
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Broadband ultrafast photoprotection by oxybenzone across the UVB and UVC spectral regions†
Lewis A.
Baker
a,
Michael D.
Horbury
a,
Simon E.
Greenough
a,
Michael N. R.
Ashfold
b and
Vasilios G.
Stavros
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
First published on 11th August 2015
Abstract
Recent studies have shed light on the energy dissipation mechanism of oxybenzone, a common ingredient in commercial sunscreens. After UVA photoexcitation, the dissipation mechanism may be understood in terms of an initial ultrafast excited state enol → keto tautomerisation, followed by nonadiabatic transfer to the ground electronic state and subsequent collisional relaxation to the starting enol tautomer. We expand on these studies using femtosecond transient electronic absorption spectroscopy to understand the non-radiative relaxation pathways of oxybenzone in cyclohexane and in methanol after UVB and UVC excitation. We find that the relaxation pathway may be understood in the same way as when exciting in the UVA region, concluding that oxybenzone displays proficient broadband non-radiative photoprotection, and thus photophysically justifying its inclusion in sunscreen mixtures.
Ultraviolet (UV) radiation is a vital requirement for the production of vitamin D. However, its benefit remains in a delicate balance given that overexposure to UV is known to be a dominant cause of cancers such as malignant melanomas.1–3 To protect against overexposure, the human body has developed an extensive network of naturally synthesised UV light absorbing biopolymers, the concentrations of which can fluctuate through the regulation of melanogenesis.4 Thus, in instances of high UV exposure, melanogenesis is up-regulated and the result is tanning of the skin. This natural photoprotective mechanism is a delayed response to overexposure from which DNA damage could have already occurred. This has led to the development of sunscreens that utilise a combination of UV chromophores to act preemptively to reduce the risk of overexposure and thus the probability of skin photodamage.5 Particularly in the current age, shifts in perceptions on tanning and increased travel and tourism have meant the correct use of sunscreens has never been more important.6,7
Oxybenzone (OB), shown in Fig. 1, has been the subject of numerous studies due to its use as a UV chromophore in many commercial sunscreens.5,10–13 It displays intense, broad absorption bands in each of the UVA (400–315 nm), UVB (315–280 nm) and UVC (280–100 nm) regions (Fig. 1) making it suitably flexible to absorb incident (broadband UV) solar radiation, and, in the case of UVC, artificial UV sources.14–16 Importantly, OB has also been shown to remain photostable after several hours of irradiation,17 although some controversy remains surrounding its suitability as a sunscreen constituent with respect to adverse dermatological effects and endocrine disruption.13,18–20 Recent ab initio electronic structure calculations on OB suggest that ultrafast dynamics are responsible for its efficacy as a sunscreen.13 Specifically, these studies identify internal conversion (IC) via a barrierless electron-driven excited state hydrogen atom transfer (ESHT) as a plausible energy deposition mechanism along the enol RO–H reaction coordinate, transforming to the keto conformer with increasing O–H bond distance. (Fig. 1, red).13 Furthermore, theoretical studies of similar species, that contain hydrogen donor (OH)–acceptor (CO) sites in close proximity, indicate similar ultrafast relaxation mechanisms may be operative.21,22 Support for the operation of such ESHT processes is provided by recent ultrafast transient absorption studies on OB following excitation in the UVA region8,9 and by earlier work which identified ESHT as a relaxation process in related molecules like 2-(2′-hydroxyphenyl)benzothiazole (HBT) and 2-(2′-hydroxy-5′-methylphenyl)benzotriazole (TINUVIN).23–25 The energy dissipation mechanism described following UVA excitation of OB involves successive ultrafast steps beginning with IC from the initially excited 1ππ* (S2) state to the lower lying 11nπ* (S1) state, followed by ultrafast enol → keto tautomerisation (ESHT) on the S1 potential energy surface (PES). A slower C–C twist then facilitates nonadiabatic coupling to form highly vibrationally excited ground state (S0) molecules, relaxing predominantly to the enol-OB conformer through a combination of ground state hydrogen transfer (GSHT) and vibrational energy transfer (VET). Other recent studies have suggested some probability for excited state OB molecules to undergo intersystem crossing (ISC) to long-lived triplet states12 as well as homolytic O–H bond fission yielding phenoxyl radicals.9 An interesting question that remains following these previous studies is whether OB dissipates electronic energy with similar efficiency when photoexcited at shorter wavelengths – a property that should be required of any organic sunscreen constituent.
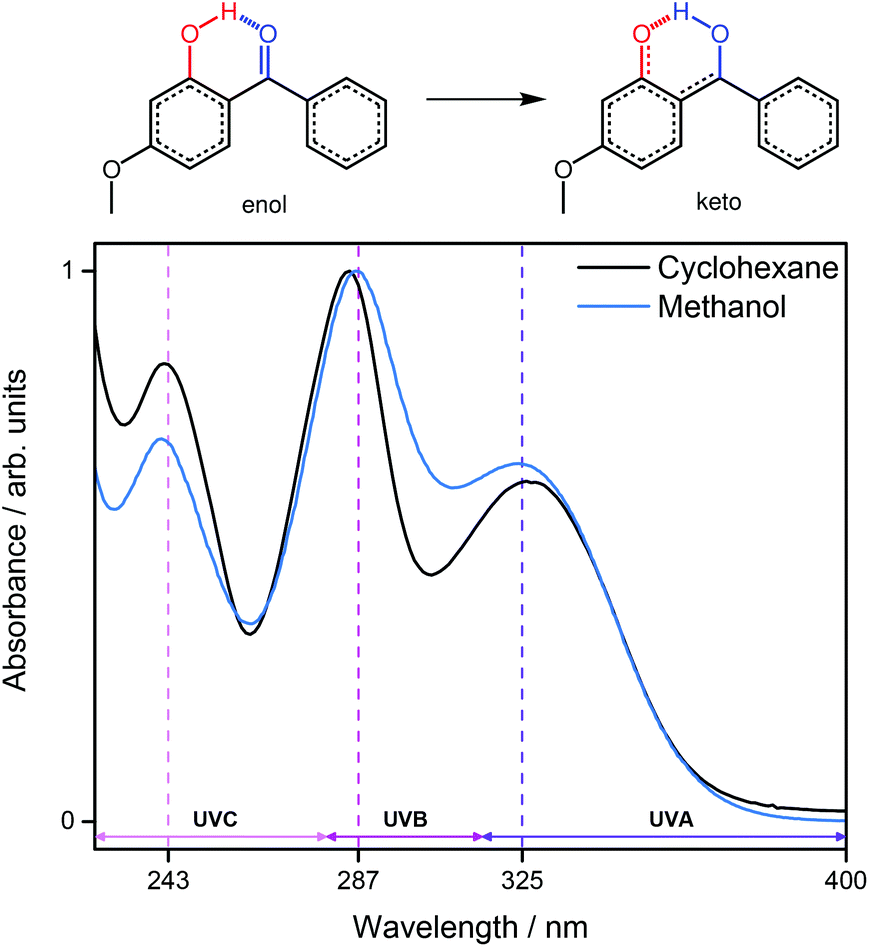 | ||
| Fig. 1 Enol-OB and keto-OB tautomers. Studies have suggested that enol-OB undergoes an ultrafast tautomerisation into a keto tautomer after UVA irradiation.8,9 The UV-visible spectrum for enol-OB in solution in cyclohexane (black) and methanol (blue) displays three absorption maxima at ca. 325 nm, 287 nm and 243 nm, each highlighted by a vertical dashed line. | ||
In this communication we utilise femtosecond pump–probe transient electronic (UV-visible) absorption spectroscopy (TEAS) to probe the ultrafast energy dissipation of OB following photoexcitation on absorption maxima within the UVB and UVC regions ca. 287 nm (4.32 eV) and ca. 243 nm (5.10 eV) respectively (Fig. 1). The observed dynamics are, again, highly suggestive of ultrafast excited state enol → keto tautomerisation, followed by IC and VET to reform the ground state enol tautomer, as documented in the case of UVA photoexcitation.8,9
For all TEAS measurements, 10 mM solutions of OB (98%, Sigma-Aldrich), in either cyclohexane (>99%, VWR) or methanol (≥99.6%, Sigma-Aldrich), were recirculated and delivered using a flow-through cell (Harrick Scientific), which is equipped with two CaF2 windows and a 100 μm thick PTFE spacer. The sample is photoexcited using 287 nm or 243 nm, ∼50 fs pump pulses with fluences of 1–2 mJ cm−2. Probe pulses are derived from a broadband white light continuum (340 to 675 nm) and set to a time delay of up to 1.7 ns relative to the pump pulse. Probe pulse polarisation is held at the magic angle (54.7°) relative to the pump polarisation. Further experimental details are reported in ref. 8 and 26. All transient absorption spectra (TAS) are chirp corrected using the KOALA package27 and reported lifetimes are determined using global fitting with uncertainties reported to a 95% confidence interval (2σ) using support plane analysis, see ESI† for details.8,28–31 All ‘static’ UV-visible spectroscopic measurements were taken using a Cary 50 UV-visible spectrophotometer with a 1 cm path length quartz cuvette, and ∼μM OB-cyclohexane and OB-methanol solutions.
Using a photoexcitation wavelength of 287 nm, the TAS of OB-cyclohexane and OB-methanol for a range of pump–probe time delays are shown in Fig. 2A and B, respectively. We consider OB-cyclohexane first. At early pump–probe time delays (<500 fs) the TAS are dominated by two features. There is an intense absorption peak centred on ∼366 nm and a broad absorption extending out to ∼650 nm. Based on the analysis of similar absorption features observed after UVA excitation, and guided by ab initio calculations, this broad absorption is assigned to the OB excited state absorption (ESA),8,9,13 likely to originate from a transition to a dense manifold of high-lying electronic states i.e. Sn ← S1.32,33 By 2 ps, the majority of the broad ESA feature has decayed away, leaving the 366 nm absorption peak. A negative feature between 340 and 350 nm is also present which we assign, through comparison with the static UV-visible of OB (Fig. 1), to a ground state bleach (GSB). By 50 ps, the 366 nm feature has almost decayed to the baseline, without full recovery of the GSB up to the maximum available pump–probe time delay of 1.7 ns. Similar dynamics are found for OB-methanol with a small observed blue-shift in the features discussed thus far.
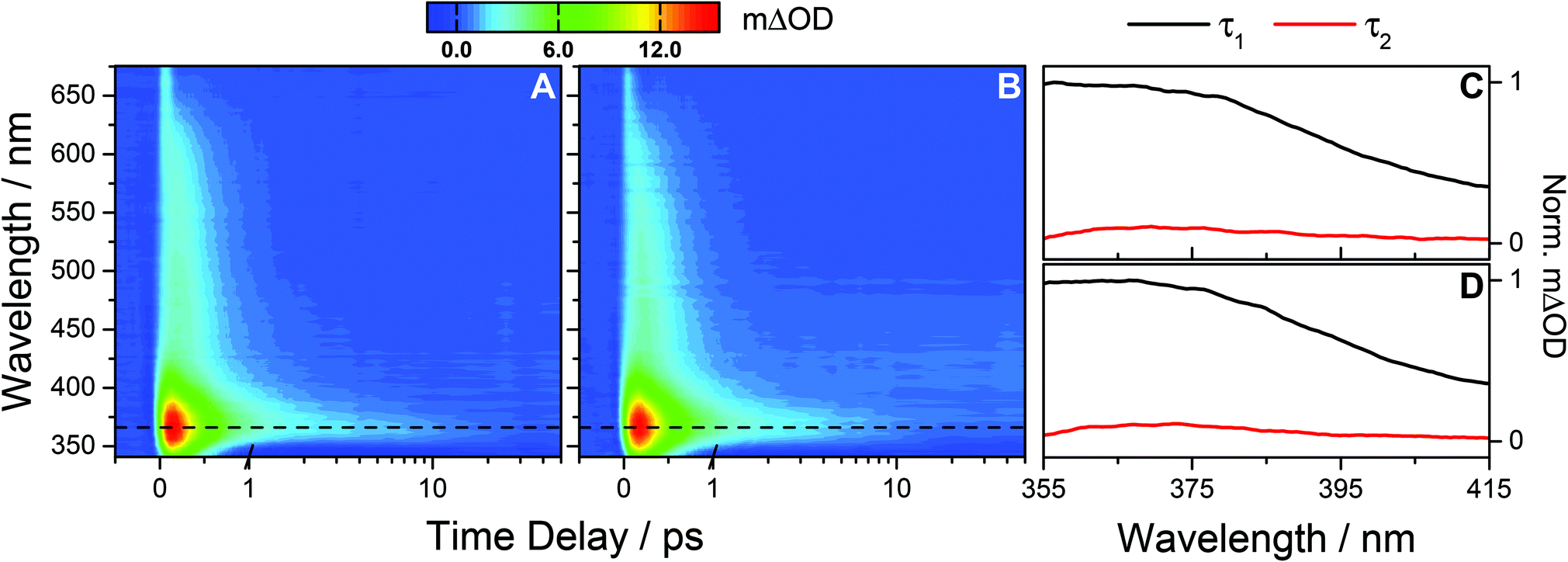 | ||
| Fig. 2 (A) Raw TAS following 287 nm photoexcitation of OB-cyclohexane displaying an absorption peak at 366 nm (dashed line) and a broad absorption extending out to ∼650 nm. The TAS for negative and early (≤1 ps) time delays are shown on a linear scale, and longer time delays (>1 ps) are displayed on a logarithmic scale. The colour map indicates the change in optical density (ΔOD). Similar observations are seen for OB-methanol (B). (C) and (D) The corresponding decay associated spectra for OB-cyclohexane and OB-methanol respectively from the bi-exponential global fit across the spectral range 355–415 nm returning a femtosecond lifetime τ1 and a picosecond lifetime τ2, as summarised in Table 1. | ||
Quantitative insight into the dynamical processes observed in the TAS is obtained by employing a global fitting procedure.28,29 To recover the dynamics observed in the TAS, two exponential fitting functions are required over the spectral range of 355 ≤ λ ≤ 415 nm. This short wavelength limit is required to avoid the GSB, and includes only the 366 nm absorption peak. Furthermore, for all UVB measurements, a broad feature around time zero, unresolvable within our instrument response (∼100 fs, see ESI†) and which shifts by ∼50 fs to longer time delays for UVC measurements (vide infra), is excluded by the omission of very early delay times (<250 fs). For OB-cyclohexane, the transient signal is fitted using two lifetimes, τ1 = 391 ± 12 fs and τ2 = 8.6 ± 2.7 ps, with corresponding decay associated spectra (DAS) as shown in Fig. 2C. For OB-methanol, the corresponding lifetimes are τ1 = 382 ± 8 fs and τ2 = 6.0 ± 1.0 ps, with corresponding DAS as shown in Fig. 2D. The dynamics of the ESA feature for λ > 415 nm is characterised by a single exponential decay function with a lifetime which closely resembles the τ1 value for OB in the corresponding solvent (see ESI†).
Analogous experiments were performed for photoexcitation at 243 nm. TAS of OB-cyclohexane and OB-methanol are shown in Fig. 3A and B respectively. We observe similar spectral features as described in the above UVB case. Again, quantitative insight into the prevailing dynamical processes is obtained through global fitting. In the case of the UVC measurements, time delays <300 fs were excluded in order to avoid the broad feature at very early time delays (vide infra). To recover the dynamics of the TAS over the spectral range of 355 ≤ λ ≤ 415 nm, three exponential functions are required; one of which is a long-lived solvent response with a lifetime ∼400 ps for cyclohexane and ∼3 ns for methanol, predetermined from fitted solvent-only TAS (see ESI†). The lifetimes of the other two processes are determined analogously to the UVB case. These are determined to be τ1 = 392 ± 10 fs and τ2 = 11.0 ± 4.4 ps for OB-cyclohexane and τ1 = 371 ± 9 fs and τ2 = 7.8 ± 1.8 ps for OB-methanol. The corresponding DAS are shown in Fig. 3C and D, respectively. The ESA feature for λ > 415 nm is described by two exponential functions; a solvent response as given above, and another with a lifetime closely mirroring the τ1 described above.
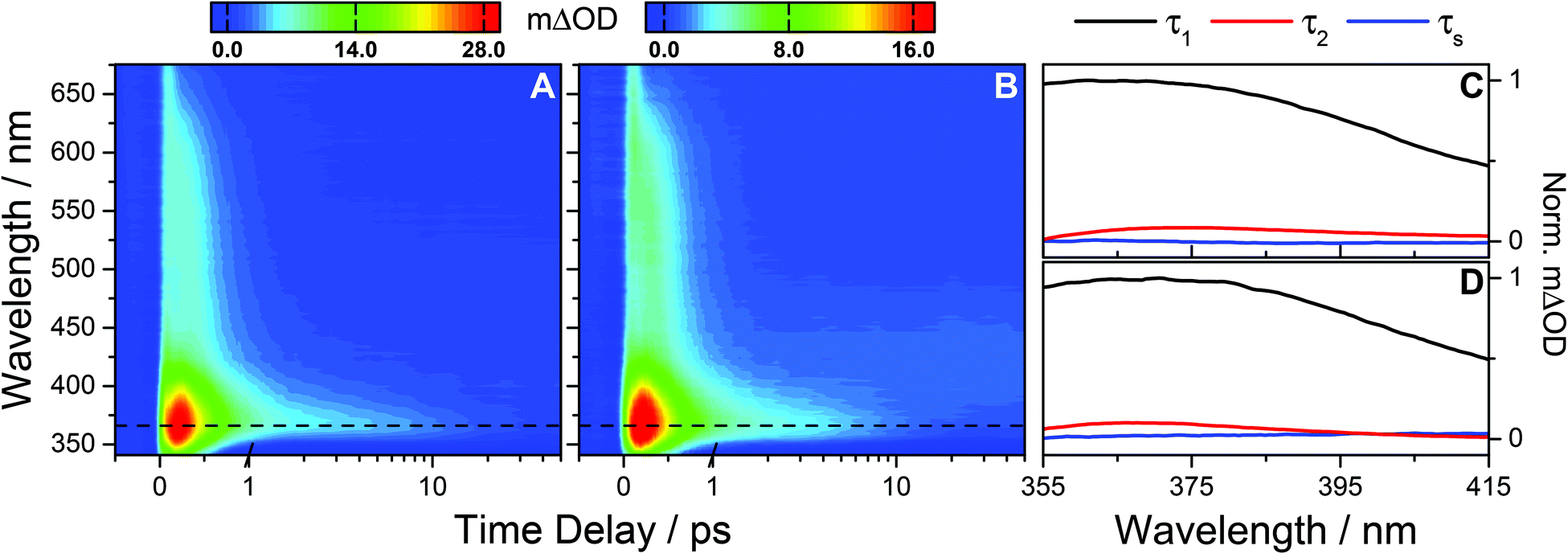 | ||
| Fig. 3 (A) Raw TAS following 243 nm photoexcitation of OB-cyclohexane displaying an absorption peak at 366 nm (dashed line) and a broad absorption extending out to ∼650 nm. The TAS for negative and early (≤1 ps) time delays are shown on a linear scale, and longer time delays (>1 ps) are displayed on a logarithmic scale. Similar observations are seen for OB-methanol (B). (C) and (D) The corresponding decay associated spectra for OB-cyclohexane and OB-methanol respectively from the bi-exponential global fit across the spectral range 355–415 nm returning a femtosecond lifetime τ1 and a picosecond lifetime τ2 as summarised in Table 1, with a solvent dependant lifetime, τs, which is ∼400 ps for cyclohexane and ∼3 ns for methanol. We note the small amplitude of τs reflecting the minor contribution of this component to the DAS. | ||
For all pump excitation wavelengths, TEAS measurements identify three dynamical features (excluding any solvent responses) which closely match those previously seen for OB photoexcited at 325 nm.8 Two of these processes, a short lived process τ1 and a longer lived process τ2, are extracted from the TAS by the global fitting of the 366 nm absorption maxima. The third feature is that previously described; a broad feature which persists over time delays 100 < Δt < 200 fs, and which appears to shift ∼50 fs to longer time delays with increasing excitation energy (see Fig. 4A). In the case of 243 nm photoexcitation, there is a suspicion of the emergence of a ‘double hump’ structure in this broad feature, that is not evident in the corresponding data obtained when exciting in the UVA or UVB regions.8 Furthermore, for all excitation wavelengths, we note that the GSB has not fully recovered, seen from the non-zero baseline out to the maximum delay time of our experiment (1.7 ns), see Fig. 4B.
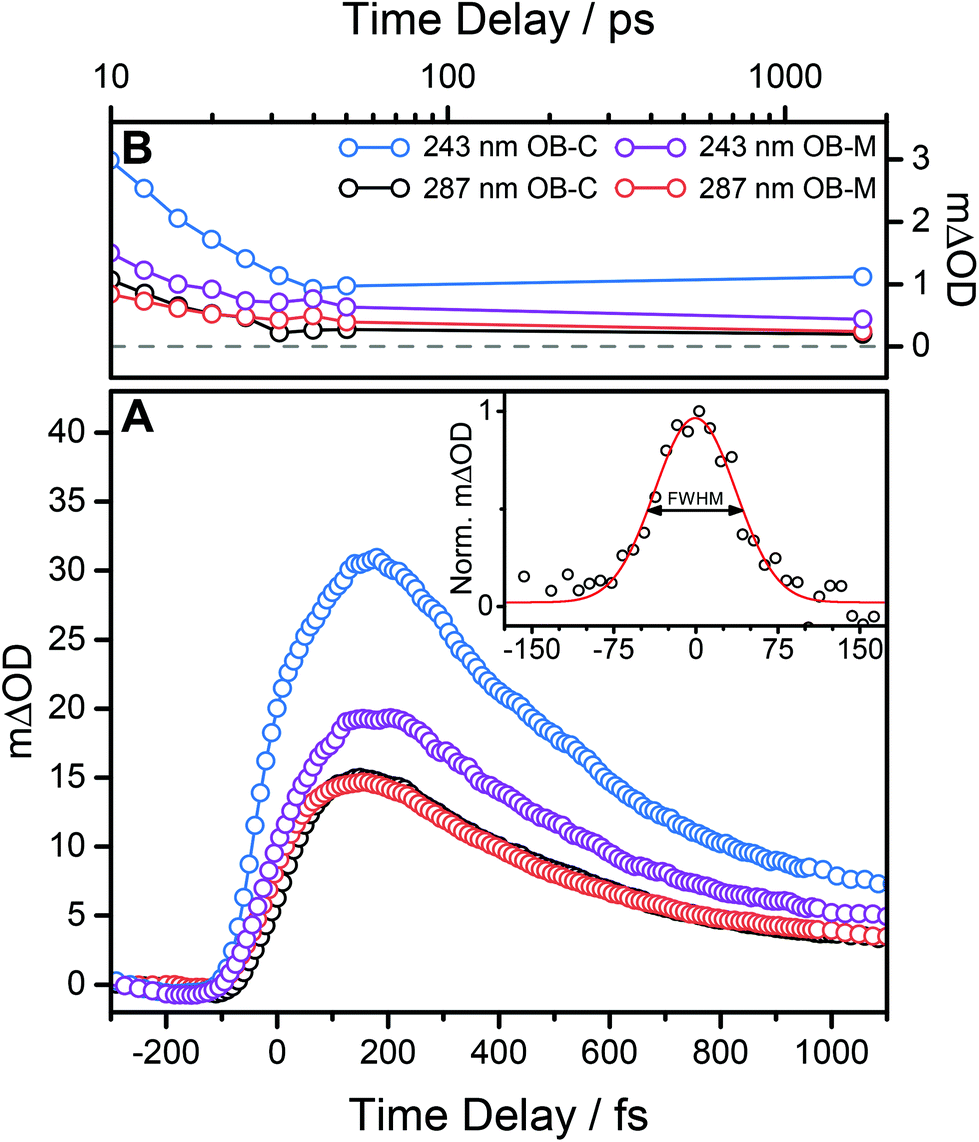 | ||
| Fig. 4 (A) 366 nm transient displaying early time dynamics of OB-cyclohexane (OB-C) and OB-methanol (OB-M) after 287 nm and 243 nm excitation. For OB excitation at 243 nm we observe a shift of the 366 nm plateau relative to the same feature following 287 nm excitation. A representative normalised 366 nm cross correlation signal is shown in the inset of panel (A) for a methanol-only scan after 243 nm photoexcitation (black circles). The full width at half maximum is determined to be 87 ± 5 fs from a Gaussian fit (red line). (B) Another observed feature for all TAS, that is also clearly shown for these transients, is the non-zero baseline out to the maximum available pump–probe time delay of 1.7 ns indicating incomplete GSB recovery, where the grey dashed line indicates zero. The transients shown are taken by integrating a 5 nm ‘slice’ about 366 nm. | ||
We can begin to rationalise these features drawing on ab initio calculations performed in previous studies.8,13 Firstly, given the similarity of the TAS measured in both cyclohexane and methanol, we conclude that the intramolecular hydrogen bond in OB is preserved in both solvents, in both the ground13 and excited states – as depicted in Fig. 1. Further support for this conclusion is provided by previous studies of HBT34 and 4-tert-butylcatechol35 for example. Both of these latter studies show markedly different dynamics when photoexcited in polar and non-polar solvents, consistent with loss of the intramolecular hydrogen bond in suitably polar solvents. The signal broadening for higher energy excitations observed in Fig. 4, may be understood as IC from a photoexcited S3 (21ππ*) state to S1 in the case of UVB excitation,13 or, in the case of UVC excitation, from a higher lying Sn state (n ≥ 4) to S1; followed by ballistic ESHT on the S1 PES. We note that ESHT may occur before IC or indeed concomitantly. This mechanism fits well with the observed shift of the very early time feature (see Fig. 4A) given that IC from higher electronic states will take longer to relax to the S1 enol-OB tautomer before ESHT occurs. In such cases, one might speculate the appearance of a double hump, as these processes begin to emerge from our experimental temporal resolution of ∼100 fs.
After this ESHT, which drives enol → keto tautomerisation, a slower rotation about the C–C bond may occur which facilitates IC back to the S0 state through a S1/S0 conical intersection (nonadiabatic transfer); similar observations have been reported previously in the case of HBT.34 We associate this process with the lifetime τ1 which is reassuringly similar to those reported for comparable systems undergoing enol → keto tautomerisation, which too require a twisted geometry rearrangement to facilitate IC.34,36–38
Thus we suggest that, following IC and formation of vibrationally hot S0 keto molecules, GSHT allows for keto → enol tautomerisation. The enol tautomer subsequently cools via VET with the surrounding solvent, which we therefore attribute to the lifetime, τ2.8 The absorption feature centred at 366 nm reflects this last step, whereby vibrationally hot molecules in the S0 state absorb the white light probe. The apparent sharpness of this feature is attributed to the spectral overlap of the (negative-going) GSB with the ESA. This model supports the determined globally fitted lifetimes, as summarised in Table 1. There is constancy in the τ1 process when OB is excited by either a UVA, UVB or UVC pump pulse. As τ1 represents the rotation around the C–C bond, this lack of change suggests that rotational motion about the C–C bond is unaffected by the increase in vibrational energy in the S1 molecules.
| Cyclohexane | Methanol | |||
|---|---|---|---|---|
| λ/nm | τ 1/fs | τ 2/ps | τ 1/fs | τ 2/ps |
| 325 | 375 ± 13 | 7.8 ± 2.8 | 368 ± 13 | 4.9 ± 1.9 |
| 287 | 391 ± 12 | 8.6 ± 2.7 | 382 ± 8 | 6.0 ± 1.0 |
| 243 | 392 ± 10 | 11.0 ± 4.4 | 371 ± 9 | 7.8 ± 1.8 |
One might anticipate that, following S1 → S0 nonadiabatic transfer, the extra vibrational energy in the vibrationally hot S0 keto tautomer would lead to an increase in VET lifetime (τ2). However, within a 2σ uncertainty, our measurements of τ2 remain constant over different excitation energies, cf. 7.8 ± 2.8 ps to 8.6 ± 2.7 ps to 11.0 ± 4.4 ps after UVA, UVB and UVC excitation respectively for OB-cyclohexane, indicating an insensitivity to this extra vibrational energy. Furthermore, we note that the τ2 lifetime is solvent dependent in that the extracted lifetimes for OB-methanol are consistently shorter than the OB-cyclohexane counterparts. This implies higher VET efficiencies for OB-methanol than OB-cyclohexane, consistent with a more strongly (polar) interacting (hydrogen bonding) solvent.39 Reassuringly, the lifetime τ2 returned from fitting a single transient at 380 nm is consistently shorter than the 366 nm counterpart. This is unsurprising given that the higher rate of VET is likely to be associated with the more vibrationally excited S0 molecules, which absorb at longer wavelengths (i.e. red-shifted from 366 nm). Further details pertaining to this can be found in the ESI.†
The aforementioned non-zero baseline, that shows incomplete GSB recovery, implies that some of the hot S0 keto molecules do not reform the enol-OB tautomer, and instead form some long-lived (>1.7 ns) photoproduct. Given previous condensed phase observations, triplet state formation is one possible assignment,12 although recent ab initio electronic structure calculations of likely triplet photoproducts coupled with transient vibrational absorption studies suggest such triplet states are negligible.8 Other studies suggest that the formation of phenoxyl radicals by O–H bond fission may explain incomplete GSB recovery,9 although we note that neither previous similar studies8 nor the current work detected any absorption attributable to the phenoxyl radical within our signal-to-noise. Guided by recent ab initio calculations and experimental studies of similar systems,8,36–38,40 we suggest the photoproduct to be a trans-keto tautomer formed from the extended rotation of the C–C bond of the vibrationally hot keto tautomer on the S0 PES. Ultimately, time-resolved infrared studies may hold the answers to the incomplete recovery of the GSB, but are beyond the scope of the present work. Finally we note that, irrespective of the initial photoexcitation energy (UVA, UVB or UVC) the observed early time dynamics reflect motion on the S1 PES, which is another manifestation of Kasha's rule.41
To conclude, we have determined a sequence of ultrafast relaxation steps by which OB molecules photoexcited in the UVB and UVC region reform the ground state enol-OB molecules with high efficiency. The non-radiative decay involves initial enol → keto tautomerisation (i.e. ESHT) followed by a nonadiabatic transfer to the ground state, a reverse keto → enol tautomerisation (i.e. GSHT) and subsequent vibrational cooling. This study extends previous work on OB relaxation dynamics after UVA excitation to reveal photostability with a similar relaxation mechanism across a broad region in the UV-visible spectrum. This indicates that OB is highly suitable as a UV absorber in sunscreens given its propensity to dissipate energy non-radiatively after photoexcitation in both UVA and UVB regions. The present results also suggest that OB is a suitable absorber in the UVC region, which could be important for industrial applications. We hope this work will serve as a stimulus for further ab initio calculations and time-resolved infrared experiments to fully characterise the energy dissipations mechanism OB displays.
Acknowledgements
The authors are grateful for technical assistance provided by Mr Wen-Dong Quan (University of Warwick). L. A. B. thanks the Engineering and Physical Sciences Research Council (EPSRC) for providing a studentship under grant EP/F500378/1, through the Molecular Organisation and Assembly in Cells Doctoral Training Centre. M. D. H. thanks the University of Warwick for an EPSRC studentship. S. E. G. thanks the Warwick Institute of Advanced Study for postdoctoral funding. M. N. R. A. thanks the EPSRC for support via program grant EP/L005913. V. G. S. thanks the EPSRC for an equipment grant (EP/J007153) and the Royal Society for a University Research Fellowship.References
- J. P. Ortonne, Photoprotective properties of skin melanin, Br. J. Dermatol., 2002, 146, 7–10 CrossRef CAS PubMed.
- R. S. Mason and J. Reichrath, Sunlight vitamin D and skin cancer, Anti-Cancer Agents Med. Chem., 2013, 13, 83–97 CrossRef CAS PubMed.
- J. Moan, A. C. Porojnicu, A. Dahlback and R. B. Setlow, Addressing the health benefits and risks, involving vitamin D or skin cancer, of increased sun exposure, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 668–673 CrossRef CAS PubMed.
- S. González, M. Fernández-Lorente and Y. Gilaberte-Calzada, The latest on skin photoprotection, Clin. Dermatol., 2008, 26, 614–626 CrossRef PubMed.
- S. Forestier, Rationale for sunscreen development, J. Am. Acad. Dermatol., 2008, 58, S133–S138 CrossRef PubMed.
- M. Norval, R. M. Lucas, A. P. Cullen, F. R. de Gruijl, J. Longstreth, Y. Takizawa and J. C. van der Leung, The human health effects of ozone depletion and interactions with climate change, Photochem. Photobiol. Sci., 2011, 10, 199–225 Search PubMed.
- J. A. Levine, M. Sorace, J. Spencer and D. M. Siegel, The indoor UV tanning industry: A review of skin cancer risk, health benefit claims, and regulation, J. Am. Acad. Dermatol., 2005, 53, 1038–1044 CrossRef PubMed.
- L. A. Baker, M. D. Horbury, S. E. Greenough, P. M. Coulter, T. N. V. Karsili, G. M. Roberts, A. J. Orr-Ewing, M. N. R. Ashfold and V. G. Stavros, Probing the Ultrafast Energy Dissipation Mechanism of the Sunscreen Oxybenzone after UVA Irradiation, J. Phys. Chem. Lett., 2015, 6, 1363–1368 CrossRef CAS PubMed.
- M. T. Ignasiak, C. Houée-Levin, G. Kciuk, B. Marciniak and T. Pedzinski, A Reevaluation of the Photolytic Properties of 2-Hydroxybenzophenone-Based UV Sunscreens: Are Chemical Sunscreens Inoffensive?, ChemPhysChem, 2015, 16, 628–633 CrossRef CAS PubMed.
- V. G. Stavros, Photochemistry: A bright future for sunscreens, Nat. Chem., 2014, 6, 955–956 CrossRef CAS PubMed.
- M. E. Burnett and S. Q. Wang, Current sunscreen controversies: a critical review, Photodermatol., Photoimmunol. Photomed., 2011, 27, 58–67 CrossRef CAS PubMed.
- R. Kumasaka, A. Kikuchi and M. Yagi, Photoexcited States of UV Absorbers, Benzophenone Derivatives, Photochem. Photobiol., 2014, 90, 727–733 CAS.
- T. N. V. Karsili, B. Marchetti, M. N. R. Ashfold and W. Domcke, Ab Initio Study of Potential Ultrafast Internal Conversion Routes in Oxybenzone, Caffeic Acid, and Ferulic Acid: Implications for Sunscreens, J. Phys. Chem. A, 2014, 118, 11999–12010 CrossRef CAS PubMed.
- T. Bintsis, E. Litopoulou-Tzanetaki and R. K. Robinson, Existing and potential applications of ultraviolet light in the food industry - a critical review, J. Sci. Food Agric., 2000, 80, 637–645 CrossRef CAS.
- Global Solar UV Index: A Practical Guide, World Health Organization, 2002 Search PubMed.
- A. R. Webb and O. Engelsen, Calculated Ultraviolet Exposure Levels for a Healthy Vitamin D Status, Photochem. Photobiol., 2006, 82, 1697–1703 CrossRef CAS PubMed.
- N. Serpone, A. Salinaro, A. V. Emeline, S. Horikoshi, H. Hidaka and J. Zhao, An in vitro systematic spectroscopic examination of the photostabilities of a random set of commercial sunscreen lotions and their chemical UVB/UVA active agents, Photochem. Photobiol., 2002, 1, 970–981 RSC.
- M. Lodén, H. Beitner, H. Gonzalez, D. Edström, U. Åkerström, J. Austad, I. Buraczewska-Norin, M. Matsson and H. Wulf, Sunscreen use: controversies, challenges and regulatory aspects, Br. J. Dermatol., 2011, 165, 255–262 CrossRef PubMed.
- V. Ambrogi, L. Latterini, F. Marmottini, M. C. Tiralti and M. Ricci, Oxybenzone Entrapped in Mesoporous Silicate MCM-41, J. Pharm. Innov., 2013, 8, 212–217 CrossRef.
- D. R. Sambandan and D. Ratner, Sunscreens: An overview and update, J. Am. Acad. Dermatol., 2011, 64, 748–758 CrossRef CAS PubMed.
- S. Perun, A. L. Sobolewski and W. Domcke, Role of Electron-Driven Proton-Transfer Processes in the Excited-State Deactivation of the Adenine-Thymine Base Pair, J. Phys. Chem. A, 2006, 110, 9031–9038 CrossRef CAS PubMed.
- A. L. Sobolewski, W. Domcke and C. Hättig, Tautomeric selectivity of the excited-state lifetime of guanine/cytosine base pairs: The role of electron-driven proton-transfer processes, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17903–17906 CrossRef CAS PubMed.
- P. F. Barbara, L. E. Brus and P. M. Rentzepis, Intramolecular Proton Transfer and Excited-State Relaxation in 2-(2-Hydroxyphenyl)benzothiazole, J. Am. Chem. Soc., 1980, 102, 5631–5635 CrossRef CAS.
- F. Laermer, T. Elsaesser and W. Kaiser, Femtosecond Spectroscopy of Excited-State Proton Transfer in 2-(2′-Hydroxyphenyl)benzothiazole, Chem. Phys. Lett., 1988, 148, 119–124 CrossRef CAS.
- C. Chudoba, S. Lutgen, T. Jentzsch, E. Riedle, M. Woerner and T. Elsaesser, Femtosecond studies of vibrationally hot molecules produced by intramolecular proton transfer in the excited state, Chem. Phys. Lett., 1995, 240, 35–41 CrossRef CAS.
- S. E. Greenough, G. M. Roberts, N. A. Smith, M. D. Horbury, R. G. McKinlay, J. M. Żurek, M. J. Paterson, P. J. Sadler and V. G. Stavros, Ultrafast photo-induced ligand solvolysis of cis-[Ru(bipyridine)2(nicotinamide)2]2+: experimental and theoretical insight into its photoactivation mechanism, Phys. Chem. Chem. Phys., 2014, 16, 19141–19155 RSC.
- M. P. Grubb, A. J. Orr-Ewing and M. N. R. Ashfold, KOALA: A program for the processing and decomposition of transient spectra, Rev. Sci. Instrum., 2014, 85, 064104 CrossRef PubMed.
- A. S. Chatterley, C. W. West, V. G. Stavros and J. R. R. Verlet, Time-resolved photoelectron imaging of the isolated deprotonated nucleotides, Chem. Sci., 2014, 5, 3963–3975 RSC.
- A. S. Chatterley, C. W. West, G. M. Roberts, V. G. Stavros and J. R. R. Verlet, Mapping the Ultrafast Dynamics of Adenine onto Its Nucleotide and Oligonucleotides by Time-Resolved Photoelectron Imaging, J. Phys. Chem. Lett., 2014, 5, 843–848 CrossRef CAS PubMed.
- S. C. Warren, A. Margineanu, D. Alibhai, D. J. Kelly, C. Talbot, Y. Alexandrov, I. Munro, M. Katan, C. Dunsby and P. M. W. French, Rapid Global Fitting of Large Fluorescence Lifetime Imaging Microscopy Datasets, PLoS One, 2013, 8, e70687 CrossRef CAS PubMed.
- T. A. Roelofs, C.-H. Lee and A. R. Holzwarth, Global target analysis of picosecond chlorophyll fluorescence kinetics from pea chloroplasts, Biophys. J., 1992, 61, 1147–1163 CrossRef CAS PubMed.
- Y. Zhang, T. A. A. Oliver, M. N. R. Ashfold and S. E. Bradforth, Contrasting the excited state reaction pathways of phenol and para-methylthiophenol in the gas and liquid phases, Faraday Discuss., 2012, 157, 141–163 RSC.
- S. E. Greenough, M. D. Horbury, J. O. F. Thompson, G. M. Roberts, T. N. V. Karsili, B. Marchetti, D. Townsend and V. G. Stavros, Solvent induced conformer specific photochemistry of guaiacol, Phys. Chem. Chem. Phys., 2014, 16, 16187–16195 RSC.
- O. F. Mohammed, S. Luber, V. S. Batista and E. T. J. Nibbering, Ultrafast Branching of Reaction Pathways in 2-(2′-Hydroxyphenyl)benzothiazole in Polar Acetonitrile Solution, J. Phys. Chem. A, 2011, 115, 7550–7558 CrossRef CAS PubMed.
- M. D. Horbury, L. A. Baker, W.-D. Quan, J. D. Young, M. Staniforth, S. E. Greenough and V. G. Stavros, Bridging the Gap between the Gas Phase and Solution Phase: Solvent Specific Photochemistry in 4-tert-Butylcatechol, J. Phys. Chem. A, 2015 DOI:10.1021/acs.jpca.5b03621.
- P. K. Verma, F. Koch, A. Steinbacher, P. Nuernberger and T. Brixner, Ultrafast UV-Induced Photoisomerization of Intramolecularly H-Bonded Symmetric β-Diketones, J. Am. Chem. Soc., 2014, 136, 14981–14989 CrossRef CAS PubMed.
- P. K. Verma, A. Steinbacher, F. Koch, P. Nuerberger and T. Brixner, Monitoring ultrafast intramolecular proton transfer processes in an unsymmetric β-diketone, Phys. Chem. Chem. Phys., 2015, 17, 8459–8466 RSC.
- M. Barbatti, A. J. A. Aquino, H. Lischka, C. Schriever, S. Lochbrunner and E. Riedle, Ultrafast internal conversion pathway and mechanism in 2-(2′-hydroxyphenyl)benzothiazole: a case study for excited-state intramolecular proton transfer systems, Phys. Chem. Chem. Phys., 2009, 11, 1406–1415 RSC.
- J. C. Owrutsky, D. Raftery and R. M. Hochstrasser, Vibrational Relaxation Dynamics in Solutions, Annu. Rev. Phys. Chem., 1994, 45, 519–555 CrossRef CAS PubMed.
- A. Dunkelberger, R. D. Kieda, B. M. Marsh and F. F. Crim, The Picosecond Dynamics of Avobenzone in Solution, J. Phys. Chem. A, 2015, 119, 6155–6161 CrossRef CAS PubMed.
- M. Kasha, Characterization of electronic transitions in complex molecules, Discuss. Faraday Soc., 1950, 6, 14–19 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Global fits for spectral ranges 355 ≤ λ ≤ 415 and 415 < λ ≤ 650. Support plane analysis on extracted lifetimes. Solvent response transient absorption spectra. Single transient fits for 366 nm and 380 nm. Cross correlation measurements are given. See DOI: 10.1039/C5PP00217F |
| This journal is © The Royal Society of Chemistry and Owner Societies 2015 |