
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrimidinone: versatile Trojan horse in DNA photodamage?
Mathias
Micheel†‡
,
Christian
Torres Ziegenbein‡
,
Peter
Gilch
* and
Gerald
Ryseck
Institut für Physikalische Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätstr. 1, D-40225 Düsseldorf, Germany. E-mail: gilch@hhu.de
First published on 8th July 2015
Abstract
(6-4) Photolesions between adjacent pyrimidine DNA bases are prone to secondary photochemistry. It has been shown that singlet excited (6-4) moieties form Dewar valence isomers as well as triplet excitations. We here report on the triplet state of a minimal model for the (6-4) photolesion, 1-methyl-2(1H)-pyrimidinone. Emphasis is laid on its ability to abstract hydrogen atoms from alcohols and carbohydrates. Steady-state and time-resolved experiments consistently yield bimolecular rate constants of ∼104 M−1 s−1 for the hydrogen abstraction. The process also occurs intramolecularly as experiments on zebularine (1-(β-D-ribofuranosyl)-2(1H)-pyrimidinone) show.
1. Introduction
2(1H)-Pyrimidinone (see Fig. 1, P) can be regarded as the basic chemical motif from which many natural compounds such as orotic acid or pyrimidine DNA/RNA bases can be derived. The P motif is strongly related to UV-induced DNA photolesions. After photoexcitation, adjacent pyrimidine bases on a DNA strand can form a pyrimidine(6-4)pyrimidinone lesion.1–3 This photolesion contains P as a moiety which is responsible for the energetically lowest transition in the absorption spectrum of the (6-4) lesion.4,5 The transition peak around 310 nm has a substantial overlap with the solar spectrum at sea level.3 Therefore, the P moiety governs the secondary photochemistry of the (6-4) lesion.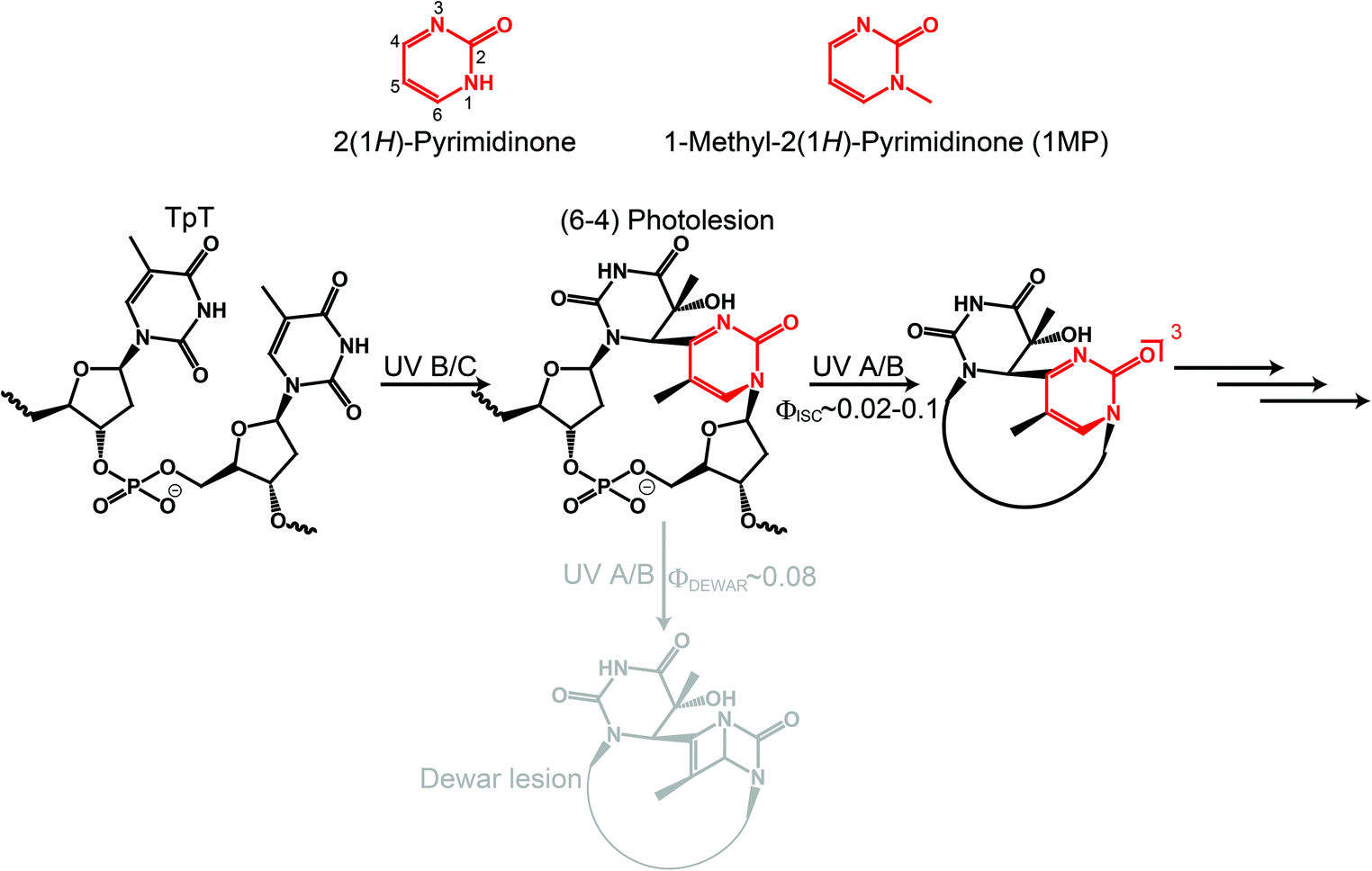 | ||
| Fig. 1 2(1H)-Pyrimidinone derivatives and their relation to DNA photolesions. | ||
It is well known that (6-4) lesions photo-convert to the corresponding Dewar valence isomers.4,6,7 A comprehensive study on a model system, a thymidine derived (6-4) dinucleotide containing a formacetal linker replacing the phosphate group (see Fig. 1), revealed that the P moiety undergoes 4π electrocyclization from the excited singlet state.7,8 The decay of this state within 100 ps is accompanied by the rise of Dewar valence isomer population. In addition to the isomerization, internal conversion (IC) and intersystem crossing (ISC) contribute to the decay of the excited singlet state. ISC results in the population of a triplet state with a quantum yield ϕisc in the range of 0.02–0.1 (all quantum yields are reported as decimal fractions). This poses the question on the role of the triplet state and possible chemical reactions therefrom.
In a recent publication Vendrell-Criado et al. studied the model system 1-(β-D-2′-deoxyribosyl)-5-methyl-2(1H)-pyrimidinone (dRMP) and gave clear evidence for the potency of its triplet excitation lesions to be the source of DNA damage not taking place at the pyrimidinone moiety.9 This “Trojan horse like” behavior has two aspects: (i) an enhanced cyclobutane pyrimidine dimer (CPD) formation in supercoiled circular DNA (pBR322) due to the presence of dRMP was observed. The triplet state energy of dRMP was estimated to be 291 kJ mol−19 in accordance with known values of 2(1H)-pyrimidinone derivatives.10 This value lies well above the one of thymine in DNA which equals 267 kJ mol−1.11,12 Thus, a triplet–triplet energy transfer (TTET) is energetically feasible and CPD formation via the triplet state of thymine could take place. In this study no quantum yield for CPD formation was reported. However, time-resolved IR spectroscopy experiments univocally give evidence that CPD is predominantly formed via the singlet channel.13,14 From the measurements an upper boundary of 0.1 for the efficiency of CPD formation via triplet excitation was deduced.14 (ii) Common lifetimes of triplet states allow for diffusional generation of singlet oxygen. As a result of oxygen quenching of 3(dRMP)* singlet oxygen is formed.9 In addition, the formation of hydroxyl radicals was detected. These two reactive oxygen species (ROS) are known to lead to oxidatively generated purine damages.15,16
In our earlier studies 1-methyl-2(1H)-pyrimidinone (1MP, see Fig. 1) served as a minimal model for the photophysics and -chemistry of (6-4) lesions.5,17 A correlation between the reaction quantum yield—defined as the consumption of 1MP upon illumination—and the nature of the solvent used was found. In solvents with a strong propensity to donate hydrogen atoms a high reaction quantum yield was observed. Photo-induced hydrogen abstractions (HA) were already reported for other pyrimidinone derivatives: besides intermolecular HA,18–21 evidence is given that HA also occurs intramolecularly. Derivatives of P were shown to abstract hydrogen in an intramolecular γ-HA22 manner as well as from a covalently linked thymine.23
According to our results, solvents with hydrogen donating properties are those containing a –CHR–O– motif.17 Carbohydrates contain such a chemical structure, in particular 2-deoxyribose (dR) as part of the DNA backbone. HA from this sugar can cause, for instance, single strand breaks (SSB) in DNA.24 In summary, (6-4) lesions could act as a much more versatile “Trojan horse” as reported heretofore.
We here present steady-state and flash-photolysis experiments which show that triplet excited pyrimidinones abstract hydrogen atoms from carbohydrates and related molecules. We rely on 1MP due to its high triplet quantum yield ϕisc of 0.5 in water5 and 0.35 in methanol.17 Quenching experiments of 31MP* by β-D-ribose find limitations because of its solubility and absorbance. Methanol, in which virtually all triplet states undergo reduction,17 serves as a quencher for time resolved measurements. The findings are finally compared to results of zebularine, 2(1H)-pyrimidinone N-glycosidically bonded to β-D-ribose. In this compound the abstraction occurs intramolecularly.
2. Materials and methods
2.1 Materials
1-Methyl-2(1H)-pyrimidinone (Specs), β-D-ribose (97%, Sigma-Aldrich) and zebularine (≥98%, Sigma-Aldrich) were used as received. The solvents water (Chromanorm, VWR chemicals), methanol (Uvasol, Merck KGaA), perdeuterated methanol (Euriso-top, 99.8%), and tetrahydrofuran (Uvasol, Merck KGaA) were of spectroscopic grade. Research grade gases (nitrogen (N50), oxygen (N48), and nitrous oxide (N25)) were supplied by Air Liquide.2.2 Product analysis
For product analysis 1MP was dissolved in methanol and spectroscopic signatures before and after illumination with monochromatic light at 310 nm were compared.For nuclear magnetic resonance (NMR) experiments, the illumination was stopped prior to complete conversion so that residual 1MP could serve as an internal standard. 1H-NMR spectra were taken with a Bruker Avance III spectrometer at 600 MHz. To record the NMR spectra the solvent methanol was removed under vacuum and the residue was dissolved in DMSO-d6.
Mass spectra (MS) were recorded with a triple–quadrupole mass spectrometer (Finnigan MAT TSQ 7000) applying electron impact ionization (EI) as well as an ion trap atmospheric pressure ionization mass spectrometer (Finnigan Thermo Quest LCQ Deca) applying electro spray ionization (ESI). MS were taken before the illumination and after complete conversion.
2.3 Steady-state illumination
Reaction quantum yields were determined as described in ref. 17. For some experiments a different light source and another absorption spectrometer was used. For these, laser diodes (UVLUX305-FW-3, Roithner Lasertechnik GmbH) with a center wavelength of 305–310 nm and a light power of 1.5–2 mW were used to illuminate the sample in a 1 cm quartz cuvette (220-QS, Hellma Analytics). Concentrations of the order of ∼0.5 mM ensured virtually complete absorption of the excitation light. After several illumination periods (with a duration on the order of ∼10–100 s) absorption spectra were recorded (Lambda 19, Perkin Elmer). Solutions were deoxygenated by purging the sealed cuvette for ∼10 min with nitrogen.2.4 Laser flash photolysis
Nanosecond transient absorption spectra were acquired with a LSK 6.0 spectrometer (Applied Photophysics) with a cross beam configuration.25 Frequency quadrupled (266 nm) pulses from a Nd:YAG laser (Innolas, Spitlight 600, 1 Hz repetition rate) served as the excitation light. Their energy was attenuated to 20 mJ and their duration was 7 ns (FWHM). A pulsed Xenon lamp (L2273, Hamamatsu) provided the probe light. After passing the sample the probe light was monochromatized by a grating spectrometer (bandwidth 4.7 nm) and detected by a photomultiplier (1P28, Hamamatsu). Its signal was digitized by an oscilloscope (Agilent Infiniium) and transferred to a computer. Time traces were recorded for detection wavelengths ranging from 200 nm to 555 nm in 5 nm steps. For spectrally resolved measurements 10 successive laser shots were averaged, for single channel measurements 100 shots were averaged for a better signal to noise ratio. Typical sample concentrations were of the order of ∼100 μM to ensure a uniform excitation along the cuvette path (sealed 111-QS, Hellma Analytics). The sample was purged with nitrous oxide to remove dissolved oxygen and to quench solvated electrons.26 To avoid the accumulation of photoproducts in the sample cell a flow system was used. The ambient temperature of 20 °C was controlled by a thermostat.3. Results
3.1 Characterization of 1MP triplet state
In our earlier study on the photophysics of 1MP in water5 we assigned the spectrum recorded at ∼1 ns after photo-excitation to the triplet state of 1MP. To back this assignment, we here study the photophysics of 1MP by means of laser flash photolysis. The earliest spectrum obtained by flash photolysis coincides with the one found as an offset spectrum in our femtosecond experiment.5 A characteristic positive absorption band peaking at approximately 430 nm, which is flanked by flat positive contributions for longer as well as shorter wavelengths, can be recognized (see Fig. 2). The laser flash instrumentation allowed extension of the spectral window down to 200 nm giving additional information. The complete ground state bleach is covered and seen as a negative contribution around 300 nm. At even lower wavelengths a strong positive absorption is observed. A global analysis, as described in ref. 5, yielded a time constant of 6.2 μs for a 1MP concentration of 95 μM. This decay leaves an offset displaying a positive band peak at 230 nm and a negative around 300 nm.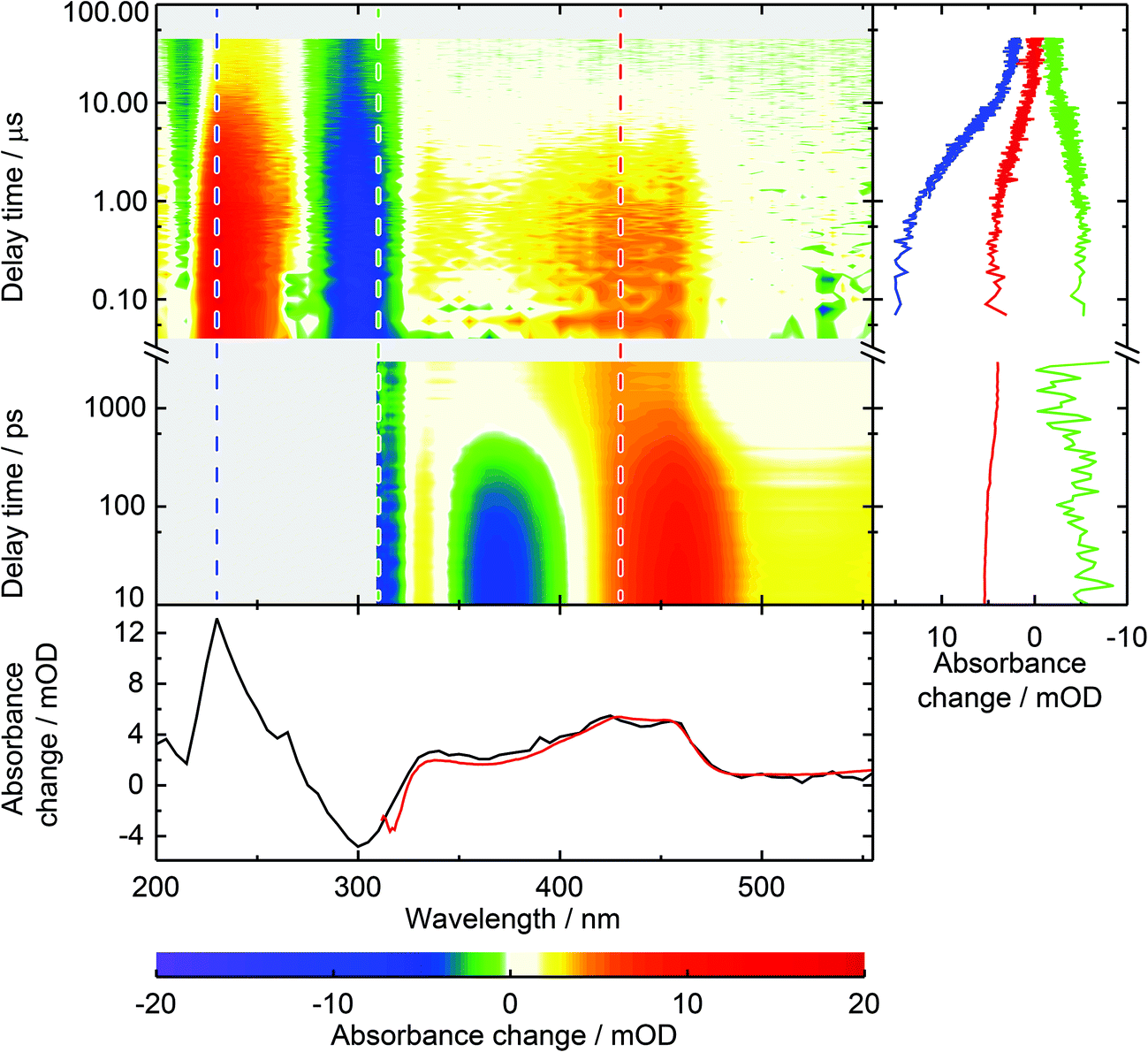 | ||
| Fig. 2 Spectrally and temporally resolved UV/Vis absorption spectra of photo-excited 1MP in water. The abscissa denotes the detection wavelength on a linear scale, the ordinate the time after photo-excitation on a logarithmic scale. The color table is chosen to display positive signals in a red hue and negative ones in blue. White indicates zero and gray a lack of data. The femtosecond transient absorption data from time 1 ps up to 3.5 ns are taken from ref. 5. Two merged data sets from laser flash photolysis measurements complement the spectra up to 50 μs, the one from 40 ns to 1 μs with a time resolution of 20 ns, the other for longer delay times with a time resolution of 100 ns. (Right) Time traces for detection wavelengths of 230 nm (blue), 310 nm (green) and 430 nm (red) are given for the covered time window. (Bottom) Decay associated spectra (DAS) from global data analysis of femtosecond transient absorption data (red) and laser flash photolysis (black). For direct comparison the flash photolysis data were scaled by 0.78. | ||
For the presented data the sample was purged with nitrous oxide to quench solvated electrons.26 Solvent only experiments under identical conditions showed that the solvated electrons result from the excitation of water with nanosecond pulses. For 1MP solutions the signal due to the solvated electrons is reduced by the inner filter effect of 1MP. Comparative measurements with nitrogen and nitrous oxide purging demonstrate that the solvated electrons partially quench 31MP*. In the experiment with nitrogen purging (data not shown) an additional time constant of ∼200 ns may be assigned to this process. In line with this, the spectral signature of *1MP3 falls noticeably as the solvated electron decays—to about half of its initial value for the applied experimental conditions. Scavenging of solvated electrons in water by nitrous oxide results in hydroxy radicals.26 These radical do not seem to affect the 31MP* decay since identical lifetimes were determined for nitrogen purging and purging with nitrous oxide.
To determine the unimolecular rate constant k0 as well as the rate constant for self-quenching ksq the dependence of the decay of 31MP* on the 1MP concentration [1MP] was monitored at a detection wavelength of 430 nm. The results of single-exponential fits to the time traces yield the corresponding non-reactive rate constants knr([1MP]) as shown in Fig. 3 (left). In the absence of oxygen quenching the rate constant should be given by
| knr([1MP]) = k0 + ksq[1MP]. | (1) |
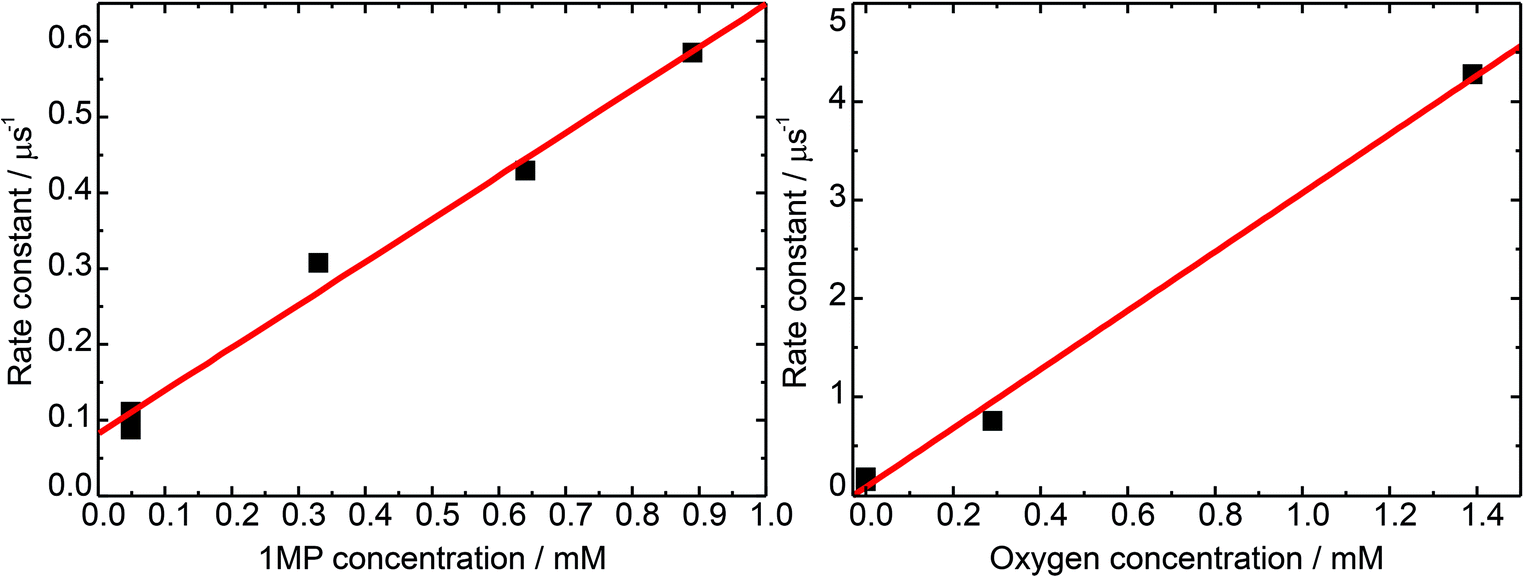 | ||
| Fig. 3 Rate constants for the triplet decay of 1MP. Rate constants are determined from exponential fits to time traces at a detection wavelength of 430 nm (squares). A linear fit to that data (lines) yields the corresponding quenching rate constants. (Left) Concentration dependence of the triplet decay in de-oxygenated solutions. (Right) Quenching by molecular oxygen at a 1MP concentration of 75 μM. | ||
A linear fit to this dependence (see Fig. 3) yielded a unimolecular rate constant k0 of (0.8 ± 0.2) × 105 s−1 (= (12.5 μs)−1). This value is comparable to the one of (9.7 μs)−1 found for the derivative dRMP9 and is in the range reported for triplet states of carbonyl compounds.27 The derived self-quenching rate constant ksq amounts to (0.57 ± 0.04) × 109 M−1 s−1, in good agreement with our earlier estimate of 0.4 × 109 M−1 s−1.5
The rate constant of triplet quenching by molecular oxygen serves as a tool to calibrate steady-state experiments for comparison with time-resolved measurements. To gain information on the rate constant of 31MP* quenching by oxygen we performed measurements with an air-saturated, an oxygen-purged, and a nitrogen-purged solution. The concentrations of oxygen in water at 20 °C amounted to 0.29 mM (air, 1 atm) and 1.38 mM (pure oxygen, 1 atm).27 The data were analyzed in the same manner as described for the self-quenching data. The derived rate constant for oxygen quenching equals kq,O2 = (3.0 ± 0.1) × 109 M−1 s−1. The diffusion-limited rate constant in water is 6.5 × 109 M−1 s−1 at 20 °C.27 The ratio of the rate constants yields a spin statistical factor28 of σ ≈ 4/9.
3.2 Reactive quenching of 1MP triplet state
In the following we examine the reactive quenching of the triplet state of 1MP. A rough estimation of the reaction rate constant in methanol yielded a value on the order of 105 M−1 s−1.17 Thus, a rather high concentration of quencher molecules is required for the quenching process to compete with the unimolecular decay of the triplet state of 1MP. This causes experimental complicacy depending on the quencher under study. Especially carbohydrates, such as β-D-ribose (R) and tetrahydrofuran considered here, exhibit non-negligible ground state and transient absorption. That is why we embark on the following strategy. We restrict ourselves to methanol for the direct observation of the quenching in time-resolved experiments as well as for the verification of the hydrogen abstraction process. The reaction of photo-excited 1MP with R is shown to occur inter- and intramolecularly by illumination experiments.A representative experiment for 1MP dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 MeOH/water solution ([1MP] = 0.4 mM) is depicted in Fig. 4 (left). The decrease of the absorption band of 1MP peaking around 300 nm goes along with the formation of a band at ∼250 nm. Such spectral changes are in line with illumination experiments on other pyrimidinone derivatives.18,23 The same behavior is seen for the illumination of zebularine in water (Fig. 4, middle). The absorption spectrum of the parent molecule resembles the one of 1MP. For every illumination step its magnitude drops and a band around 250 nm increases. Both photo-reactions, the intermolecular one of 1MP with MeOH as well as the intramolecular one of zebularine, do not differ substantially according to their spectral imprints. In addition, the reaction quantum yields of the bimolecular reaction of 1MP with R seem to continue the outcome seen for the bimolecular reaction of 1MP with MeOH (Fig. 4, right). We, thus, suppose that these photoreactions obey the same mechanism.
:10 MeOH/water solution ([1MP] = 0.4 mM) is depicted in Fig. 4 (left). The decrease of the absorption band of 1MP peaking around 300 nm goes along with the formation of a band at ∼250 nm. Such spectral changes are in line with illumination experiments on other pyrimidinone derivatives.18,23 The same behavior is seen for the illumination of zebularine in water (Fig. 4, middle). The absorption spectrum of the parent molecule resembles the one of 1MP. For every illumination step its magnitude drops and a band around 250 nm increases. Both photo-reactions, the intermolecular one of 1MP with MeOH as well as the intramolecular one of zebularine, do not differ substantially according to their spectral imprints. In addition, the reaction quantum yields of the bimolecular reaction of 1MP with R seem to continue the outcome seen for the bimolecular reaction of 1MP with MeOH (Fig. 4, right). We, thus, suppose that these photoreactions obey the same mechanism.
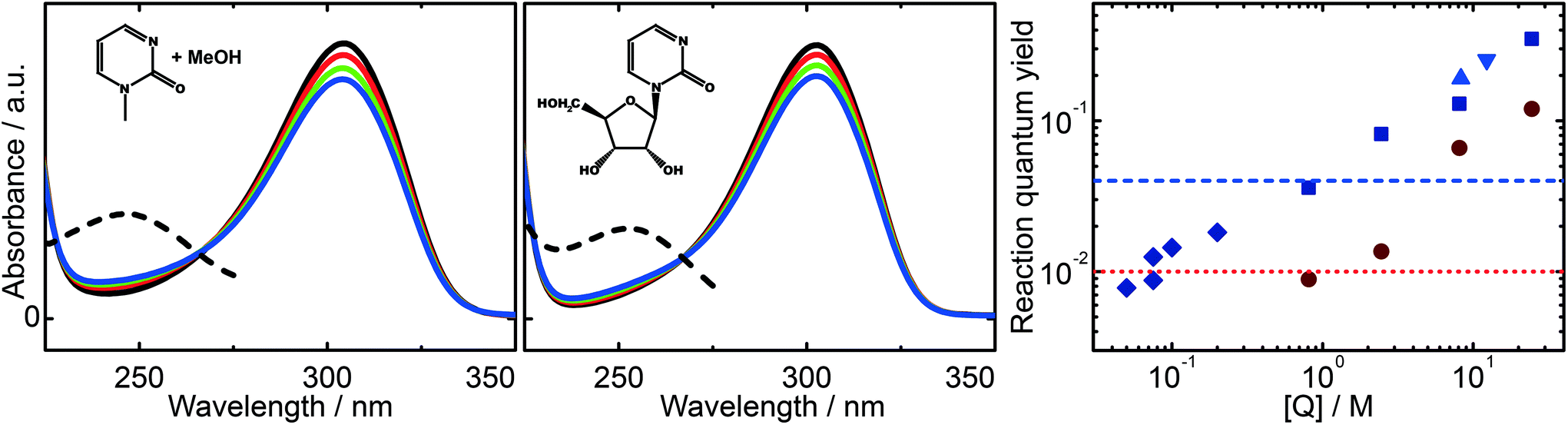 | ||
| Fig. 4 Representative spectral changes resulting from illumination of 1MP in methanol (left) and zebularine in water (middle) as well as quantum yields derived from these changes (right). Subsequent illumination steps correspond to a light dose of ∼1 photon per molecule. The product spectra obtained by extrapolation to complete consumption are given by dashed lines. A compilation of reaction quantum yields for different quencher concentrations is found on the right (log–log plot). Blue points depict values derived for nitrogen-purged solutions, red ones for air-saturated solutions. Values are given for methanol (squares), THF (triangles) and β-D-ribose (diamonds). The horizontal dashed lines specify the reaction quantum yields of zebularine in nitrogen-purged (blue) and air-saturated (red) aqueous solution. | ||
In a recent study we postulated that a photo-induced hydrogen abstraction reaction occurs.17 To prove this hypothesis we here rely on analysis of the product formed. The highest reaction rate can be achieved in pure methanol. Therefore, we recorded the nuclear magnetic resonance spectrum (NMR) and the mass spectrum (MS) of the photo-product of 1MP in methanol.
In these experiments we will not detect the initial product of the HA which is a radical (Fig. 5). Presumably, the hydrogen atom is first attached to the carbonyl oxygen of 1MP. This is often assumed for carbonyl triplets.29,30 The hydroxyl substituted carbon centered radical may then undergo a lactam–lactim like tautomerization31,32 resulting in the restoration of the carbonyl group and the formation of an NH-group. This radical may then recombine with the quencher radical or dimerize. The NH moiety present in the final products should show up around 8 ppm in a 1H-NMR spectrum.18,23,33 Such resonances are commonly very broad.33 Both features are found for the photo-product formed in our illumination experiments (Fig. 6, left). Illumination of 1MP in perdeuterated methanol results in a decreased signal at 8 ppm supporting this assignment. Inspection of other resonances in the NMR spectrum points to a mixture of photo-products. This matches the proposed radical mechanism.34 No attempts were made to isolate the products.
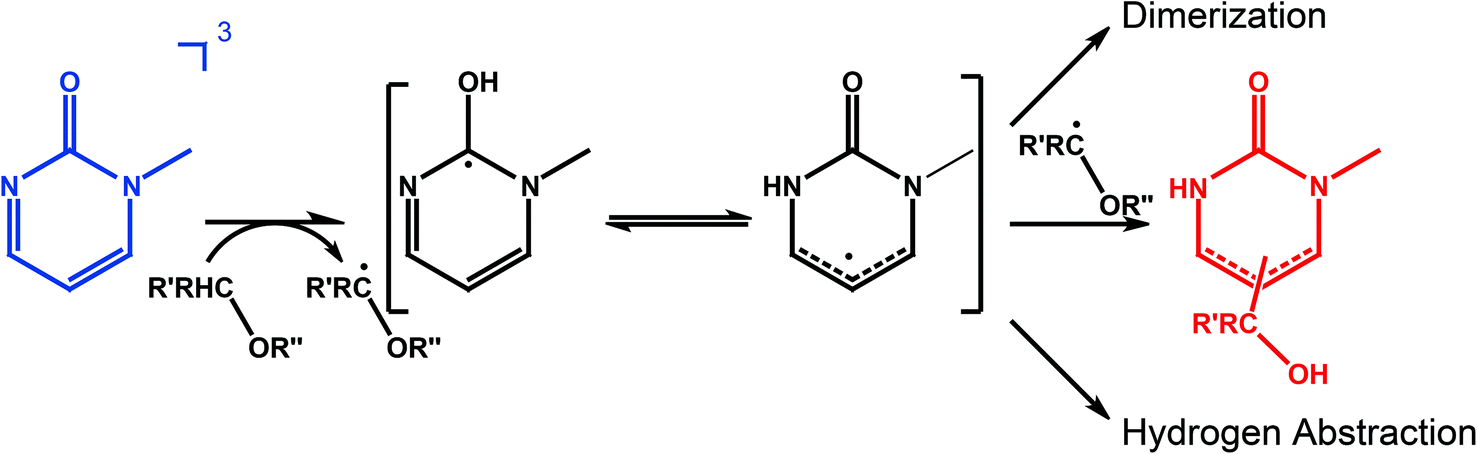 | ||
| Fig. 5 Scheme summarizing HA by 31MP*. 1MP in the triplet state abstracts a hydrogen atom from the quencher bearing a –CRH–O– chemical motif. The so formed radical can tautomerize forming a NH group. Recombination with the quencher radical terminates the reaction chain. For high concentrations also dimerization could occur. | ||
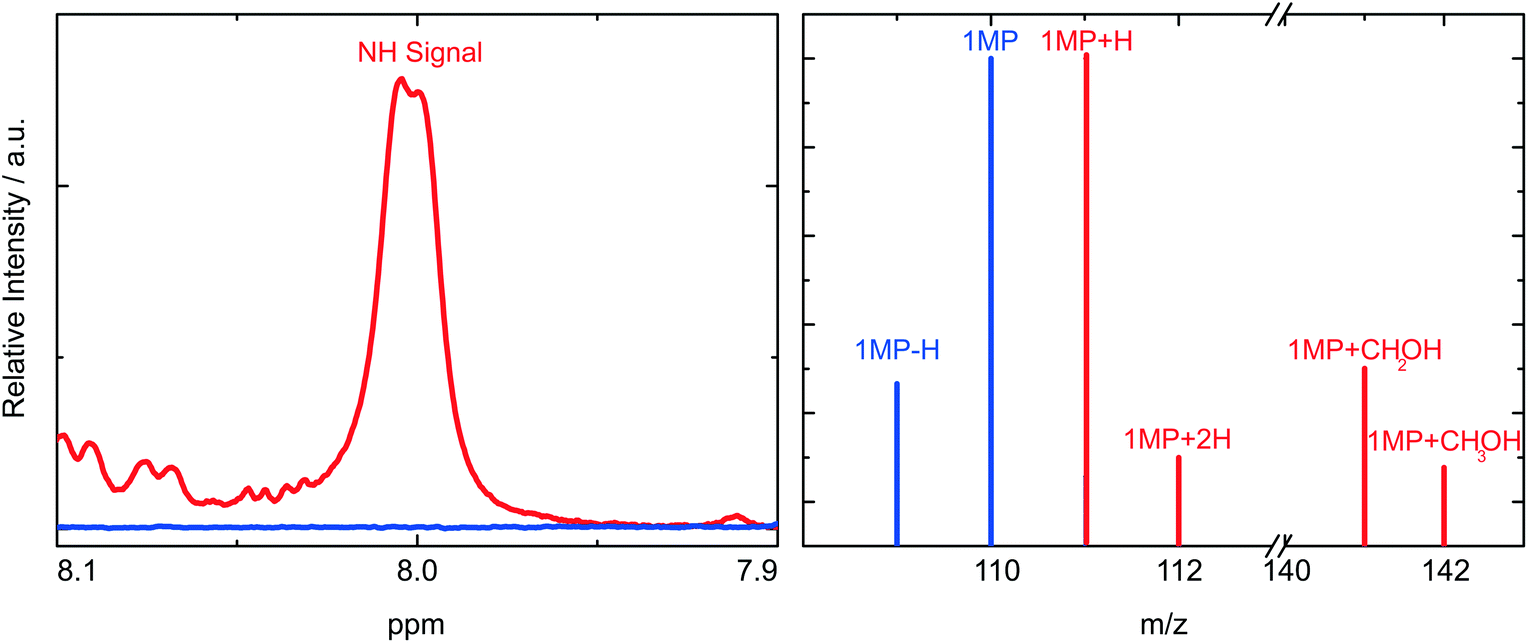 | ||
| Fig. 6 Determination of the chemical motifs present in the photo-product of 1MP in methanol. Measurements of 1MP are colored blue, those of the photoproduct red. (Left) The product displays a broad signal around 8 ppm in the NMR spectrum. (Right) The mass spectrum of the product exhibits strong peaks at m/z = 111 (1MP + H) and m/z = 141 (1MP + CH2OH). Slightly weaker ones at m/z = 112 (1MP + 2H) and m/z = 142 (1MP + CH3OH). | ||
To supplement this finding we performed MS measurements to determine the final product. In the mass spectrum (Fig. 6, right) a peak for the 1MP + H fragment shows up at m/z = 111. This could be due to the fragmentation of the dimer. Other strong MS peaks at m/z = 141 and m/z = 142 are in accordance with the addition of MeOH to the parent 1MP. MS evidence for the MeOH addition was observed after illumination of 1MP solutions purged with nitrogen as well as ones purged with oxygen (∼1 atm). Such a photoinduced alcohol addition to a pyrimidinone derivative has already been reported.18 The NMR and MS measurements give evidence that the quenching of the triplet state of 1MP by MeOH is due to a hydrogen abstraction mechanism followed by an addition of the ˙CH2–OH radical.
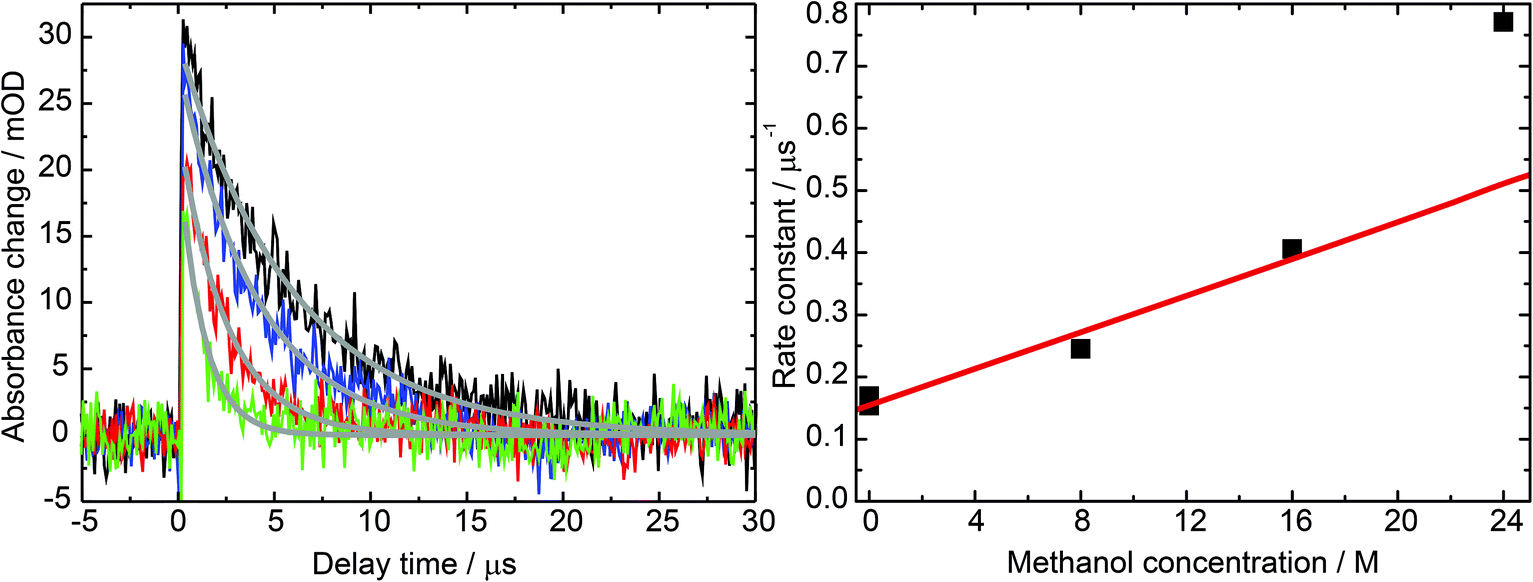 | ||
| Fig. 7 Impact of the methanol concentration on the 31MP* lifetime. (Left) Laser flash photolysis of 1MP in water (black), methanol (green) and methanol/water mixtures with a ratio of 1:2 (blue) and 2:1 (red). For all experiments the concentration of 1MP was around 100 μM. The detection wavelength was set to 430 nm. Exponential fits are given by gray lines. (Right) The rate constants derived from these fits are plotted in dependence on the methanol concentration (black squares). The red fit line illustrates the linearity. The last data point was excluded from the linear fit, see text. | ||
Exponential fits to the time traces yielded time constants varying from 5.9 μs (pure water) to 1.3 μs (pure methanol). Analysis of the kinetic data according to
| k([MeOH];[1MP]) = knr([1MP]) + krq[MeOH] | (2) |
:1 methanol/water mixture for varying temperatures yield an activation energy on the order of ∼2000 cm−1.
4. Discussion
1MP dissolved in MeOH shows a reaction quantum yield ϕr equal to the triplet quantum yield ϕisc pointing to an effective chemical quenching via the triplet channel. This was attributed to a hydrogen abstraction (HA) mechanism by 31MP*.17 Here, this assumption was proven by product analysis based on NMR and MS spectroscopy. The MS data further show that the addition of MeOH to 1MP occurs in the presence and absence of oxygen. In methanol this addition ought to involve the radical ˙CH2OH as an intermediate. Since this radical is known to be scavenged by oxygen with a rate constant close to the diffusion limit35,36 this is surprising. It could indicate that the 1MP + H radical and ˙CH2OH undergo efficient geminate recombination30 with which the reaction with oxygen cannot compete.We now turn to the comparison of kinetic parameters derived from steady-state and time-resolved measurements. The fraction ηT of triplet states abstracting hydrogen atoms reads
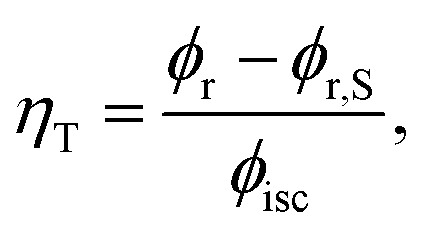 | (3) |
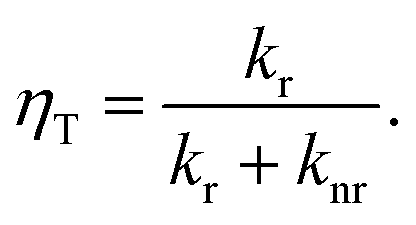 | (4) |
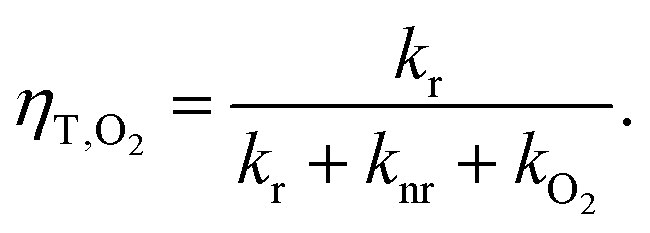 | (5) |
After combining this relation with eqn (4) the reactive rate constant can be calculated according to
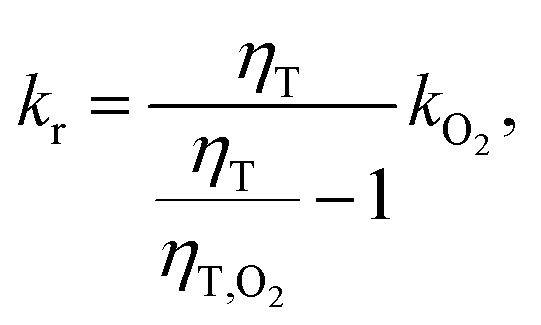 | (6) |
We have shown that the spectral changes for illumination of zebularine are in line with the experiments on the photo-conversion of 1MP in MeOH. It can be, thus, expected that the very same mechanism is operative. We will now show that the pertinent rate parameters also are very similar. The rate constant kuni for the reaction, which is now a uni-molecular one, was obtained from the data in Fig. 4 using eqn (6). It amounts to 2.2 × 104 s−1. The comparison of this rate constant with the one for the bimolecular reaction can be achieved based on the theory of the encounter complex.37,38 The bimolecular reaction of 1MP triplet with the sugar (R) can be described by two step kinetics:
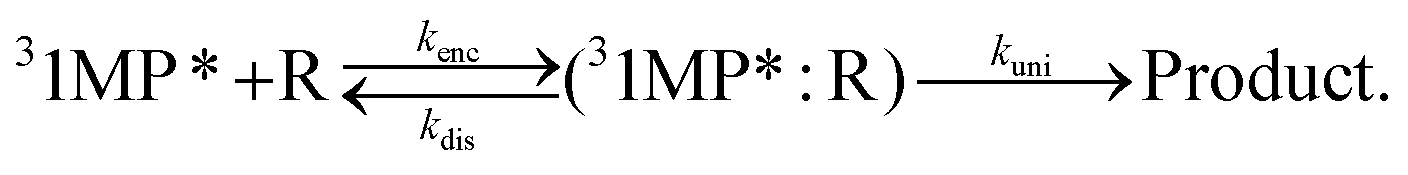 | (7) |
An encounter complex is formed (rate constant kenc) which can undergo a chemical reaction with a rate constant kuni or dissociate (rate constant kdis). In the case of a reaction-limited process the bimolecular rate constant krq is given by
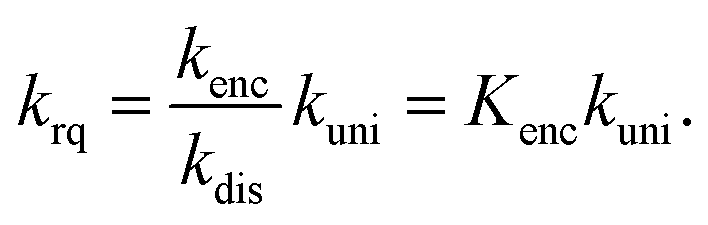 | (8) |
Eqn (8) and the rate constants given above yield an equilibrium constant Kenc of ∼0.7 M−1. In the simplest approximation37 the equilibrium constant Kenc of the encounter complex is given by its volume Venc times Avogadro's constant NA:
| Kenc = VencNA. | (9) |
For an independent rough estimate of Venc we relied on the structure of zebularine assuming that this system adequately represents the encounter complex. The ground state of zebularine was optimized (MM2 force field, ChemBio3D v14) and its molecular volume Venc = 0.173 nm3 derived as described in ref. 39 (Connolly's solvent excluded volume). The estimated equilibrium constant then equals ∼0.1 M−1. This value being smaller than that for the encounter complex is to be expected since in zebularine the moieties are covalently linked. The higher value for the encounter complex corresponds to a distance between the moieties which is by a factor of ∼2 larger.
The feasibility of photo-induced HA from the sugar-phosphate backbone in (6-4) lesions is hereby shown. The question of its potency compared to other triplet deactivation mechanisms, for instance TTET, remains open. Vendrell-Criado and coworkers determined the rate constant for TTET from the pyrimidinone moiety to thymidine to be 1.6 × 107 M−1 s−1.9 This value is three orders of magnitudes higher than the rate constant of HA derived here. Assuming that after TTET from pyrimidinone to thymine a fraction of ≤0.114 of the thymine triplets leads to CPD lesions, the ratio of damage due to HA and TTET would be 1/100 or higher. From the data published in ref. 9 it emerges that, in addition to TTET-induced CPD formation, the presence of excited pyrimidinone leads to SSB. It might be that these SSB are triggered by HA.
5. Conclusions
The present findings give strong evidence that intramolecular hydrogen abstraction occurs in triplet excited zebularine. The zebularine moiety closely resembles the structural motifs found in natural (6-4) lesions. In addition to Dewar valence isomerization and TTET, hydrogen abstraction may play a part in the secondary photochemistry of (6-4) lesions. Thus, the (6-4) damage could be seen as a much more versatile hotspot for secondary photochemistry as already reported in the literature.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (projects GI349/3-2 and GI349/5-1) is gratefully acknowledged.References
- J. Cadet, C. Anselmino, T. Douki and L. Voituriez, J. Photochem. Photobiol., B, 1992, 15, 277–298 CrossRef CAS.
- P. H. Clingen, C. F. Arlett, L. Roza, T. Mori, O. Nikaido and M. H. L. Green, Cancer Res., 1995, 55, 2245–2248 CAS.
- J. Cadet, A. Grand and T. Douki, in Photoinduced Phenomena in Nucleic Acids II, ed. M. Barbatti, A. C. Borin and S. Ullrich, Springer International Publishing, 2015, vol. 356, pp. 249–275 Search PubMed.
- D. G. E. Lemaire and B. P. Ruzsicska, Photochem. Photobiol., 1993, 57, 755–769 CrossRef CAS PubMed.
- G. Ryseck, T. Schmierer, K. Haiser, W. Schreier, W. Zinth and P. Gilch, ChemPhysChem, 2011, 12, 1880–1888 CrossRef CAS PubMed.
- J. S. Taylor and M. P. Cohrs, J. Am. Chem. Soc., 1987, 109, 2834–2835 CrossRef CAS.
- K. Haiser, B. P. Fingerhut, K. Heil, A. Glas, T. T. Herzog, B. M. Pilles, W. J. Schreier, W. Zinth, R. de Vivie-Riedle and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 408–411 CrossRef CAS PubMed.
- B. P. Fingerhut, T. T. Herzog, G. Ryseck, K. Haiser, F. F. Graupner, K. Heil, P. Gilch, W. J. Schreier, T. Carell, R. de Vivie-Riedle and W. Zinth, New J. Phys., 2012, 14, 065006 CrossRef.
- V. Vendrell-Criado, G. M. Rodriguez, M. C. Cuquerella, V. Lhiaubet-Vallet and M. A. Miranda, Angew. Chem., Int. Ed., 2013, 52, 6476–6479 CrossRef CAS PubMed.
- G. Wenska, B. Skalski and S. Paszyc, J. Photochem. Photobiol., A, 1991, 57, 279–291 CrossRef CAS.
- F. Bosca, V. Lhiaubet-Vallet, M. C. Cuquerella, J. V. Castell and M. A. Miranda, J. Am. Chem. Soc., 2006, 128, 6318–6319 CrossRef CAS PubMed.
- M. C. Cuquerella, V. Lhiaubet-Vallet, F. Bosca and M. A. Miranda, Chem. Sci., 2011, 2, 1219–1232 RSC.
- W. J. Schreier, J. Kubon, N. Regner, K. Haiser, T. E. Schrader, W. Zinth, P. Clivio and P. Gilch, J. Am. Chem. Soc., 2009, 131, 5038–5039 CrossRef CAS PubMed.
- B. M. Pilles, B. D. Bucher, L. Z. Liu, P. Clivio, P. Gilch, W. Zinth and W. J. Schreier, J. Phys. Chem. Lett., 2014, 5, 1616–1622 CrossRef CAS.
- L. F. Batista, B. Kaina, R. Meneghini and C. F. Menck, Mutat. Res., Rev. Mutat. Res., 2009, 681, 197–208 CrossRef CAS PubMed.
- J. Cadet and J. R. Wagner, Cold Spring Harbor Perspect. Biol., 2013, 5, a012559 Search PubMed.
- G. Ryseck, T. Villnow, S. Hugenbruch, K. Schaper and P. Gilch, Photochem. Photobiol. Sci., 2013, 12, 1423–1430 CAS.
- K.-H. Pfoertner, Helv. Chim. Acta, 1975, 58, 865–871 CrossRef CAS PubMed.
- T. Nishio and Y. Omote, J. Chem. Soc., Perkin Trans. 1, 1988, 957–960 RSC.
- T. Nishio, M. Kato and C. Kashima, Liebigs Ann. Chem., 1989, 611–615 CrossRef CAS PubMed.
- T. Nishio, Liebigs Ann. Chem., 1992, 71–73 CrossRef CAS PubMed.
- T. Nishio, S. Kameyama and Y. Omote, J. Chem. Soc., Perkin Trans. 1, 1986, 1147–1150 RSC.
- S. Y. Wang and D. F. Rhoades, J. Am. Chem. Soc., 1971, 93, 2554–2556 CrossRef CAS.
- W. K. Pogozelski and T. D. Tullius, Chem. Rev., 1998, 98, 1089–1108 CrossRef CAS PubMed.
- S. Fröbel, L. Buschhaus, T. Villnow, O. Weingart and P. Gilch, Phys. Chem. Chem. Phys., 2015, 17, 376–386 RSC.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS PubMed.
- Handbook of Photochemistry, ed. S. L. Murov and I. Carmichael and G. L. Hug, Marcel Dekker, Inc., New York, Basel, Hong-Kong, 1993 Search PubMed.
- J. Saltiel and B. W. Atwater, in Spin-Statistical Factors in Diffusion-Controlled Reactions, ed. D. H. Volman, G. S. Hammond and K. Gollnick, John Wiley & Sons, Inc., 1988, pp. 1–90 Search PubMed.
- P. Klán and J. Wirz, Photochemistry of Organic Compounds: From Concepts to Practice, Wiley-Blackwell, Chichester, 2009 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Sciences Books, Sausalito, California, 2010 Search PubMed.
- A. Katritzky and J. Lagowski, in Advances in Heterocyclic Chemistry, ed. A. Katritzky, Academic Press, 1963, vol. 1, pp. 311–338 Search PubMed.
- A. Lledós and J. Bertran, Tetrahedron Lett., 1981, 22, 775–778 CrossRef.
- M. Hesse, H. Meier, B. Zeeh, R. Dunmur and M. Murray, Spectroscopic methods in organic chemistry, Thieme, Stuttgart, 2008 Search PubMed.
- M. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, And Structure, Wiley-Interscience, 2007 Search PubMed.
- G. E. Adams and R. L. Willson, Trans. Faraday Soc., 1969, 65, 2981–2987 RSC.
- T. Schaefer, D. van Pinxteren and H. Herrmann, Environ. Sci. Technol., 2015, 49, 343–350 CrossRef CAS PubMed.
- S. Benson, Physical Models for Solution Reactions, in Foundation of Chemical Kinetics, McGraw-Hill Book Company Inc., 1960, ch. 15 Search PubMed.
- S. K. Upadhyay, Chemical kinetics and reaction dynamics, Springer Science & Business Media, 2007 Search PubMed.
- M. L. Connolly, J. Am. Chem. Soc., 1985, 107, 1118–1124 CrossRef CAS.
Footnotes |
| † Present address: Leibniz-Institute of Photonic Technology Jena (IPHT), Albert-Einstein-Str. 9, D-07745 Jena, Germany. |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2015 |