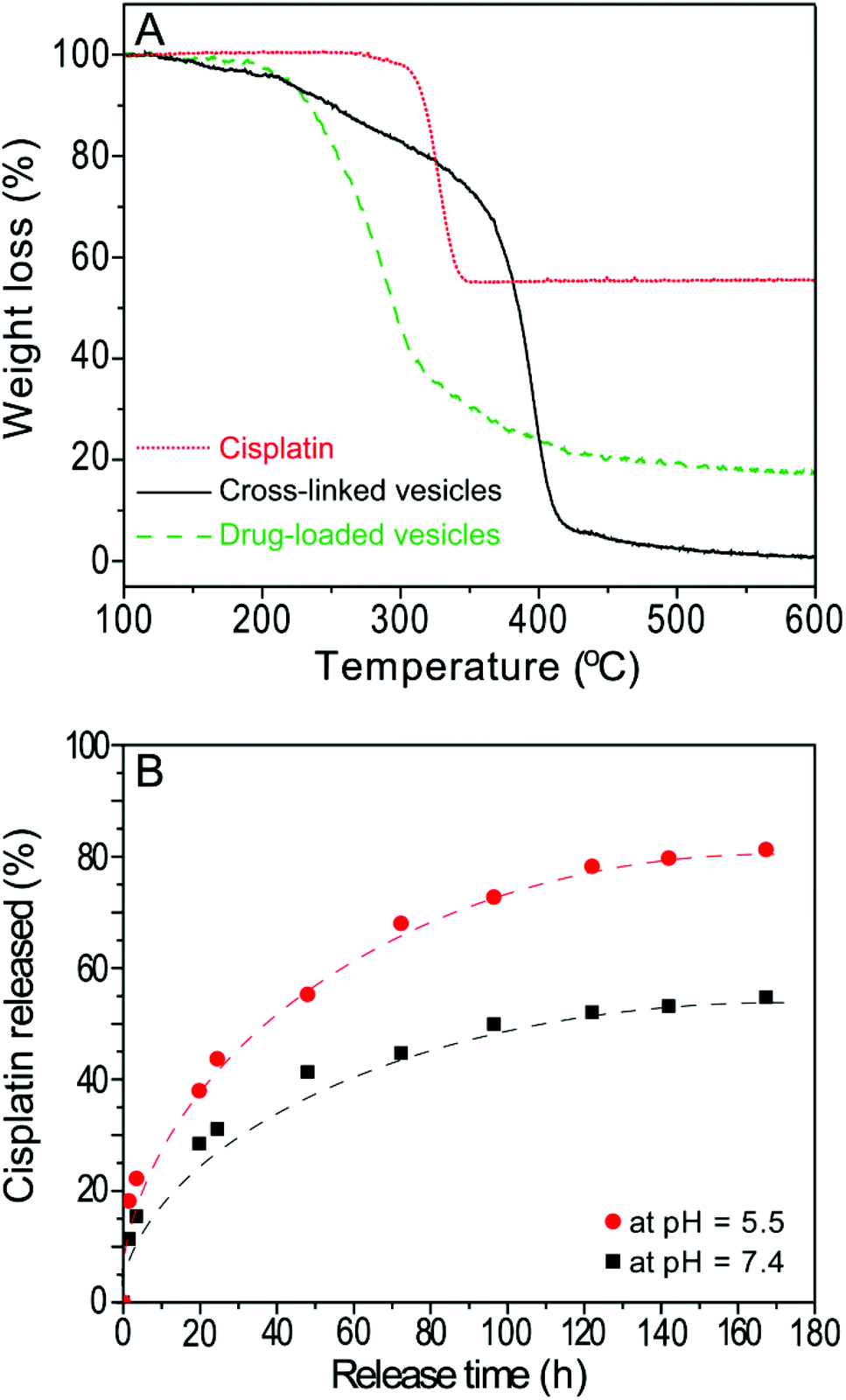 | ||
| Fig. 4 (A) TGA traces for cisplatin, cross-linked vesicles and drug-loaded vesicles. (B) Release profile of cisplatin from drug-loaded vesicles in saline solution (0.9 wt.% NaCl) at pH 5.5 (●) and pH 7.4 (■). The line was fitted according to equation: % CDDP released = Aexp(−t/τ) + y0. | ||
The in vitro release of the CDDP was performed using a well-established method.20,30 The release of cisplatin, CDDP, from the drug-loaded vesicles can be initiated in the presence of saline solution, which can lead to ligand exchange of the platinum complex thus converting it from a carboxylato to a chloride complex.32 In order to investigate the cooperative effect of degradation of the drug carrier in acidic condition and the chloride-mediated release of CDDP from the drug-loaded vesicles, drug release was performed in 0.9 wt% NaCl at pH 5.5 as well as pH 7.4 for comparison (Fig. 4B). Over one week, the release profile reaches a plateau at ∼50% at pH 7.4, while up to 80% of the drugs are released at pH 5.5 over the same time period. The value presented in this study is higher than that of cross-linked micelles (up to 70% higher)12,30 and non-crosslinked micelles (PLGA-b-mPEG, ranging from 30 to 70% higher)32 The release rates can be described by a simple model assuming first-order kinetics12 with different release constants (τ) of 47.8 h and 39.1 h at pH 5.5 and pH 7.4, respectively. This observation indicates that more drugs will be released under an endosomal environment within the cell, compared to the physiological pH observed during circulation in the body. This result also demonstrates that the degradation of vesicles increases the drug release rate, thus, the actual release process in vivo may combine diffusion of the drug (out of the polymeric vesicles), chemically controlled de-complexation of the drug-polymer complex and degradation of the polymer vesicles.
Cell culture
The cytotoxicity of parental linear-brush diblock copolymer poly(ONB-diacid35-b-ONB-PEG5), unconjugated cross-linked vesicle, degraded cross-linked vesicle, drug-loaded cross-linked vesicle, degraded drug-loaded crosslinked vesicle and free drug was evaluated over a 48 h time period using a simple endpoint viability assay (Fig. 5). Cervical cancer cells (HeLa) were exposed to varied concentrations of investigated polymer materials and free drug ranging from 10−6 to 10−2 mol Pt L−1. The IC50 values were determined using the logarithmic concentration.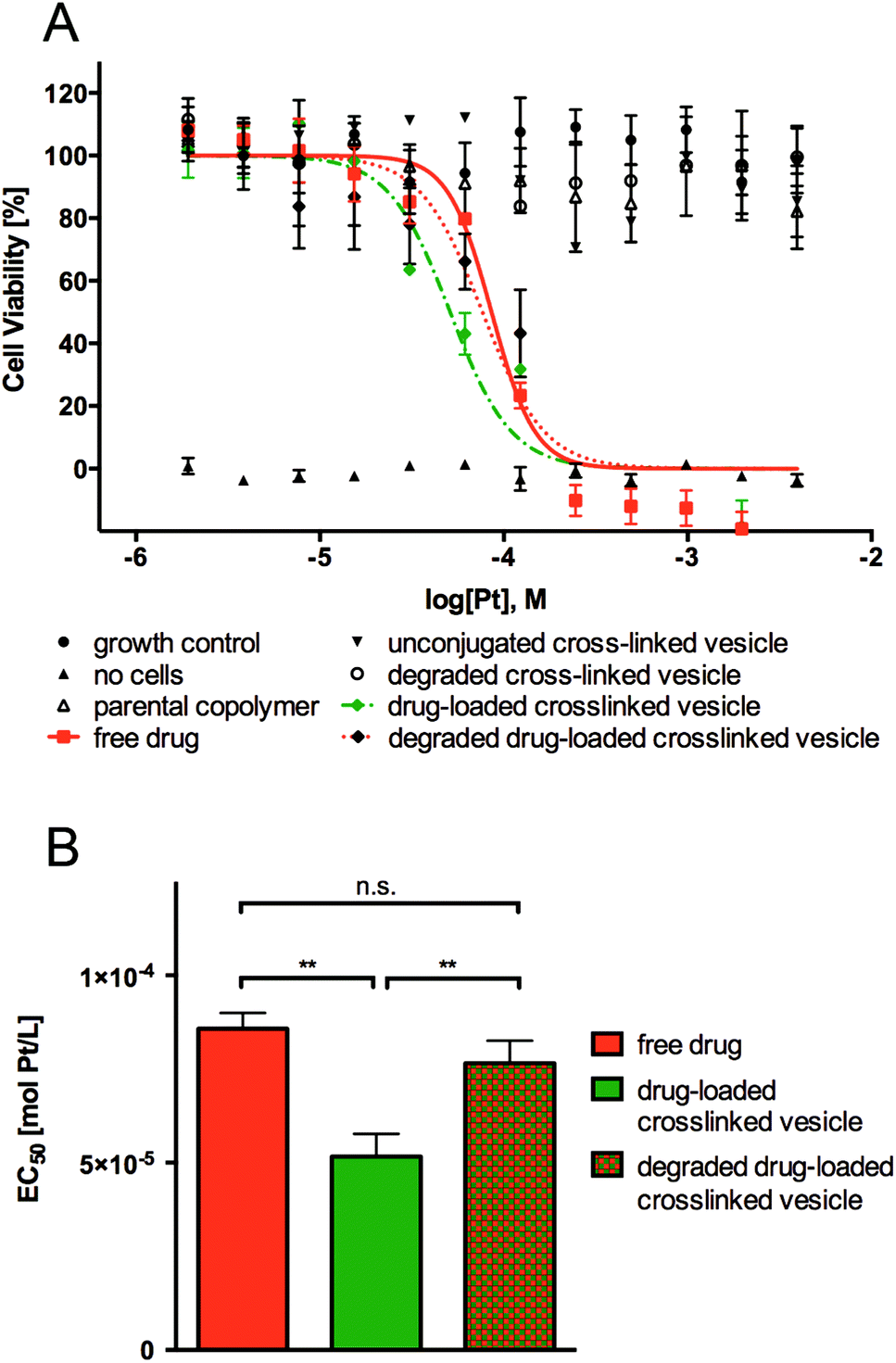 | ||
| Fig. 5 (A) Cell viability of HeLa cells after 48 h of exposure to parental linear-brush diblock copolymer (poly(ONB-diacid)35-b-poly(ONB-PEG)5), unconjugated cross-linked vesicle, degraded cross-linked vesicle, drug-loaded cross-linked vesicle, degraded drug-loaded cross-linked vesicle and free drug. (B) EC50 values for free drug, drug-loaded cross-linked vesicle and degraded drug-loaded cross-linked vesicle (**p < 0.001, n = 27–33). | ||
As shown in Fig. 5, no observed toxicity of the parental linear-brush diblock copolymer, unconjugated cross-linked vesicle and degraded cross-linked vesicle was detected after 48 h exposures to cancer cells. As expected, the free drug and the degraded drug-loaded vesicle showed comparable cytotoxicity, with IC50 values of 85 ± 4.2 μmol Pt L−1 and 81 ± 6 μmol Pt L−1, respectively. It is worth to note that the drug-loaded vesicle showed a lower IC50 value of 50 ± 6.0 μmol Pt L−1 (thus higher cytotoxicity), even though in total only 50% of the drug would have been released from the drug carrier during the 48 h the assay was conducted (see Fig. 4B, pH = 5.5 curve). Considering 80% cisplatin release from the drug-loaded vesicles is to be expected upon one week, the IC50 is in reality even lower than that. This result suggests that the polymer vesicles are a highly efficient means of delivering cisplatin to cancer cells. Internalization of drug-loaded vesicles/carriers (rather than diffusion of the free drug) is often more efficient since the uptake mechanism differs from that of the corresponding free drug. Free drugs are required to diffuse through the cell membrane, which often is an inefficient process due to multidrug resistance (MDR) efflux pumps.33 Endocytotic uptake of drug-loaded vesicles, which can overcome MDR mechanisms, is thought to cause greater drug efficiency due to the avoidance of MDR efflux pumps.
In order to confirm that the developed drug-loaded vesicles are indeed internalized by cells and may thus deliver their payload intracellularly, we conducted cellular uptake studies with unconjugated (non-drug loaded), cross-linked vesicles using the same cell type. For this purpose, a fluorescent tag, Alexa Fluor®594 (AF594), was introduced into the cross-linked vesicles via reaction with the di-acid functionalities in the bilayer membrane, and HeLa cells were exposed to such AF594-tagged vesicles for 24 h. As shown in Fig. 6, the red fluorescence associated with the vesicles is clearly observed within the cells’ cytoplasm rather than being located outside or co-located with the cell wall after this time period, which indicates successful cellular uptake. The observed fluorescence is further well distributed within the cytoplasm rather than being observed inside discrete intracellular vesicles (endosomes/lysosomes). Lastly, treated cells appear with normal morphology (i.e., no cell shrinking, membrane blebbing, chromatin condensation, nucleus fragmentation, apoptotic bodies or membrane permeability are observed), confirming the benign nature of the empty, cross-linked vesicles already discussed in Fig. 5.
 | ||
| Fig. 6 Confocal laser scanning microscopy (CLSM) images of HeLa cells 24 h after the addition of AF594-tagged vesicles. (A) Cell nuclei were stained using 4′,6-diamidino-2-phenylindole (DAPI) staining. (B) AF594-tagged vesicles are internalized within cells. (C) Composite image. | ||
Conclusions
In summary, this work presented an improved approach to fabricate acid-degradable, polymeric vesicles for the efficient delivery of cisplatin drug. The vesicles were synthesized via the self-assembly of linear-brush diblock copolymers and subsequent cross-linking using an acid-degradable cross-linker. The utilization of hydrolysed anhydrides allowed for the straightforward, efficient and stable conjugation of cisplatin to the cross-linked polymer vesicles. Moreover, the cross-linked drug carriers are designed to degrade at lysosomal acid condition (pH ∼ 5.5), which leads to augmented drug release compared to pH 7.4. The activity of the cisplatin loaded into the cross-linked vesicle was improved in comparison to the free drug (and the degraded drug-loaded vesicle control) and this is potentially due to the different uptake mechanism of the drug. These initial biological evaluations demonstrated the feasibility of this novel drug delivery system and further studies along with an in vivo analysis of the current system are ongoing.Acknowledgements
The authors acknowledge the Australian Research Council under the Future Fellowship Scheme (FT110100411, G.G.Q.). Q.F. and K.L. are the recipients of an Australian Research Council Super Science Fellowship (FS110200025). Q.F. also wishes to acknowledge the receipt of 2013 Early Career Researcher Grants from The University of Melbourne.Notes and references
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- J. Reedijk, Eur. J. Inorg. Chem., 2009, 10, 1303–1312 CrossRef.
- E. Gianasi, M. Wasil, E. G. Evagorou, A. Keddle, G. Wilson and R. Duncan, Eur. J. Cancer, 1999, 35, 994–1002 CrossRef CAS.
- E. Soussan, S. Cassel, M. Blanzat and I. Rico-Lattes, Angew. Chem., Int. Ed., 2009, 48, 274–288 CrossRef CAS PubMed.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef CAS.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS.
- H. Wang, L. Tang, C. Tu, Z. Song, Q. Yin, L. Yin, Z. Zhang and J. Cheng, Biomacromolecules, 2013, 14, 3706–3712 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- V. T. Huynh, P. d. Souza and M. H. Stenzel, Macromolecules, 2011, 44, 7888–7900 CrossRef CAS.
- V. T. Huynh, G. Chen, P. d. Souza and a. M. H. Stenzel, Biomacromolecules, 2011, 12, 1738–1751 CrossRef CAS PubMed.
- L. Liao, J. Liu, E. C. Dreaden, S. W. Morton, K. E. Shopsowitz, P. T. Hammond and J. A. Johnson, J. Am. Chem. Soc., 2014, 136, 5896–5899 CrossRef CAS PubMed.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- R. Cheng, F. Meng, C. Deng, H.-A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. van-Nostrum, J. Controlled Release, 2007, 120, 131–148 CrossRef CAS PubMed.
- J. Xu, Q. Fu, J. M. Ren, G. Bryant and G. G. Qiao, Chem. Commun., 2013, 49, 33–35 RSC.
- V. T. Huynh, S. Binauld, P. L. d. Souza and M. H. Stenzel, Chem. Mater., 2012, 24, 3197–3211 CrossRef CAS.
- R. B. Woodward and H. Baer, J. Am. Chem. Soc., 1948, 70, 1161–1166 CrossRef CAS.
- D. Le, V. Montembault, J.-C. Soutif, M. Rutnakornpituk and L. Fontaine, Macromolecules, 2010, 43, 5611–5617 CrossRef CAS.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS.
- B. Schechter, A. Neumann, M. Wilchek and R. Arnon, J. Controlled Release, 1989, 10, 75–87 CrossRef CAS.
- C. W. Bielawskia and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef PubMed.
- Q. Fu, J. M. Ren and G. G. Qiao, Polym. Chem., 2012, 3, 343–351 RSC.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS.
- B. S. Lele and A. S. Hoffman, J. Controlled Release, 2000, 69, 237–248 CrossRef CAS.
- P. Ivopoulos, M. Sotiropoulou, G. Bokias and G. Staikos, Langmuir, 2006, 22, 9181–9186 CrossRef CAS PubMed.
- V. T. Huynh, J. Y. Quek, P. L. d. Souza and a. M. H. Stenzel, Biomacromolecules, 2012, 13, 1010–1023 CrossRef CAS PubMed.
- R. Tong and J. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4830–4834 CrossRef CAS PubMed.
- K. Avgoustakis, A. Beletsi, Z. Panagi, P. Klepetsanis, A. G. Karydas and D. S. Ithakissios, J. Controlled Release, 2002, 79, 123–135 CrossRef CAS.
- C. Vauthier, C. Dubernet, C. Chauvierre, I. Brigger and P. Couvreur, J. Controlled Release, 2003, 93, 151–160 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the DLS and TEM measurements of polymer vesicles, determination of the platinum conjugation efficiency and platinum loading content. See DOI: 10.1039/c4py01123f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |