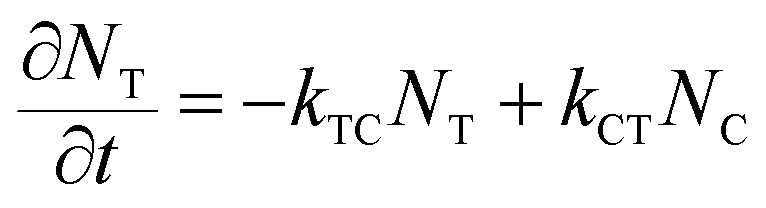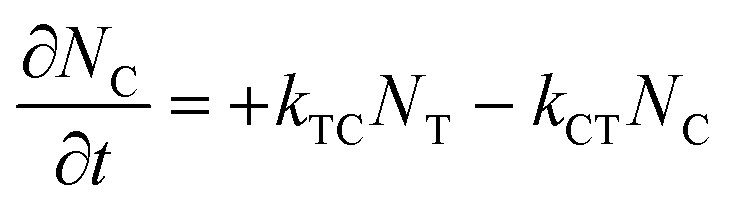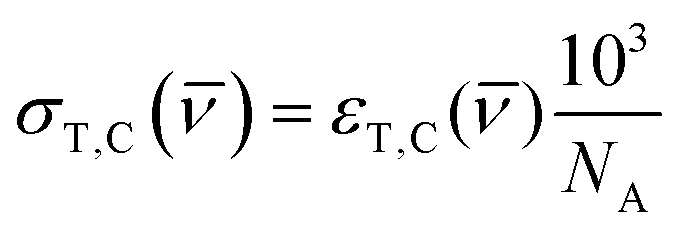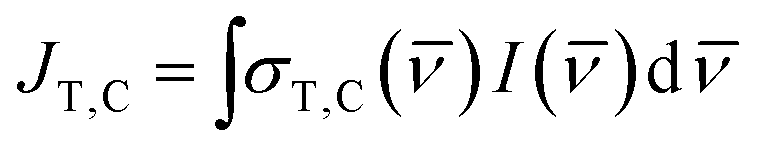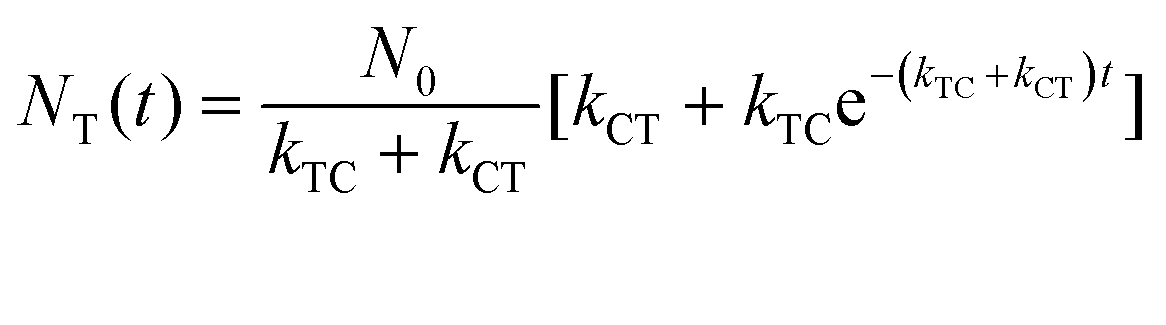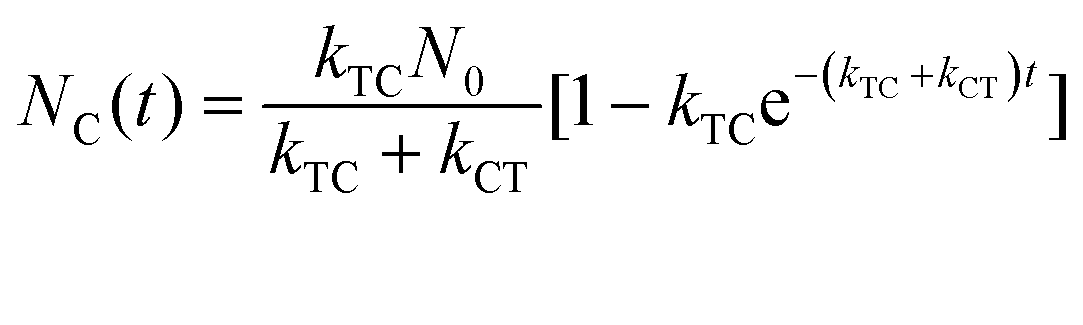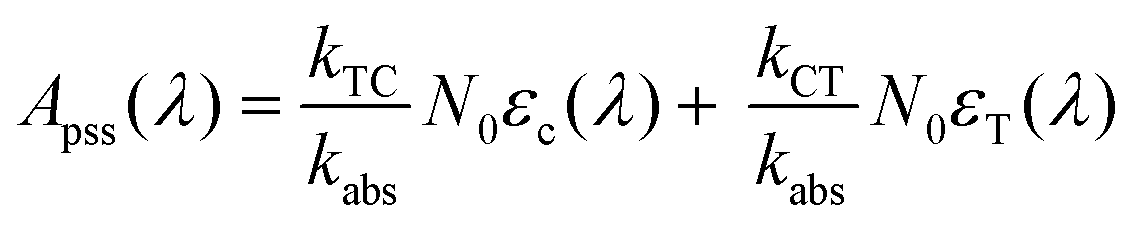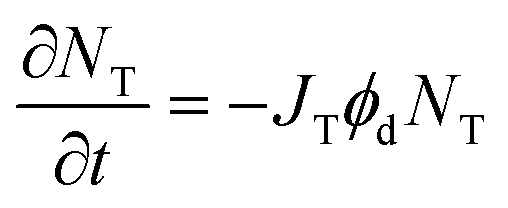Photochemical degradation of the UV filter octyl methoxycinnamate in solution and in aggregates†
Received
19th February 2015
, Accepted 22nd May 2015
First published on 26th May 2015
Abstract
The photodegradation of the ultraviolet (UV) filter octyl methoxycinnamate (OMC) is investigated in both dilute solution and in aggregated form. In dilute solution, the ratio of trans and cis isomers achieved at the photostationary state is solvent-dependent because of variations in the isomerization quantum yield. The two isomeric forms at the photostationary state are highly resistant to further photodegradation and no other UVA-absorbing species are formed. Aggregation of OMC, either in a neat film or in aqueous colloidal suspensions, leads to irreversible photodegradation of the molecule and the formation of multiple photoproducts. In addition to previously identified photoproducts like the UVB-absorbing cis and trans isomers and photodimers, we find photoproduct species whose absorption extends into the UVA. Characterization of the photophysical properties of these species indicates that they have long-lived excited-states (τf > 1 ns, 400 nm), unlike the isomeric forms of OMC (τf < 30 ps, 266 nm), and that excitation at 405 nm can sensitize the formation of singlet oxygen. These results show that the environment of OMC affects the photochemistry of the molecule and that the environmental conditions must be taken into account when considering the molecule's stability. In particular, aggregation of OMC molecules results in complex photochemistry that can produce species whose absorption extends into UVA and are capable of generating reactive oxygen species.
Introduction
The use of commercial sunscreens to provide protection from damaging ultraviolet (UV) solar radiation is an important public health issue. Sunscreens use UV filter molecules to absorb UV photons at the skin surface before they can penetrate into the deeper epidermis and dermis, where UV absorption can cause inflammation, immune suppression, reactive oxygen species generation and DNA damage.1–5 The efficacy of a sunscreen is determined not only by its ability to absorb UV photons, but also by its photostability under irradiation.6–13 If a sunscreen degrades and loses its ability to absorb UV photons after sun exposure, its protective effect will be short-lived. This issue has recently been recognized by new FDA regulations that require that photostability be taken into account when the Sunburn Protection Factor (SPF) of a sunscreen is calculated.14 A second area of concern is the potential of the sunscreen or its degradation products to generate highly reactive and biologically damaging species like singlet oxygen (1O2).15 Because of these concerns, determining the factors that affect the photostability of UV filter molecules has a direct impact on public health.
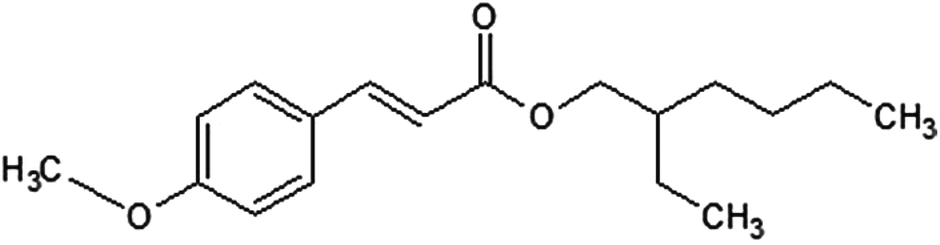
Octylmethoxycinnamate (OMC) is a FDA-approved UV filter commonly used in commercial sunscreens. Its chemical structure is shown in Fig. 1. It is used as a UVB (280 nm–320 nm) filter although the low-energy tail of its absorption spectrum extends into the UVA (320 nm–400 nm). The main photochemical reaction pathway for the trans isomer in solution is isomerization to the cis form. Because cis-OMC has a lower extinction coefficient (ε) than its trans-OMC counterpart, the overall absorptivity decreases under UV irradiation (εcis = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 at 291 nm; εtrans = 24000 at 310 nm).16 Steady-state, time-resolved, Raman and two-photon fluorescence imaging experiments have all been used to investigate different aspects of OMC's excited state dynamics, ground state structures, and reactive oxygen species sensitization.15,17–19 A wide variance in the amount of loss of OMC absorptivity during irradiation has been reported in the literature. In solution, OMC has been reported to degrade irreversibly, losing up to 90% of its absorbance.7 Other workers have found that it forms a photostationary state between its trans and cis isomers with only a 30–50% loss of absorption.13,16,20,21 In aqueous solution and in neat films, the production of additional photoproducts like truxilllic and truxinic dimers has also been reported.22–25 In actual sunscreen formulations, where OMC is mixed with other organic molecules, absorbance losses range from 10% to 90% or more.6,13,26,27 These reports used different experimental conditions like sample type (dilute solution, concentrated films, dissolved creams), lamp sources, irradiance, and solvent.
600 at 291 nm; εtrans = 24000 at 310 nm).16 Steady-state, time-resolved, Raman and two-photon fluorescence imaging experiments have all been used to investigate different aspects of OMC's excited state dynamics, ground state structures, and reactive oxygen species sensitization.15,17–19 A wide variance in the amount of loss of OMC absorptivity during irradiation has been reported in the literature. In solution, OMC has been reported to degrade irreversibly, losing up to 90% of its absorbance.7 Other workers have found that it forms a photostationary state between its trans and cis isomers with only a 30–50% loss of absorption.13,16,20,21 In aqueous solution and in neat films, the production of additional photoproducts like truxilllic and truxinic dimers has also been reported.22–25 In actual sunscreen formulations, where OMC is mixed with other organic molecules, absorbance losses range from 10% to 90% or more.6,13,26,27 These reports used different experimental conditions like sample type (dilute solution, concentrated films, dissolved creams), lamp sources, irradiance, and solvent.
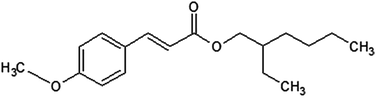 |
| | Fig. 1
trans-OMC. | |
Commercial sunscreens are composed of multiple UV filters in order to provide higher SPF values and broad-spectrum (UVB + UVA) photoprotection, and thus no sunscreen would contain OMC as the sole UV filter. However, the study of individual sunscreen components is necessary in order to unravel the factors that determine the overall efficacy of more complicated mixtures. In order to gain a better understanding of the factors that affect the photostability of OMC, herein we present results on the photodegradation kinetics of OMC in dilute solutions, neat films, and in aqueous suspensions of aggregates. In addition to clarifying the isomerization kinetics in solution, we find that aggregation of OMC leads to rapid irreversible degradation to form a complex mixture of photoproducts. A considerable fraction of colloidal OMC forms previously identified species like the cis isomer and the truxuillic and truxinic photodimers, whose absorption spectra all shift to higher energies. In this work, we also show that additional photoproducts are formed whose absorption shifts to lower energies where there is greater solar UVA overlap. These red-shifted UVA-absorbing photoproducts generate 1O2 under 405 nm irradiation. The results demonstrate the importance of considering the role of aggregation in determining the fate of UV filter molecules under solar irradiation.
Experimental
Reagents
Octylmethoxycinnamate (98%) was provided by Merck Consumer Care Inc. (Memphis, TN). All solvents (cyclohexane, ethanol, and methanol; >99%) were obtained from Aldrich and used without further purification.
UV Irradiation
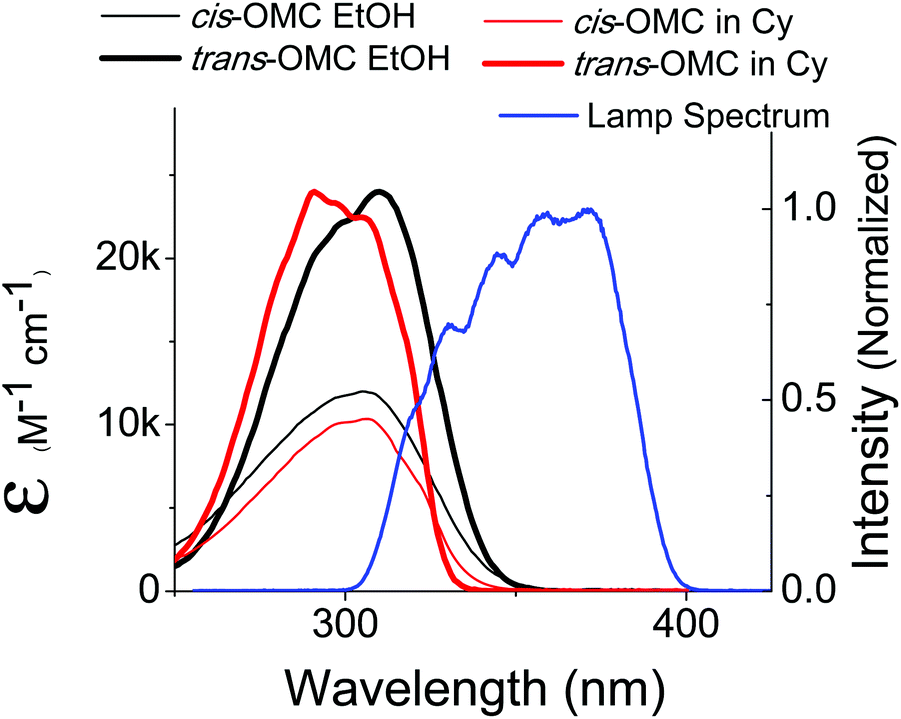
A solar simulator (Model 16S, 150 W, Solar Light Company, Glenside, PA) was used to generate a UVB and UVA spectral output that mimics the solar spectrum. The output of this lamp was coupled through an optical fiber whose spectral output is shown in Fig. 2. The power distribution was measured to be 30 mW UVA and 1 mW UVB, close to the actual UVA:UVB solar ratio of 34:1 as determined from the standard solar spectrum (ASTM G173-03).28 The optical fiber was placed in front of the quartz sample cuvettes (pathlength: 1 cm) so that the UV output beam incident on the sample had a diameter of 1 cm.
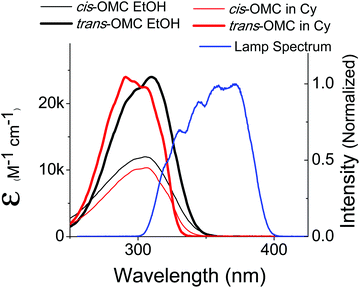 |
| | Fig. 2 The absorption spectra of trans-OMC (solid thin) and cis-OMC (solid thick) in ethanol (black) and cyclohexane (red). The spectral overlap of the solar simulated lamp is shown in blue. | |
Isolation of the cis-OMC isomer
14 mg of OMC was dissolved in 10 mL CH2Cl2 and subsequently irradiated for 600 s with the solar simulator in a 1 cm pathlength quartz cuvette. The solution was stirred continuously during irradiation. A silica column was used to separate the trans and cis isomers using 100% CH2Cl2 as the eluent.
Photostability studies in dilute solutions
The concentration of OMC was 8 μM in all solvents, corresponding to a peak optical density <0.2. This low absorption value prevented inner-filter effects and guaranteed that the entire sample experienced the same light intensity to within 20%. Each solution had an identical volume (3.4 mL) and was stirred constantly during irradiation. Photoisomerization quantum yield values were determined from solar-simulated photostability data and confirmed from single-wavelength photostability data. The solar simulator dose control system was used to set the UV dose at time (t) = 0, 6, 12, 18, 24, 30, 56 and 109 (±0.2 seconds), where 56 s is equivalent to 1 minimal erythemal dose (MED) measured as 22 mJ cm−2 UVB, and 660 mJ cm−2 UVA. Single-wavelength data were acquired using a Optical Building Blocks Kiloarc (1000 W Hg/Xe) lamp coupled to a monochrometer and fiber optic output (1 cm diameter) at 330 nm (4 mW) at similar time points. At each time point, the absorption spectrum was recorded by a Cary-50 absorption spectrophotometer. Experiments were performed in duplicate. Absorption spectra of the neat solvents were recorded and showed no absorption >250 nm; they were subtracted from the sample spectra.
Photostability studies in colloidal suspensions and thin films
To make suspensions of colloidal OMC particles, the re-precipitation method was used. 200 μL of 10 mM OMC in methanol was quickly injected into a rapidly stirred solution of 10 mL deionized water. The solution was stirred uncovered for 45 minutes to allow evaporation of the methanol. Dilute solutions (8 μM) were created from this stock solution, such that the peak UV absorbance was <0.2 at λmax = 328 nm. Solutions were stirred and irradiated following the identical procedure used for the dilute solution experiments to t = 6000 s. To avoid dramatic changes in the absorption spectrum due to further aggregation, freshly made aggregate solutions were used immediately. Experiments were performed in duplicate.
To create thin films, the OMC oil was spread onto a fused silica microscope slide and sandwiched beneath a second slide. We use the term “neat film” to describe the OMC oil sandwiched in this manner. Samples were irradiated without stirring over a 1 cm spot. The sample was never moved such that the absorption spectrum was taken on the irradiated spot. Experiments were performed in duplicate.
Mass spectral analysis
Mass spectra were acquired on pre-and post-irradiated thin films of OMC using a Waters GCT high resolution mass spectrometer (see ESI†).
Singlet oxygen detection
Detection of singlet oxygen-sensitization by OMC-photoproducts was achieved by monitoring the fluorescence intensity of the 1O2-specific fluorescence probe Singlet Oxygen Sensor Green (SOSG, Invitrogen) in the presence of pre- or post-UV irradiated colloidal suspensions of OMC. SOSG is weakly fluorescent upon excitation at 504 nm, but upon reaction with 1O2 in aqueous solutions, forms a fluorescent photoproduct (λmax = 525 nm).29 An aqueous suspension of OMC was created as described above and divided into two samples. A 2 ml aliquot was transferred to a 1 cm quartz cuvette and irradiated with the solar simulator to deliver a dose of 3.6 J UVB and 110 J UVA to create the UVA-absorbing photoproducts. Subsequently, SOSG (final concentration 2 μM) was transferred to the sample, and the sample was irradiated at 405 nm using a 40 mW diode laser. The fluorescence of SOSG was monitored using a Spex Fluorolog (λ excitation 504 nm). Both SOSG and pre-UV aggregates with SOSG were used as controls. Experiments were performed in duplicate.
Fluorescence experiments: steady-state spectra and time-resolved fluorescence lifetimes
Fluorescence spectra of dilute solutions, colloidal suspensions and thin films of pre- and post-UV irradiated OMC, as described above, were acquired in front-face mode on a Spex Fluorolog. Identical samples were used to collect the fluorescence lifetime (τf) data. In brief, an 800 nm, 150 fs pulse from a Ti:sapphire regenerative amplifier (40 kHz, Spitfire, Spectra-Physics) was frequency doubled to 400 nm, and focused using a 10 cm lens on the sample (solutions: 1 cm cuvette; thin films: 100 μm cuvette). The laser power was set to be at or below 75 μW at the sample surface. Fluorescence lifetime data were collected in front face detection using a picosecond streakscope streak camera (Hamamatsu G4334) with a time resolution of 15 ps. Scattered IR and 400 nm light were removed by placing two 420 nm long wave pass filters before the input of the streak camera.
HPLC
HPLC was performed on UV-irradiated thin films of OMC in an attempt to isolate the UVA-absorbing photoproduct(s). The OMC oil was transferred to a 100 μm pathlength cuvette, and placed before the output of the solar simulator such that the beam diameter irradiated the entire sample for 60 min until the sample was bright yellow. Separation of photoproducts was achieved using the protocol described in Macmanus et al.25 Irradiated OMC was dissolved in 100% MeOH. A Waters 2695 HPLC with photodiode array 995 was coupled with an Apollo C18 5μ 250 mm × 10 mm (Alltech) column. The injection volume was 100 μL and the flow rate was 4 mL min−1. A gradient of methanol and pH 5 sodium acetate buffer was used.
Results and discussion
OMC in dilute solution
The absorption spectra of dilute solutions of OMC in polar ethanol and non-polar cyclohexane are shown in Fig. 2. Note that the absorption spectrum for both isomers of OMC in methanol overlaps that in ethanol. The graph is in units of molar absorptivity for both trans-OMC and cis-OMC isomers, which are calculated from the product of the ε(λ) values and our experimental normalized absorption spectrum of each isomer in each solvent (Fig. 2).16 The absorption spectrum of trans-OMC is affected by solvent polarity, with λmax = 291 nm in cyclohexane shifting to 310 nm in ethanol and methanol. The λmax of cis-OMC is unaffected by solvent polarity, with λmax = 305 nm in cyclohexane or ethanol; however, the spectrum is narrower in cyclohexane and has a lower ε(λ) than in ethanol. Fig. 2 also shows the normalized solar simulator lamp spectrum. The molecule in both isomeric forms has a greater degree of overlap with the lamp spectrum in polar solvents like ethanol than in non-polar environments like cyclohexane.
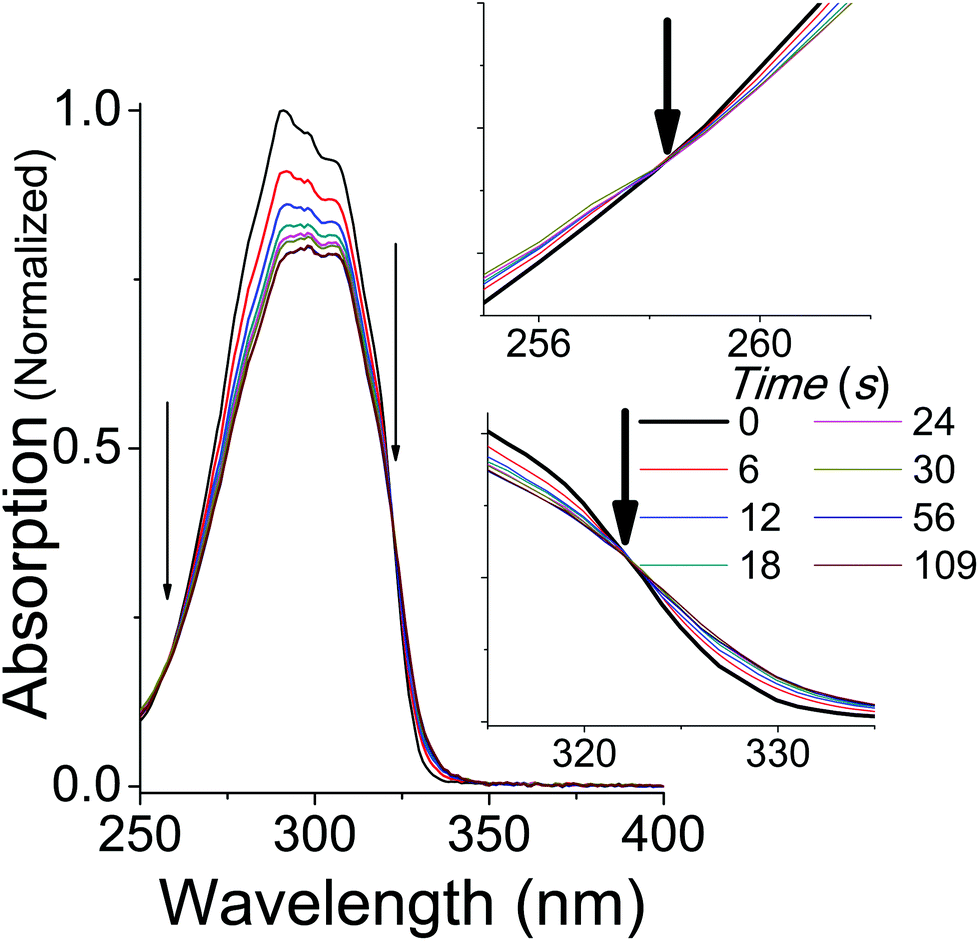
Upon irradiation with UVB + UVA, the absorption spectrum of trans-OMC in cyclohexane rapidly decreases (Fig. 3) until a photostationary state is achieved. As the UVB + UVA dose increases, the absorption intensity reaches a minimum at t = 56 s (22 mJ cm−2 UVB (1 MED), 660 mJ cm−2 UVA), and then does not dramatically change under further irradiation to 112 s (2 MED). At this point, the primary photochemical reaction is a photostationary state between trans-OMC and its cis-OMC counterpart. Two isosbestic points are visible at 255 nm and 322 nm (Fig. 3), consistent with a two-species photoisomerization reaction.30 Similar spectral changes were seen of OMC in methanol (ESI†).
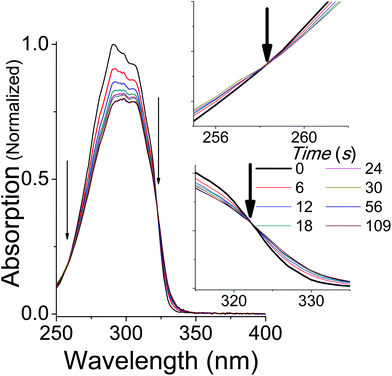 |
| | Fig. 3 The absorption spectrum of trans-OMC in cyclohexane under solar simulated UV irradiation. Isosbestic points at 255 nm and 322 nm are marked by arrows and enhanced in the inset. | |
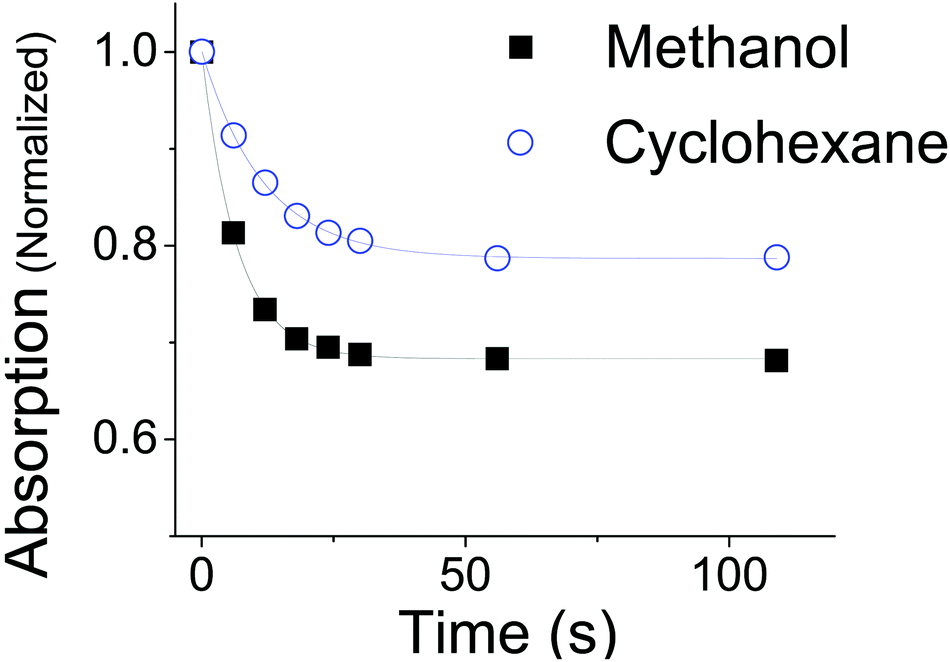
A plot of the absorption at the λmax for OMC in cyclohexane and methanol as a function of irradiation time is seen in Fig. 4. Kinetic data taken in ethanol were identical to those in methanol to within a few percent. The absorbance values are normalized both to the starting concentration intensity at t = 0 s and at the λmax for each solvent (λmax = 310 in methanol; λmax = 291 nm in cyclohexane). The data show that the absorption intensity at the photostationary state in cyclohexane is greater than that in methanol, indicating that non-polar environments help trans-OMC retain more of its absorbance. In cyclohexane, 79% ±1% of the absorptivity is retained, whereas only 68% ±2% of OMC absorptivity is retained in polar methanol (Fig. 4). Once this plateau is reached, the absorption remains remarkably stable, with less than a 2% decrease in absorptivity between 112 s (0.3 kJ) and 1120 s (34 kJ) of solar-simulated UV exposure.
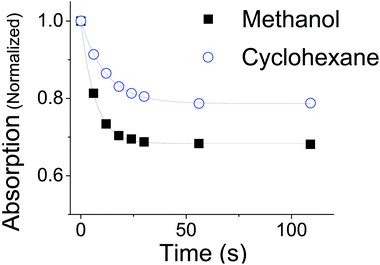 |
| | Fig. 4 The absorption of trans-OMC at λmax in methanol (black, λmax = 310 nm) or cyclohexane (blue, λmax = 291 nm) at increasing UV irradiation. The data points are fit to the exponential described in eqn (8). | |
Analysis
In order to quantitatively analyze the photodegradation kinetics in the different solvents, we use the simple two-state model outlined below. At any given frequency (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) ) and time (t), the absorption intensity (A) is equivalent to the sum of the trans-OMC and cis-OMC isomer contributions to the overall absorption intensity as described in eqn (1), below.
) and time (t), the absorption intensity (A) is equivalent to the sum of the trans-OMC and cis-OMC isomer contributions to the overall absorption intensity as described in eqn (1), below.| | | A(,t) = εT()NT(t)L + εC()NC(t)L | (1) |
where εT() and εC() are the molar absorptivity values for trans- and cis-OMC isomers, respectively, at frequency ; NT(t) and NC(t) are the concentrations for trans- and cis-OMC, respectively; and L is the pathlength (1 cm). The εT() and εC() values were calculated using previously determined values of εT and εC at max and our experimentally obtained trans- and cis-OMC absorption spectra.16 With εT() and εC() known at each wavelength, we need to determine NT(t) and NC(t) to calculate the absorbance change. The change in concentration of NT(t) and NC(t) is dependent upon the rate of conversion between the two isomers, and can be described as| | 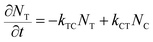 | (2) |
| | 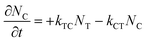 | (3) |
where kTC is the rate of conversion from trans-OMC to cis-OMC and kCT is the rate of conversion from cis-OMC to trans-OMC. These rates are the product of three factors: (1) the absorption cross-section of the trans (σT()) or cis isomer (σC()); (2) the UV lamp intensity (I()); and (3) the quantum yield of conversion for trans-to-cis (ϕTC) or cis-to-trans-OMC (ϕCT). The values of eqn (4) and (5) describe this relationship. The absorption cross section is obtained from the experimental absorption coefficients using the relation| | 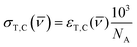 | (4) |
where NA is Avogadro's number. Since we are using a broadband solar simulator, we integrate over all frequencies to take into account the varying I() and σ() values. We define the spectral overlap between the trans-OMC absorption with the solar simulated spectrum (JT) or the cis-OMC absorption spectrum with the solar simulator spectrum (JC) in eqn (5):| | 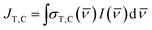 | (5) |
As eqn (5) indicates, JT,C requires integration of the product of the lamp intensity and the absorption cross-section. This requires that each experimentally obtained spectrum be interpolated over the same frequency interval (Δ = 100 cm−1). The rate constants are defined as
With these parameters defined, we solve eqn (2) and (3) for NT and NC using the initial starting concentration of a dilute solution (NT(t = 0) = N0).
| | 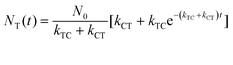 | (7a) |
| | 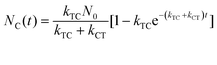 | (7b) |
An experimental plot of absorption (A(λ)) against time can be fit to the exponential equation
| | | A(λ,t) = A0(λ)e−kabst + Apss(λ) | (8) |
where
and
Apss(
λ) is equivalent to the absorbance at
t = ∞.
| | 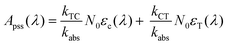 | (10) |
Fitting the data in Fig. 4 to eqn (8) allows us to extract both kabs and Apss. We then use eqn (9) and (10) to solve for kTC and kCT. With the experimental determination of JT and JC, eqn (6a) and (6b) can then be solved for the photoisomerization quantum yields ϕTC and ϕCT, which are given in Table 1. The results obtained using browadband lamp illumination were the same to within experimental error as those obtained for narrowband irradiation at 330 nm using a filtered Xenon lamp (Table 1). The origin of the different quantum yield values in different solvents is an interesting question. We find that ϕTC in methanol is greater than ϕTC in cyclohexane, while ϕCT has the opposite trend. A simple argument based on viscosity would imply that both yields should be lower in the more viscous cyclohexane (methanol: 0.59 cP; cyclohexane: 1.02 cP).31,32 A more likely explanation is that the increased polarity of the cis-OMC isomer leads to its increased stability in more polar solvents. In this case, we expect a larger ϕTC and smaller ϕCT in the more polar methanol, as experimentally observed.
Table 1 Quantum yield values calculated for the photoisomerization reaction of OMC in dilute solution
| Solvent |
ϕ
TC (δ)a |
ϕ
CT (δ)a |
|
Standard deviation.
|
| Methanol (solar irradiation) |
0.37 (0.01) |
0.47 (0.06) |
| Methanol: single wavelength (330 nm) |
0.41 (0.05) |
0.52 (0.04) |
| Cyclohexane (solar irradiation) |
0.28 (0.01) |
0.60 (0.00) |
Morliere et al. have previously determined ϕTC and ϕCT values for OMC in several solvents, finding values almost a factor of 2 higher than those reported in Table 1. Their results were based upon numerical analysis of OMC degradation in optically dense solutions, where εC(λ), ϕTC and ϕCT were all variable parameters.20 In the current work, the use of low optical density solutions allows us to fit the data using the analytical formula in eqn (8). In addition, we use an experimental value for εC(λ), which leaves only ϕTC and ϕCT as undetermined parameters in our analysis. We believe that our approach results in more reliable quantum yield values for trans–cis-OMC photoisomerization in dilute solution. For example, our values result in the sum (ϕTC + ϕCT) being 0.84 in methanol and 0.88 in cyclohexane. Both values are <1, consistent with a common transition state, as typically observed for cis–trans photoisomerization reactions.33 The results of Moliere resulted in the sum of photoisomerization reaction yields (ϕTC + ϕCT) > 1, which those authors acknowledged to be unusual.
OMC in aqueous suspensions and neat films
The photophysics of OMC in dilute solution can be understood in terms of a classical cis–trans photodynamic equilibrium. Once this equilibrium is established, the OMC absorbance is quite stable. The situation changes drastically when OMC molecules are allowed to aggregate. We studied two routes to form aggregated OMC: an aqueous suspension of nano- and microparticles, and a neat film of OMC oil sandwiched between two fused silica substrates.
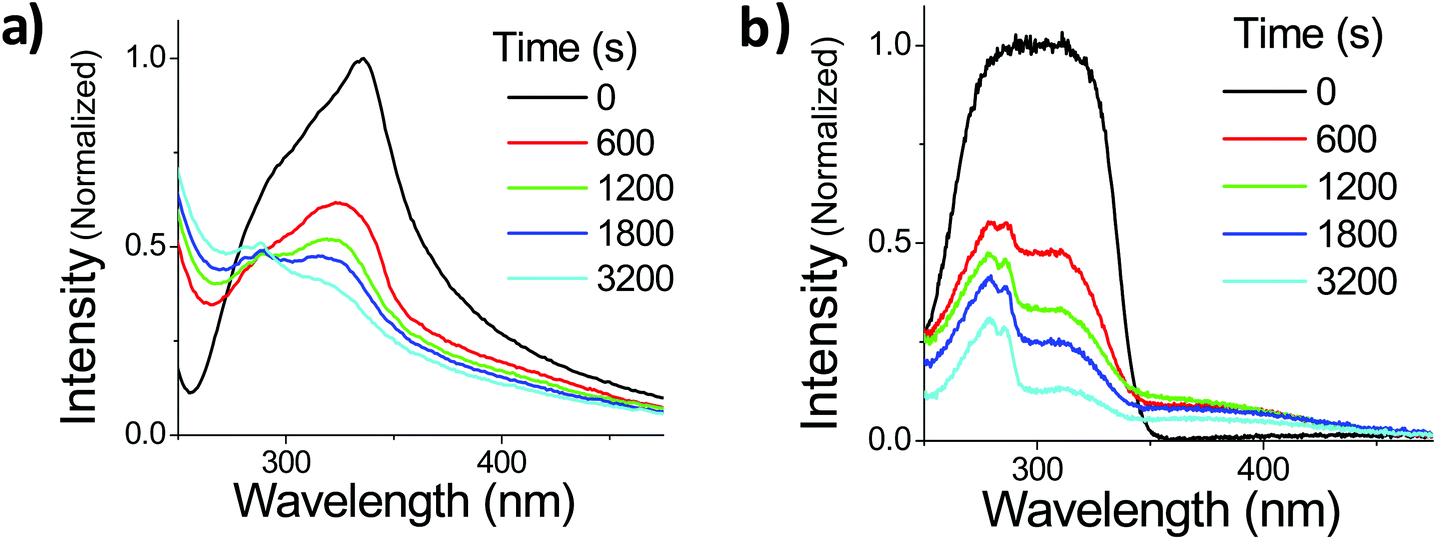
Suspensions of OMC particles can be prepared using the reprecipitation method, resulting in a cloudy, highly scattering solution formed even at μM concentrations. Fig. 5a shows the absorption spectra of freshly made OMC aggregates in water. It is important to use freshly made samples to minimize the formation of larger aggregates that increase the scattering background. The aggregates have a spectral shape different from that of OMC in dilute solution, with a long tail in the red, consistent with light scattering by aggregate particles in solution. The absorption spectrum is also red-shifted further into the UVA (λmax = 328 nm) compared to dilute solutions of OMC in methanol (λmax = 310 nm) or cyclohexane (λmax = 291 nm) and thus has greater overlap with the UVA portion of the solar radiation. The absorption shift is consistent with aggregation of the trans-OMC molecules leading to a more polarizable dielectric environment and a solvatochromic shift. But it is also possible that this shift reflects excitonic interactions between the OMC molecules in aggregated form, as opposed to monomeric OMC in dilute solution.34
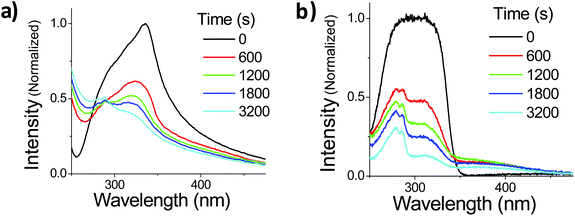 |
| | Fig. 5 The normalized absorption spectra of OMC aggregates in H2O (a) or OMC film (b) under UV irradiation. Note that although the film spectrum at t = 0 s appears to be distorted, its un-normalized peak absorbance is below the saturation limit of the spectrometer (o.d. = 1.04 @ 300 nm) and thus it is a true representation of the film absorption shape. | |
Also shown in Fig. 5a is the changing absorption spectrum of OMC aggregates under UV irradiation, which continuously decreases up to 3200 s (108.5 kJ cm−2 solar simulated UV). Note that there is a scattering offset in the absorbance of the particles. In order to confirm that these spectral changes are a general phenomenon in aggregated OMC, we repeated the experiments on ultrathin films of OMC oil spread onto a fused silica microscope slide. This sample has the advantage of being more optically uniform, with a lower scattering background that allowed us to identify new absorption peaks more easily. Fig. 5b displays the evolution of the absorption spectrum of a film of OMC under UV irradiation. The film absorption starts as a broad single peak, but as the irradiation proceeds, three distinct peaks can be discerned at about 280 nm, 310 nm, and 400 nm. It is clear that other photoproducts are being formed besides cis-OMC. The 280 nm peak can be assigned to the various dimers of OMC formed by the [2 + 2] photocyclization reaction between two OMC molecules.35 The origin of the peak at 400 nm is less clear. Analysis by HPLC yielded a complicated chromatograph with multiple peaks detected at 400 nm, at retention times beyond that exhibited by the two OMC isomers. Absorption and mass spectral analysis shows that each HPLC peak appears to be a mixture of compounds that absorb in the UVA, as well as UVB and UVC (ESI†). Some dimer is present in each sample; however, additional species with masses greater than the dimer also are present, indicating that a complicated photochemistry beyond simple isomerization, as well as dimerization, occurs when OMC is allowed to aggregate. Although the sample exhibited a strong yellow color visually, separation of the UVA-absorbing photoproducts by HPLC led to <1 mg of these photoproducts, making it difficult to characterize the structures of these molecules. Finally, we note that the 400 nm feature itself is subject to photodegradation and disappears after ∼1 hour of irradiation.
For irreversible photodegradation, the two-state model given by eqn (2) and (3) is no longer necessary, and instead we can assume that the OMC absorption decays irreversibly via a first-order rate process,
| | 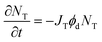 | (11) |
where
ϕd is the quantum yield of the irreversible photo-induced decomposition process and
JT is the integrated spectral overlap term defined previously in
eqn (5). Note that
eqn (11) does not distinguish between
cis- and
trans-OMC, since both absorb in this wavelength range.
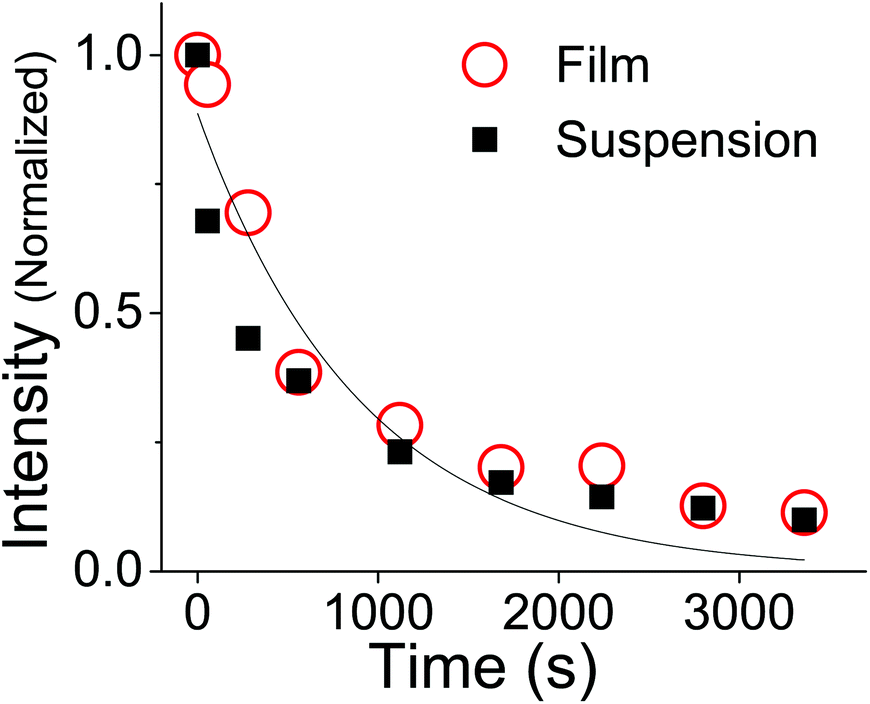
Fig. 6 shows that the absorption decays of the aqueous suspension and neat thin film samples of OMC are identical. After subtraction of a scattering background, the decays can be fit to an exponential whose rate constant is
JTϕd in
eqn (11). Extracting the quantum yield
ϕd is complicated by the fact that we do not know the absorption coefficient of the aggregates. Aggregation can lead to changes in absorption strength depending on the intermolecular interactions between aggregate molecules, and thus we cannot confirm
JTvia experiment. However, if we assume that the aggregates have the same absorption strength as monomeric
trans-OMC,
ελmax = 24
000 M
−1 cm
−1 and
JT can be calculated. The quantum yield for photodegradation (
ϕd) was then calculated using
eqn (11) and found to be 6.2 (±0.6) × 10
−4. If we follow MacManus-Spencer
et al. and use a lower value for the absorption coefficient of aggregated
trans-OMC (
ελmax = 8200 M
−1 cm
−1), the calculated
ϕd value increases to 1.9 (±0.3) × 10
−3. This value is similar to the value for
trans-OMC reported by MacManus-Spencer
et al. (
ϕd = 1.8 × 10
−3) using a
p-nitroanisole/pyridine actinometer.
21 The agreement between the two values of
ϕd is reasonably good considering that completely different experimental methods were used. In any case, given our uncertainty about the true value of
ελmax, all these values should be considered tentative. But it is certain that the irreversible formation of the [2 + 2] photodimers and other photoproducts leads to very different photodegradation kinetics in the aggregated state than in dilute solution. Our calculation of
ϕd shows that photodegradation yield for
trans-OMC in aggregated form is roughly two orders of magnitude greater than typical
ϕd values for molecules that are defined as photostable, such as laser dyes, which typically have
ϕd values of 10
−6.
36–38
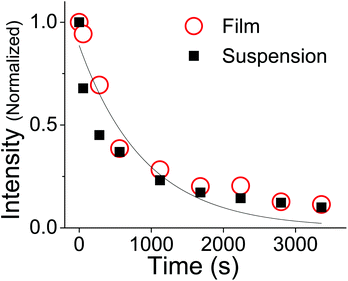 |
| | Fig. 6 The change in absorption of OMC aggregates at 328 nm under UV irradiation in a film (circles) or as a suspension in H2O (squares). The exponential fit (solid line) assumes there is no offset at long times, and thus neglects the possibility that photoproduct absorption is present at the same wavelength. | |
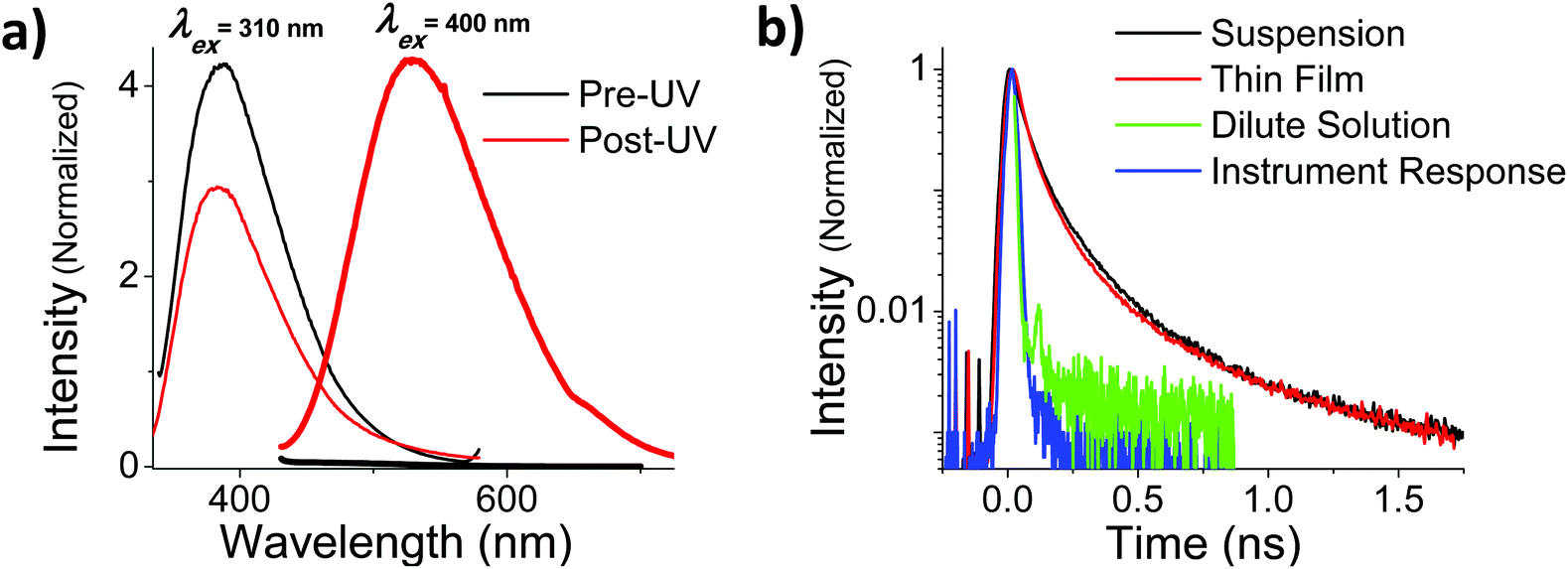
In addition to the more rapid degradation, aggregated OMC also produces a number of new photoproducts that contribute to a characteristic absorption in the UVA, as can be seen from Fig. 5b. We were interested in the photophysical behaviour of the 400 nm species, since this absorption has strong overlap with the solar UVA spectrum and might be expected to play a role in subsequent photochemical reactions. In both films and aqueous particle suspensions, photoexcitation of this feature gives rise to a broad emission centered at 540 nm that is shifted by more than 100 nm from the 400 nm emission that arises from the un-irradiated film after excitation at 310 nm. The excitation spectrum of the 540 nm emission shows a well-resolved peak at 395 nm in both the film and the particles. Fig. 7a summarizes the fluorescence emission and excitation spectra for a neat film after 560 s of UV exposure. Fig. 7b shows the fluorescence decay of the irradiated films and particle suspensions, measured after excitation at 400 nm. The aggregated samples exhibit identical decays that extend well into the nanosecond time regime, in contrast to what is seen for the cis and trans isomers of OMC, which have τf values on the order of the instrument response, i.e. less than 30 ps. The multi-exponential nature of the decay is consistent with the presence of multiple photoproducts, as we concluded based on the presence of multiple peaks in the HPLC chromatograph and the mass spectra of the UVA-absorbing photoproducts (ESI†).
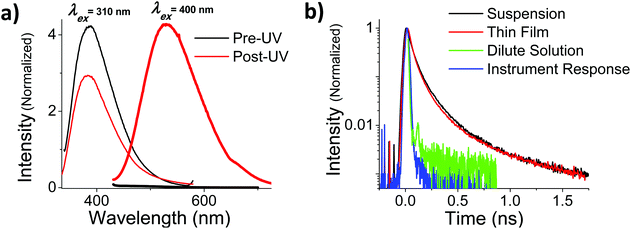 |
| | Fig. 7 (a) Fluorescence emission spectra of a thin film of OMC before (black) and after (red) solar-simulated UV irradiation for 560 s. Samples were excited at 310 nm, the λmax of trans-OMC absorption, and 400 nm, the λmax of UVA-absorbing photoproducts. The fluorescence following excitation at 400 nm is negligible for pre-UV irradiated samples (thick black line 420 nm–700 nm), and increases dramatically once the sample is irradiated with solar simulated UV (thick red line 420 nm–700 nm. (b) Time resolved fluorescence of both colloidal suspensions (black) and thin films (red) of post-UV irradiated OMC excited at 400 nm. The instrument response (blue) and τf of pre-UV irradiated OMC in cyclohexane (green) are much shorter than the τf of the UVA-absorbing photoproducts. | |
We now discuss possible origins of the UVA absorbing photoproducts. In solution, there is general agreement that cis- and trans-OMC are the dominant species generated by UV radiation.13,20,21 In thin films and aqueous-based solutions, the formation of truxillic and truxinic photodimers has also been observed.22–24 McManus-Spencer et al. have identified additional photoproducts that include 4-methoxybenzaldehyde and 2-ethylhexanol, which are likely formed by photohydrolysis.25 All these photoproducts absorb at higher energies than OMC due to loss of conjugation, which decreases their ability to absorb UVB radiation. The formation of the 400 nm absorbing species shown in this work suggests that photodegradation of OMC can also produce transient species whose absorption is shifted to lower energy, into the UVA region. This observation suggests that photodegradation can lead to products with increased conjugation. Cinnamate derivatives are known to undergo a photo-Fries rearrangement that can lead to a yellowing of the sample. But Subramanian et al. showed that this pathway requires a phenolic linkage that is not present in OMC.35 The observation of MacManus-Spencer et al. that OMC can fragment suggests the possibility that these radical fragments could react with neighboring OMC molecules in a variety of ways, before reacting with water to form hydrolysis products. Such larger molecules would have red-shifted absorption spectra, but would also be subject to further photodecomposition, as observed. More detailed structural characterization of OMC's photoproducts will be necessary to understand their mechanism of formation.
The relatively long excited state lifetime of the 400 nm absorbing species suggests that they could participate in excited state chemical reactions or sensitization events. This contrasts with monomeric OMC which has a very short-lived excited state lifetime, releasing its absorbed photon energy through isomerization and internal conversion. In order to assess the ability of the UVA absorbing photoproducts to initiate photochemical reactions, we tested whether irradiation at 405 nm could sensitize singlet oxygen production. Using the fluorescence probe molecule Singlet Oxygen Sensor Green (SOSG), we tested the ability of aqueous OMC particles to generate 1O2 both pre- and post-UV irradiation. Both the unirradiated OMC particles and SOSG by itself show a decline in fluorescence during irradiation by a diode laser at 405 nm, most likely due to photobleaching of the SOSG by the 405 nm laser. Under the same conditions, the irradiated particles lead to a steady rise in SOSG fluorescence as the SOSG molecules are oxidized by the 1O2 (Fig. 8). These results demonstrate that the photoproducts of aggregated OMC are capable of sensitizing singlet oxygen production under UVA light exposure. Due to the likely presence of a mixture of photoproducts with unknown absorption coefficients, we did not attempt to quantify the 1O2 yield.
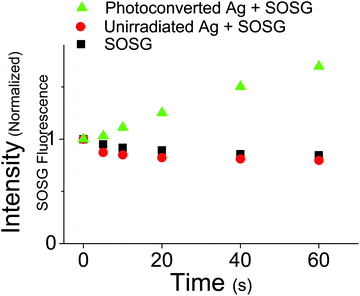 |
| | Fig. 8 Fluorescence intensity at different time points following irradiation at 405 nm of SOSG (SOSG, black squares), pre-UV irradiated OMC aggregates + SOSG (Unirradiated Aggregates + SOSG, red circles), and post-UV irradiated OMC aggregates + SOSG (photoconverted Aggregates + SOSG, green triangles). The latter data show that 1O2 is generated by the UVA absorbing OMC photoproduct(s). | |
Monomeric OMC has a triplet energy ET = 239 kJ mol−1 and has been shown to sensitize 1O2 formation in solution, which requires ET ≥ 94 kJ mol−1.39–41 The UVA absorbing photoproducts would presumably have lower triplet energies than OMC due to their increased conjugation. The singlet state energy of the photoproducts decreases by about 25%, as judged by the shift of the absorption from 310 nm to 400 nm. If their triplet state energies parallel the shift in singlet energies, we would estimate that ET decreases to about 180 kJ mol−1, sill high enough to sensitize 1O2 formation. The dominant relaxation pathways of monomeric OMC are internal conversion and isomerization.40 Although the intersystem crossing rate of OMC is not known, it is assumed to be small as phosphorescence is weak in rigid matrixes at 77 K in ethanol and requires heavy atom enhancement for detection.40,41 The intersystem crossing rate of the photoproducts is thus a matter of conjecture, but their longer fluorescence lifetime indicates that the triplet yield could be higher than in monomeric OMC, since intersystem crossing no longer has to compete with rapid isomerization and internal conversion.
Finally, we briefly comment on the relevance of our results for sunscreen design. UV filters, including OMC, have been reported to penetrate into the lower stratum corneum as well as deeper into the dermis.42,43 For example, Freitas et al. recently reported that up to 10% of applied OMC penetrates below the stratum corneum, and as we noted in previous work, if UV light reaches penetrated UV filters (including OMC), ROS can be sensitized.15,43 The mechanism by which penetrated OMC could generate ROS was not identified. The data in this paper show that OMC aggregates can form photoproducts that can sensitize 1O2, providing a possible mechanism for OMC-sensitized ROS in the lower epidermis.15 It is noted that in general, cutaneous penetration decreases with molecular weight, suggesting that penetration of aggregates is less likely; however, it is possible that OMC aggregates form in deeper epidermal or dermal layers after the monomer penetrates.42–44 We should emphasize that photo-instability of UV filters and the formation of photoproducts are not necessarily concomitant with photo-toxicity, and sunscreens containing OMC have been deemed safe by FDA.45 Additionally, it should be noted that there exist strategies to prevent OMC aggregation, as well as prevent ROS formation through inclusion of antioxidants and quenchers of photoexcited states.46–48
Conclusions
The data presented in this paper show that the environment plays a critical role in determining the photoresponse of OMC to UVB and UVA. In dilute solution, the dominant photochemical response in an organic solvent is photoisomerization, where a photostationary state is achieved between the two trans-OMC and cis-OMC isomers. Some additional photodegradation may occur after the photostationary state is reached, but the subsequent decrease in absorption intensity is very small (<2%). The lower absorbance of the cis-OMC isomer compared to trans-OMC leads to a decrease in the overall absorption of the OMC in solution as the two isomers reach a photostationary state, but typically the solution retains 60–70% of its original absorbance. As compared to monomeric OMC, aggregated OMC undergoes both photoisomerization and irreversible photodegradation to a complex mixture of photoproducts. Further research to establish the chemical identities of these photoproducts is clearly warranted and will be the subject of future work. While the rapid loss of UV protection is a problem, the ability of some of these photoproducts to sensitize 1O2 under UVA irradiation is also cause for concern. The complex photochemistry of OMC in aggregated form suggests that sunscreen formulators may want to consider strategies to reduce aggregation in commercial sunscreen products.
Acknowledgements
We thank Dr Tom Meyer and Donathan Beasley of Bayer Consumer Care, Memphis, TN, for helpful discussions. This work was supported by the UC Office of the President, under a University of California Discovery Grant (#191739). The UCR ACIF mass spectrometer is made possible by a grant from the NSF (CHE-0742001). KMH has woked as a consultant for Merck Consumer Care recently acquired by Bayer Consumer Care.
References
- L. L. Hruza and A. P. Pentland, J. Invest. Dermatol., 1993, 100, 355–415 CrossRef PubMed.
- M. Norval, Prog. Biophys. Mol. Biol., 2006, 92, 108–118 CrossRef CAS PubMed.
- A. Sarasin, Mutat. Res., 1999, 428, 5–10 CrossRef CAS.
- R. B. Setlow and J. K. Setlow, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 1250–1257 CrossRef CAS.
- H. E. Kanavy and M. R. Gerstenblith, Semin. Cutaneous Med. Surg., 2011, 30, 222–228 CrossRef CAS PubMed.
- R. M. Sayre, J. C. Dowdy, A. J. Gerwig, W. J. Shields and R. V. Lloyd, Photochem. Photobiol., 2005, 81, 452–456 CrossRef CAS.
- N. Serpone, A. Salinaro, A. V. Emeline, S. Horikoshi, H. Hidaka and J. Zhao, Photochem. Photobiol. Sci., 2002, 1, 970 CAS.
- R. Stokes and B. Diffey, Int. J. Cosmet. Sci., 1999, 21, 341–351 CAS.
-
C. A. Bonda, in Sunscreens: Regulation and Commercial Development, ed. N. A. Shaath, Taylor & Francis, Boca Raton, 2005, pp. 321–349 Search PubMed.
- B. Diffey, R. P. Stokes, S. Forestier, C. Mazailier and A. Rougier, Eur. J. Dermatology, 1997, 7, 226–228 Search PubMed.
-
Comprehensive Series in Photochemical and Photobiological Sciences: Biophysical and Physiological Effects of Solar Radiation on Human Skin, ed. P. U. Giacomoni, RSC Publishing, Cambridge, 2007, vol. 10 Search PubMed.
-
Comprehensive Series in Photosciences: Sun Protection in Man, ed. P. U. Giacomoni, Elsevier, Amsterdam, 2001 Search PubMed.
- S. P. Huong, V. Andrieu, J.-P. Reynier, E. Rocher and J.-D. Fourneron, J. Photochem. Photobiol., A, 2007, 186, 65–70 CrossRef CAS PubMed.
-
Sunscreen Drug Products for Over-the-Counter Use; Final Rule and Proposed Rules, ed. Jeffrey Shuren, FDA Department of Health and Human Services, 2011, vol. 76, pp. 35620–35665 Search PubMed.
- K. M. Hanson, E. Gratton and C. J. Bardeen, Free Radicals Biol. Med., 2006, 41, 1205–1212 CrossRef CAS PubMed.
- S. Pattanaargson, T. Munhapol, P. Hirunsupachot and P. Luangthongaram, J. Photochem. Photobiol., A, 2004, 161, 269–274 CrossRef CAS.
- R. Krishnan and T. M. Nordlund, J. Fluoresc., 2008, 18, 203–217 CrossRef CAS PubMed.
- T. M. Karpkird, S. Wanichweacharungruang and B. Albinsson, Photochem. Photobiol. Sci., 2009, 8, 1455–1460 CAS.
- L. Beyere, S. Yarasi and G. R. Loppnow, J. Raman Spectrosc., 2003, 34, 743–750 CrossRef CAS PubMed.
- P. Morliere, O. Avice, T. S. E. Melo, L. Dubertret, M. Giraud and R. Santus, Photochem. Photobiol., 1982, 36, 395–399 CrossRef CAS PubMed.
- S. Pattanaargson and P. Limphong, Int. J. Cosmet. Sci., 2001, 23, 153–160 CrossRef CAS PubMed.
- J. K. Broadbent, B. S. Martincigh, M. W. Raynor, L. F. Salter, R. Moulder, P. Sjoberg and K. E. Markides, J. Chromatogr. A, 1996, 732, 101–110 CrossRef CAS.
- D. Dondi, A. Albini and N. Serpone, Photochem. Photobiol. Sci., 2006, 5, 835–843 CAS.
- R. Rodil, M. Moeder, R. Altenburger and M. Schmitt-Jansen, Anal. Bioanal. Chem., 2009, 395, 1513–1524 CrossRef CAS PubMed.
- L. A. MacManus-Spencer, M. L. Tse, J. L. Klein and A. E. Kracunas, Environ. Sci. Technol., 2011, 45, 3931–3937 CrossRef CAS PubMed.
- A. Deflandre and G. Lang, Int. J.
Cosmet. Sci., 1988, 10, 53–62 CrossRef CAS PubMed.
- D. Moyal, J. L. Refregier and A. Chardon, Photodermatol. Photoimmunol. Photomed., 2002, 18, 14–22 CAS.
- American Society for Testing and Materials International. ASTM G173-032012 Standard Tables for Reference Solar Spectral Irradiance http://www.astm.org/Standards/G173.htm.
- C. Flors, M. J. Fryer, J. Waring, B. Reeder, U. Bechtold, P. M. Mullineaux, S. Nonell, M. T. Wilson and N. R. Baker, J. Exp. Bot., 2006, 57, 1725–1734 CrossRef CAS PubMed.
- E. Lippert and W. Luder, J. Phys. Chem., 1962, 66, 2430–2434 CrossRef CAS.
- S. P. Velsko and G. R. Fleming, J. Chem. Phys., 1982, 76, 3553–3562 CrossRef CAS PubMed.
- S. K. Kim, S. H. Courtney and G. H. Fleming, J. Phys. Chem., 1989, 159, 544–548 Search PubMed.
- J. Saltiel, J. D'agostino, E. D. Megarity, L. Metts, K. R. Neuberger, M. Wrighton and O. C. Zafiriou, Org. Photochem., 1973, 3, 1–113 CAS.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- P. Subramanian, D. Creed, a. C. Griffin, C. E. Hoyle and K. Venkataram, J. Photochem. Photobiol., A, 1991, 61, 317–327 CrossRef CAS.
- F. J. Kao, Y. M. Wang, J. C. Chen, P. C. Cheng, R. W. Chen and B. L. Lin, Opt. Commun., 2002, 201, 85–91 CrossRef CAS.
- C. Eggeling, J. Widengren, R. Rigler and C. A. M. Seidel, Anal. Chem., 1998, 70, 2651–2659 CrossRef CAS PubMed.
- R. J. Dillon and C. J. Bardeen, J. Phys. A: Chem., 2001, 115, 1627–1633 CrossRef PubMed.
- J. M. Allen and C. J. Gossett, Chem. Res. Toxicol., 1996, 9, 605–609 CrossRef CAS PubMed.
- A. Kikuchi, S. Yukimaru, N. Oguchi, K. Miyazawa and M. Yagi, Chem. Soc. Jan, 2010, 39, 633–635 CAS.
- H. Gonzenbach, T. J. Hill and T. G. Truscott, J. Photochem. Photobiol., B, 1992, 16, 377–379 CrossRef CAS.
- E. Chatelain, B. Gabard and C. Surber, Skin Pharmacol. Physiol., 2003, 16, 28–35 CrossRef CAS PubMed.
- J. V. Freitas, F. S. G. Praca, M. V. L. B. Bentley and L. R. Gaspar, Int. J. Pharm., 2015, 484, 131–137 CrossRef CAS PubMed.
- W. J. Pugh, I. T. Degim and J. Hadgraft, Int. J. Pharm., 2000, 197, 203–211 CrossRef CAS.
- J. F. Nash and P. R. Tanner, Photodermatol. Photoimmunol. Photomed., 2014, 30, 88–95 CrossRef CAS PubMed.
- K. M. Hanson, C. J. Bardeen, D. G. Beasley and T. A. Meyer, Cosm. Toil., 2011, 126, 710–718 CAS.
- C. Puglia, F. Bonina, L. Rizza, P. Blasi, A. Schoubben, R. Perrotta, M. S. Tarico and E. Damiani, J. Pharm. Sci., 2012, 101, 301–311 CrossRef CAS PubMed.
- G. T. Wondrak, M. K. Jacobson and E. L. Jacobson, J. Pharmacol. Exp. Ther., 2005, 312, 482–491 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectrometry spectral data. See DOI: 10.1039/c5pp00074b |
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence