
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of aza and carbocyclic β-carbolines for the treatment of alcohol abuse. Regiospecific solution to the problem of 3,6-disubstituted β- and aza-β-carboline specificity†
V. V. N.
Phani Babu Tiruveedhula
a,
Kashi Reddy
Methuku
a,
Jeffrey R.
Deschamps
b and
James M.
Cook
*a
aDepartment of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, Milwaukee, WI 53201, USA. E-mail: capncook@uwm.edu
bCenter for Bimolecular Science and Engineering, Naval Research Laboratory, Code 6930, Washington, D. C. 20375, USA
First published on 3rd September 2015
Abstract
A novel two step protocol was developed to gain regiospecific access to 3-substituted β- and aza-β-carbolines, 3-PBC (1), 3-ISOPBC (2), βCCt (3), 6-aza-3-PBC (4) and 6-aza-3-ISOPBC (5). These β-carbolines (1–3) are potential clinical agents to reduce alcohol self-administration, especially 3-ISOPBC·HCl (2·HCl) which appears to be a potent anti-alcohol agent active against binge drinking in a rat model of maternally deprived (MD) rats. The method consists of two consecutive palladium-catalyzed reactions: a Buchwald–Hartwig amination followed by an intramolecular Heck-type cyclization in high yield.
Introduction
β-Carbolines, aza-β-carbolines and their derivatives are important targets in synthetic chemistry.1 In addition, they are found in a large number of natural products, many of which demonstrate novel biological activity, especially in regard to the reduction of alcohol self-administration [binge drinking (BD)]. This is proposed to be due to the activity at the benzodiazepine site of the GABAA receptor.2 Surprisingly, BD kills six people a day, most of which are men, and approximately 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people die from alcohol related issues annually making it the third leading preventable cause of death in the United States.3 In 2006, this alcohol misuse cost the US government approximately $223.5 billion dollars.3 BD (Blood-alcohol level ≥0.08 g% in a 2 hour period) is one form of excessive drinking and because of it, alcohol addiction and dependence remain a significant public health concern.4 Maternal separation and early life events can cause profound neurochemical and behavioral alterations in childhood that persist into adulthood, enhance the risk to develop alcohol use disorders and excessive drinking.5–7 Consequently, the development of clinically safe and cost effective therapeutic agents to reduce alcohol addiction and dependence remain essential for the future treatment of alcoholism.8,9
000 people die from alcohol related issues annually making it the third leading preventable cause of death in the United States.3 In 2006, this alcohol misuse cost the US government approximately $223.5 billion dollars.3 BD (Blood-alcohol level ≥0.08 g% in a 2 hour period) is one form of excessive drinking and because of it, alcohol addiction and dependence remain a significant public health concern.4 Maternal separation and early life events can cause profound neurochemical and behavioral alterations in childhood that persist into adulthood, enhance the risk to develop alcohol use disorders and excessive drinking.5–7 Consequently, the development of clinically safe and cost effective therapeutic agents to reduce alcohol addiction and dependence remain essential for the future treatment of alcoholism.8,9
One influence on alcohol abuse is known to be mediated by GABAA receptors, the major inhibitory chloride ion gated channels with γ-aminobutyric acid (GABA) as the endogenous ligand in the central nervous system. It plays a vital role in several neuronal disorders including anxiety, epilepsy, insomnia, depression, bipolar disorder, schizophrenia, as well as mild cognitive impairments and Alzheimer's disease.10–15 The pentameric structure of the GABAA receptor is made up of 2 α, 2 β and 1 γ subunits, with a higher distribution of the α1-subunit in the mesolimbic system of the ventral pallidum (VP) possibly playing an important role in regulating alcohol abuse.16–20 However, the precise neuromechanisms of regulating alcohol-seeking behavior remain unknown. In addition to the ventral pallidum, there is now compelling evidence that the GABAA receptors within the striatopallidal and extended amygdala system are involved in the ‘acute’ reinforcing actions of alcohol.21–23
To evaluate the role of the α1 receptor in regulating alcohol reinforcement, the orally active β-carbolines 3-propoxy-β-carboline hydrochloride 1·HCl (3-PBC·HCl) and β-carboline-3-carboxylate-tert-butyl ester 3 (βCCt), the mixed benzodiazepine (BDZ) agonist-antagonists with binding selectivity at the α1 Bz/GABAA receptor were developed (see Fig. 1).18,24,25Behavioral studies in several species (e.g., rats, mice, primates) show that these ligands were BDZ antagonists, at the α1 Bz/GABAA subtype exhibiting competitive binding-site interactions with BDZ agonists over a broad range of doses.18,24,26 In studies which involved the α1 subtype, they were shown to selectively reduce alcohol-motivated behaviors and more importantly, 3-PBC·HCl significantly reduced alcohol self-administration and reduced craving in baboons.26 β-Carbolines 1·HCl and 3 displayed mixed weak agonist-antagonist profiles in vivo in alcohol preferring (P) and high alcohol drinking (HAD) rats.18,26–28 Therefore, in addition to their use to study the molecular basis of alcohol reinforcement, α1 Bz β-carboline ligands which display mixed pharmacological antagonist-agonist activity in alcohol P and HAD rats may be capable of reducing alcohol intake while eliminating or greatly reducing the anxiety associated with habitual alcohol, abstinence or detoxification.18,28–30 Consequently, these types of ligands may be ideal clinical agents for the treatment of alcohol dependent individuals.
 | ||
| Fig. 1 Structures of 3-PBC (1), 3-ISOPBC (2), βCCt (3), 6-aza-3-PBC (4) and 6-aza-3-ISOPBC (5). | ||
Results and discussion
Previously, the β-carbolines 1 and 3 have been synthesized from DL-tryptophan. The overall yield of 1 (via 6 steps) as reported previously was 8%, while the combined yield of 3 (5 steps) was 35%. A few key steps occurred in low yields which was something of which we sought to improve on31–34 in a continued effort to find more potent subtype selective ligands for GABAA receptors. This interest resulted in a short and concise synthesis of 1 and 3. In 2011, a palladium catalyzed two-step protocol for the synthesis of 1, and 3 as well as analogs of 1 was reported.35 In the search for a more potent subtype selective ligand for the GABAA receptor, with the knowledge that many 3-substituted β-carbolines and more water soluble aza-β-carbolines might exhibit greater subtype selectivity at α1β2/3γ2 BZR/GABAergic receptors,31–33,36–38 the ligands 3-ISOPBC (2), 6-aza-3-PBC (4), and 6-aza-3-ISOPBC (5) were designed (see Fig. 1) and synthesized using a two-step protocol (Scheme 1). | ||
| Scheme 1 Synthesis of substituted carboline analogues. Reagents and conditions: (a) Pd(OAc)2, X-Phos, Cs2CO3, toluene, 100–140 °C, 15–24 h (b) Pd(OAc)2, (t-Bu)3P·HBF4, K2CO3, DMA, 120 °C, 16 h. | ||
As shown in Scheme 1, bromopyridines 6a–c39,40 were reacted with anilines 4a–b in toluene at 100–140 °C in the presence of 5 mol% Pd(OAc)2 and 7.5 mol% X-Phos to obtain the corresponding diarlyamines 7a–e in moderate to good yields. Unfortunately, the intramolecular Heck cyclization [Pd(OAc)2, (t-Bu)3·HBF4, K2CO3, DMA, 120 °C] of 7a–e afforded both the β-carbolines 1–5 (individually) and their regioisomeric δ-carbolines 9a–e, respectively. Carbolines 2, 3, 9a, and 9d were subjected to X-ray crystallographic analysis (see Fig. 2, Scheme 4, and the ESI†) to confirm the regiochemistry. Although this protocol permitted synthesis of β-carbolines on gram scale for in vivo studies, occasionally the first step in the Buckwald–Hartwig coupling failed to give complete conversion into the carboline. This complicated purification for the diarylamine was difficult to purify via column chromatography because the diarylamine and one of the starting anilines had almost identical Rf values. Furthermore, in the case of the water soluble aza-β-carboline the yields (51%) were very poor and importantly, since the second step was not regiospecific, this required careful purification to remove the unwanted δ-carboline present in 30 to 62.5% yield (Scheme 1). Interestingly, the in vivo results (unpublished) for 3-isopropoxy-β-carboline hydrochloride 2·HCl (3-ISOPBC·HCl) carried out in maternally deprived rats for binge drinking decreased dramatically this self-administration compared to 1·HCl without affecting the overall activity of the rats (i.e. no sedation). This important finding led to the interest in a regiospecific synthesis of 3-ISOPBC (2) on large scale.
 | ||
| Fig. 2 ORTEP view of the crystal structure of substituted carbolines 3, 9a, and 9d (displacement ellipsoids are at the 50% level (β-carboline numbering not followed). | ||
The revised synthetic strategy for the regiospecific synthesis of 2 began with the protection of the intermediate amine 7b (Na–H) with bulkier groups such as tert-butyloxycarbonyl (Boc) 10 or a fluorenylmethylenoxy group (Fmoc) 11, which might block the formation of the PdII π-complex that is required to obtain the undesired regioisomeric δ-carboline. The Boc protected amine 10 was easily accessible by treating the amine 7b with di-tert-butyl dicarbonate (Boc)2O and 4-(dimethylamino)pyridine (DMAP) in good yield (85%). The Fmoc protected amine 11 was synthesized under solvent free conditions by reaction of the amine 7b and Fmoc-Cl by microwave irradiation at 80 °C in moderate yield (65%, Scheme 2).41 Once protected, diarylamines 10 and 11 were subjected to a palladium catalyzed Heck-type cyclization using similar conditions to those from above. Unfortunately, both reactions afforded the deprotected regioisomers 3-ISOPBC (2) and δ-isomer 9b in approximately the same 2:1 ratio, as compared to cyclization with the previously unprotected diarylamine 7b (see Scheme 1 above). It was felt that deprotection of the carbamate occurred once the indole ring had formed (Scheme 2) which provided the better indole leaving group. To test the thermal stability of the carbamate starting materials, diarylamines 10 and 11 were heated at 120 °C in DMA; they were stable to these conditions. In addition, the cyclization with PdCl2(PPh3)2 as a palladium source was also attempted using standard Heck-type reaction conditions with a milder base (NaOAc), but this failed to give the cyclized product. We also explored the reaction by varying the water content using NaOAc·3H2O as a base; however, there was no cyclization (Scheme 2).
 | ||
| Scheme 2 Synthesis of the carbamate protected analogs from intermediate 7b. Reagents and conditions: (a) (Boc)2O, DMAP, THF, rt, 24 h; (b) Fmoc-Cl, 80 °C, microwave, 1 h; (c) Pd(OAc)2, (t-Bu)3P·HBF4, K2CO3, DMA, 120 °C, 16 h; (d) PdCl2(PPh3)2, NaOAc·3H2O, DMA, 120 °C, 14 h; (e) PdCl2(PPh3)2, NaOAc, DMA, 120 °C, 14 h. | ||
The second approach rested on the important switch of the chlorine atom from the benzene ring to the pyridine ring in amine 7b. Retrosynthetically, it was envisioned that the core structure of 3,6-disubstituted β-carboline A could be obtained from diarylamine Bvia an intramolecular Heck cyclization and it was anticipated that diarylamine B could arise from a substituted aniline C and a substituted pyridine derivative Dvia a Buchwald–Hartwig amination (Scheme 3).
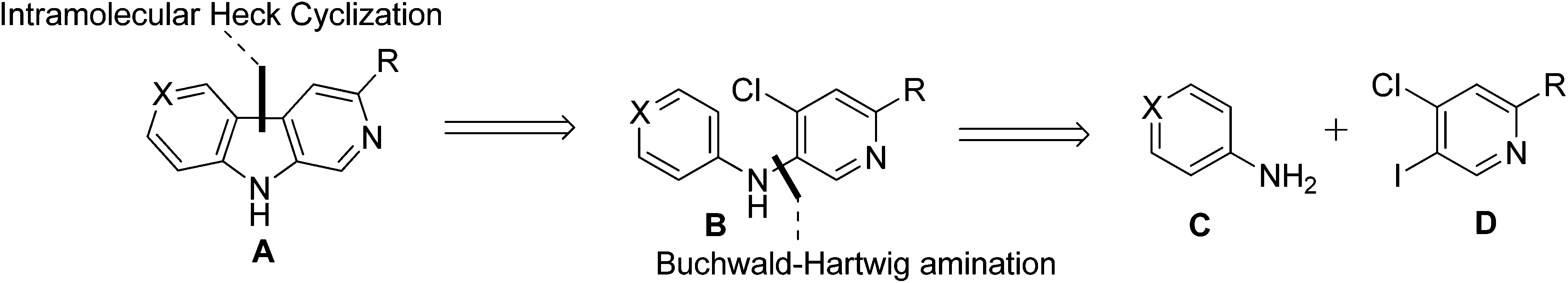 | ||
| Scheme 3 Retrosynthetic analysis of 3,6-disubstituted β-carbolines. | ||
At this point it was decided to explore the regioselective palladium catalyzed Buchwald–Hartwig coupling between aniline and pyridine 1442 for the synthesis of diarylamine 16 (Table 1). With the previous history in mind,35 the initial attempt was made with 5 mol% Pd(OAc)2, 7.5 mol% X-Phos and Cs2CO3 (1.5 equiv.) in toluene at 110 °C which gave only 18% of the diarylamine 16 with a large excess of unreacted starting material even after heating for 24 hours (Table 1, entry 1). However, the catalyst based on the combination of Pd2(dba)3, Xantphos and Pd(OAc)2, Xantphos with Cs2CO3 in toluene and dioxane gave the desire product diarylamine 16 in up to 62% yield (Table 1, entries 2–3). The ligand Xantphos has been shown to be efficient in cross coupling reactions of C–N bond formation because of a wider bite angle,43 which facilitates the reductive elimination. In addition, the excess base may also play a role in the improvement of the yield.43 In recent years rapid synthesis with microwave technology has attracted a considerable amount of attention for C–N bond formation.44–46 All three previous cyclizations were attempted with microwave irradiation (for 1 hour) in order to decrease the duration of the reaction time, as well as increase the selectivity under similar reaction conditions. However, the results were the same except that in the Xantphos-based ligand systems the cyclizations were completed in 1 hour. During continuation of the study of this selective amination, recent reports from Buchwald and co-workers47 demonstrated air- and moisture-stable palladacyclic precatalysts, when employed with aryl iodides and heteroaryliodides were attractive substrates in Pd-catalyzed C–N cross-coupling reactions. This process works by preventing formation of the stable bridging iodide dimers and also using a solvent system in which iodide salts were insoluble. These complexes easily undergo deprotonation and reductive elimination to generate LPd(0) along with relatively inert indoline (for generation of 1) or carbazole (for generation of 2 and 3). These conditions also permit the successful coupling of aryl iodides with amines at ambient temperature.47–50
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd source | Ligand | Base (equiv.) | Solvent | Temp (time) | Yieldb (%) |
| a 14 (0.1 mmol), aniline (0.12 mmol), Pd (3 mol%), ligand (3 mol%), base, and solvent (1 mL). b Isolated yields. c Pd (5 mol%), ligand (7.5 mol%). d Aniline (0.1 mmol). e 90% of diaminated product [6-isopropoxy-N3,N4-diphenylpyridine-3,4-diamine] was observed. | ||||||
| 1 | Pd(OAc)2 | X-Phos | Cs2CO3 (1.5) | Toluene | 110 °C (24 h) | 18c |
| 2 | Pd2(dba)3 | Xantphos | Cs2CO3 (2) | Dioxane | 110 °C (6 h) | 51 |
| 3 | Pd(OAc)2 | Xantphos | Cs2CO3 (4) | Toluene | 110 °C (6 h) | 62 |
| 4 | BrettPhos Pd G3 | BrettPhos | Cs2CO3 (1.5) | Toluene | 110 °C (14 h) | 45 |
| 5 | BrettPhos Pd G3 | BrettPhos | Cs2CO3 (3) | Toluene | 110 °C (5 h) | 66 |
| 6 | BrettPhos Pd G3 | BrettPhos | NaOt-Bu (1.5) | Toluene | 110 °C (5 h) | 52 |
| 7 | BrettPhos Pd G3 | BrettPhos | Cs2CO3 (5) | Toluene | 110 °C (5 h) | 0e |
| 8 | Pd2(dba)3 | Xantphos | Cs2CO3 (5) | Toluene | 110 °C (3 h) | 74 |
| 9 | Pd(OAc)2 | rac-BINAP | Cs2CO3 (5) | Toluene | 110 °C (5 h) | 80 |
| 10 | Pd(OAc)2 | rac-BINAP | K2CO3 (5) | Toluene | 110 °C (24 h) | 22 |
| 11 | Pd(OAc) 2 | rac-BINAP | Cs 2 CO 3 (5) | Toluene | 110 °C (5 h) | 92 |

The first attempt in this modification was to use the Buchwald 3rd generation palladacycle precatalyst (BrettPhos Pd G3) with the BrettPhos ligand in the presence of Cs2CO3 or NaOt-Bu in toluene at room temperature. This failed to give the desired product and there was no consumption of starting material. Following this attempt, the temperature was raised to reflux, with the addition of 3 equivalents of Cs2CO3 and the reaction went to completion within 5 hours. However, it only gave the desired amine 16 in 66% yield (Table 1, entry 5). When the same experiment was performed using only 1.5 equiv. of Cs2CO3 the process took a longer time to go to completion with an isolated yield of 45% of the desired amine 16. This was accompanied by the diaminated product [6-isopropoxy-N3,N4-diphenylpyridine-3,4-diamine] in ∼18% yield (Table 1, entry 4). Unfortunately, when the stronger base NaOt-Bu was employed comparable results to the above reaction (Table 1, entry 4) were obtained accompanied by more decomposed material [TLC(silica gel; Table 1, entry 6)]. The use of excess base (Cs2CO3) gave only the unwanted diaminated product in 90% yield (Table 1, entry 7). It was found the Pd(OAc)2, rac-BINAP and K2CO3 combination, unfortunately, did not lead to full conversion even after heating for 24 hours (Table 1, entry 10). Interestingly, the catalyst system Pd2(dba)3 and Xantphos with a large excess of base [Cs2CO3 (5 equiv.)] gave 74% yield of 16, whereas the catalyst system Pd(OAc)2, rac-BINAP under similar reaction conditions yielded 80% (Table 1, entry 8 and 9) of the desired amine 16. Remarkably, these data indicated a large excess of mild base was essential to obtain good yields, as well as selectivity. Furthermore, a rate-limiting interphase deprotonation of the Pd(II)-amine complex intermediate has occured in the catalytic cycle.51–53 Encouraged by these promising results, efforts turned toward lowering the aniline loading from 1.2 equivalents to 1 equivalent for regioselectivity. In doing so we achieved selective amination of pyridine 14 with aniline. Interestingly, neither a 4- nor 4,5-diaminated pyridine product was obtained. Using this catalyst-base combination in refluxing toluene, the desired cross-coupling proceeded smoothly to provide the desired anilinopyridine 16 in excellent yield (92%, Table 1, entry 11). Interestingly, the same reaction conditions gave good yields in the case of the more polar starting 4-amino pyridine (Scheme 4); however, the temperature was necessarily increased to 140 °C to increase the solubility of the starting material, 4-amino pyridine. In contrast, when a polar solvent such as DMA was employed, the result was either inferior yields and/or deiodination of pyridine 16, as mentioned above.
 | ||
| Scheme 4 Regiospecific synthesis of β-carbolines (1–2) and aza-β-carbolines (4–5). Reagents and conditions: (a) Pd(OAc)2, rac-BINAP, Cs2CO3, toluene, 110–140 °C, 5–6 h (b) Pd(OAc)2, (t-Bu)3P·HBF4, K2CO3, DMA, 120 °C, 16 h. | ||
Once the diarylamines 15–18 were in hand in good to excellent yields, the previously applied Heck-type conditions [Pd(OAc)2, (t-Bu)3·HBF4, K2CO3, DMA, 120 °C] were employed for cyclization. Gratifyingly, this catalyst system gave excellent yields of 91–92% and 90–92% for β-carbolines 1–2 and aza-β-carbolines 4–5, respectively (Scheme 4). The switch of the chlorine position from the benzene ring to the pyridine ring worked regiospecifically and completely eliminated the corresponding unwanted δ regioisomer. This completely eliminated the difficult chromatography required to separate β and δ carbolines. The 3-ISOPBC 2 has now been prepared on 15–25 gram scale for studies in vivo (Scheme 5) and it is very easy to scale up to 50–100 gram level. Finally, the overall yield increased from 43% to 84% compared to the previous syntheses.33,35
 | ||
| Scheme 5 Large-scale regiospecific synthesis of β-carboline 3-ISOPBC (2). Reagents and conditions: (a) Pd(OAc)2, rac-BINAP, Cs2CO3, toluene, 110 °C, 15 h (b) Pd(OAc)2, (t-Bu)3P·HBF4, K2CO3, DMA, 120 °C, 16 h. | ||
Conclusions
In conclusion, a novel two-step regiospecific route to the four anti-alcohol agents of biological interest, 3-PBC (1), 3-ISOPBC (2), 6-aza-3-PBC (4) and 6-aza-3-ISOPBC (5), has been developed. The process provided improved yields when compared to the earlier reported syntheses.33,35 This two-step protocol consists of the combination of a regioselective Buchwald–Hartwig amination and an intramolecular Heck-type cyclization. The first step, regioselective arylamination, was achieved by using a Pd-BINAP catalytic system in combination with a large excess of Cs2CO3, while the latter intramolecular Heck-type cyclization went smoothly with Pd(OAc)2 in combination with the air-stable monodentate ligand (t-Bu)3·HBF4 and K2CO3. These conditions permit the presence of base sensitive functional groups in the substrates. Regiospecific synthesis of β- and aza-β-carbolines was achieved by simply changing the chlorine position from the benzene ring to the pyridine derivatives. Importantly, these reactions are capable of scale-up to multigram quantities and were performed on 25 gram scale level for in vivo biology. We observed similar results except in the case of the Buchwald–Hartwig amination step, where it required an increase of the catalyst loading from 3 to 6 mol% whenever the starting material was not consumed. This new process reported here provides the material necessary to study alcohol self-administration and reduction thereof in MD rats and in primates. This regiospecific two-step synthetic protocol increased the overall yield from 43% to 84% in the case of β-carbolines 1–2 and from 16% to 66% for aza-β-carbolines 4–5 respectively, and negated the need for a difficult chromatographic step.Experimental
General considerations
All reactions were carried out in oven-dried, round-bottom flasks or in resealable screw-cap test tubes or heavy-wall pressure vessels under an argon atmosphere. The solvents were anhydrous unless otherwise stated. Stainless steel syringes were used to transfer air-sensitive liquids. Organic solvents were purified when necessary by standard methods or purchased from commercial suppliers. Anhydrous solvents of toluene, dioxane and N,N-dimethylacetamide (DMA) were subjected to the freeze–thaw method to render them oxygen free to execute the Buckwald–Hartwig coupling and intramolecular Heck reactions. All chemicals purchased from commercial suppliers were employed as is, unless stated otherwise in regard to purification. Silica gel (230–400 mesh) for flash chromatography was utilized to purify the analogues. The 1H and 13C NMR data were obtained on an NMR spectrometer (300 MHz/500 MHz) instrument with chemical shifts in δ (ppm) reported relative to TMS. The HRMS were obtained on a LCMS-IT-TOF mass spectrometer by Dr Mark Wang.General procedure for the Buchwald–Hartwig coupling reaction between substituted anilines and substituted pyridines: representative procedure for the synthesis of N-(2-chlorophenyl)-6-propoxypyridin-3-amine (7a)
A heavy-wall pressure tube was equipped with a magnetic stir bar and fitted with a rubber septum. It was then charged with 5-bromo-2-propoxypyridine 6a (1.3 g, 6 mmol), Pd(OAc)2 (67.4 mg, 0.3 mmol), X-Phos (214 mg, 0.45 mmol) and Cs2CO3 (2.34 g, 7.2 mmol). The vessel was evacuated and backfilled with argon (this process was repeated a total of 3 times). The 2-chloroaniline 4a (0.8 g, 6.3 mmol) and freeze–thawed toluene (20 mL) was injected into the tube with a degassed syringe under a positive pressure of argon. The rubber septum was replaced with a screw-cap by quickly removing the rubber septum under the flow of argon and the sealed tube was introduced into a pre-heated oil bath at 110 °C. The reaction mixture was maintained at this temperature for 15 h. At the end of this time period, the pressure tube was allowed to cool to rt. The reaction mixture was filtered through a short pad of celite, and the pad was washed with ethyl acetate (until no more product could be obtained; ≈100 mL; TLC, silica gel). The combined organic fractions were washed with water (100 mL), brine (100 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography (silica gel, 20:1 hexanes/ethyl acetate) to afford 7a (0.64 g, 81%) as a pale yellow oil: 1H NMR (300 MHz, CDCl3) δ 8.04 (d, J = 2.6 Hz, 1H), 7.47 (dd, J = 8.8, 2.8 Hz, 1H), 7.33 (dd, J = 7.9, 1.4 Hz, 1H), 7.12–7.02 (m, 1H), 6.84 (dd, J = 8.2, 1.3 Hz, 1H), 6.74 (dd, J = 11.5, 5.1 Hz, 2H), 5.88 (br, 1H), 4.24 (t, J = 6.7 Hz, 2H), 1.90–1.72 (m, 2H), 1.04 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 161.2, 142.1, 141.9, 135.3, 131.0, 129.7, 127.6, 120.1, 119.5, 113.5, 111.4, 67.9, 22.4, 10.6; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C14H16ClN2O: 263.0951, found: 263.0958.
N-(2-Chlorophenyl)-6-isopropoxypyridin-3-amine (7b)
Following the general procedure, 5-bromo-2-isopropoxypyridine 6b (0.44 g, 2.0 mmol) with 2-chloroaniline 4a (0.268 g, 2.1 mmol), Pd(OAc)2 (22.4 mg, 0.1 mmol), X-Phos (71.4 mg, 0.15 mmol), and Cs2CO3 (0.78 g, 2.4 mmol) were heated to 110 °C in toluene. After flash chromatography (silica gel, 20:1 hexane/ethyl acetate), the process afforded 7b (0.215 g, 82%) as a pale yellow oil: 1H NMR (300 MHz, CDCl3) δ 8.06 (d, J = 2.7 Hz, 1H), 7.47 (dd, J = 8.7, 2.8 Hz, 1H), 7.34 (dd, J = 7.9, 1.3 Hz, 1H), 7.13–7.04 (m, 1H), 6.87 (dd, J = 8.2, 1.2 Hz, 1H), 6.81–6.67 (m, 2H), 5.90 (br, 1H), 5.36–5.24 (m, 1H), 1.39 (d, J = 6.2 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 160.6, 142.3, 142.0, 135.3, 130.7, 129.6, 127.6, 120.1, 119.5, 113.5, 111.9, 68.2, 22.1; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C14H16ClN2O: 263.0951, found: 263.0935.
tert-Butyl 5-[(2-chlorophenyl)amino]picolinate (7c)
Following the general procedure, tert-butyl 5-bromopicolinate 6c (5 g, 19.4 mmol) with 2-chloroaniline 4a (2.6 g, 20.3 mmol), Pd(OAc)2 (0.22 g, 0.97 mmol), X-Phos (0.69 g, 1.45 mmol), and Cs2CO3 (7.59 g, 23.3 mmol) was heated to 110 °C in toluene. After flash chromatography (silica gel, 5:1 hexanes/ethyl acetate), this process afforded 7c (5.02 g, 85%) as an off-white solid; mp 148–149 °C: 1H NMR (300 MHz, CDCl3) δ 8.54 (d, J = 2.7 Hz, 1H), 8.00 (d, J = 8.6 Hz, 1H), 7.45 (dd, J = 8.1, 1.7 Hz, 2H), 7.39 (dd, J = 8.1, 1.2 Hz, 1H), 7.27–7.20 (m, 1H), 7.02 (td, J = 7.9, 1.4 Hz, 1H), 6.45 (br, 1H), 1.65 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 163.8, 141.5, 141.3, 139.3, 137.3, 130.3, 127.7, 125.9, 124.6, 123.7, 122.6, 118.8, 81.9, 28.2; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C16H17ClN2O2Na: 327.0876, found: 327.0857.
N-(3-Chloropyridin-4-yl)-6-propoxypyridin-3-amine (7d)
Following the general procedure for 24 h at 140 °C, 5-bromo-2-propoxypyridine 6a (13.45 g, 62.50 mmol) was heated with 4-amino-3-chloropyridine 4b (8.0 g, 62.5 mmol), Pd(OAc)2 (697 mg, 3.1 mmol), X-Phos (1.46 g, 3.1 mmol) and Cs2CO3 (40.6 g, 125 mmol) in refluxing toluene to yield the crude diaza material 7d. After flash chromatography (silica gel, 1:1 ethyl acetate/hexane), this afforded the pure diaza material 7d (8.29 g, 51%) as a white solid; mp 71.6–72.6 °C: 1H NMR (300 MHz, CDCl3): δ 8.35 (s, 1H), 8.10–8.07 (m, 2H), 7.48 (dd, J = 6.0, 3.0 Hz, 1H), 6.80 (d, J = 6.0 Hz, 1H), 6.60 (d, J = 6.0 Hz, 1H), δ 6.45 (br, 1H), 4.25 (t, J = 6.9, Hz, 2H), 1.87–1.75 (m, 2H), 1.03 (t, J = 7.2, Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 162.3, 148.8, 148.3, 148.2, 144.0, 136.56, 128.2, 117.1, 111.7, 106.9, 68.0, 22.3, 10.5; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H15ClN3O 264.0904, found 264.0893.
N-(3-Chloropyridin-4-yl)-6-isopropoxypyridin-3-amine (7e)
Following the general procedure for 24 h at 140 °C, 5-bromo-2-isopropoxypyridine 6b (8.09 g, 37.20 mmol) was heated with 4-amino-3-chloropyridine 4b (4.74 g, 37.20 mmol), Pd(OAc)2 (419 mg, 1.87 mmol), X-Phos (608 mg, 1.87 mmol), Cs2CO3 (15.25 g, 46.80 mmol) in refluxing toluene to afford a crude solid which was purified by flash chromatography (silica gel, 1:1 ethyl acetate/hexane) to furnish a white solid 7e (5.20 g, 52.4%); mp 76–78 °C: 1H NMR (300 MHz, CDCl3): δ 8.35 (s, 1H), 8.10–8.06 (m, 2H), 7.46 (dd, J = 6.0, 3.0 Hz, 1H), 6.74 (d, J = 6.0 Hz, 1H), 6.61 (d, J = 6.0 Hz, 1H), 6.42 (br s, 1H), 5.35–5.23 (m, 1H), 1.36 (d, J = 6.0 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 161.7, 148.7, 148.3, 144.0, 136.5, 127.9, 117.0, 112.2, 106.9, 68.5, 22.0; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H15ClN3O 264.0904, found 264.0909.
General procedure for the intramolecular Heck cyclization: representative procedure for the synthesis of 3-propoxy-9H-pyrido[3,4-b]indole (3-PBC, 1) and 2-propoxy-5H-pyrido[3,2-b]indole (9a)
A heavy-wall pressure tube was equipped with a magnetic stir bar and fitted with a rubber septum and loaded with N-(2-chlorophenyl)-6-propoxypyridin-3-amine 7a (526 mg, 2.0 mmol), Pd(OAc)2 (44.8 mg, 0.2 mmol), (t-Bu)3P·HBF4 (116 mg, 0.4 mmol) and K2CO3 (552 mg, 4.0 mmol). The vessel was evacuated and backfilled with argon (this process was repeated a total of 3 times) and degassed DMA (8 mL) was injected into the tube with a degassed syringe under a positive pressure of argon. The rubber septum was replaced with a screw-cap by quickly removing the rubber septum under the flow of argon and the sealed tube was introduced into a pre-heated oil bath at 120 °C. The reaction mixture was maintained at this temperature for 16 h. At the end of this period, the reaction mixture was allowed to cool to rt. The dark brown mixture which resulted was then passed through a short pad of celite. The celite pad was further washed with ethyl acetate (150 mL) until no more product (TLC; silica gel) was detected in the eluent. The combined filtrate was washed with water (100 mL × 3), brine (100 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography (silica gel, 5:1 hexanes/ethyl acetate) to afford 3-PBC (1) (235 mg, 52%) as an off white solid. mp 120.5–121.5 °C (lit.35 mp 119.3–120.5 °C): 1, 1H NMR (300 MHz, CDCl3) δ 8.66 (br, 1H), 8.42 (s, 1H), 8.05 (d, J = 7.9 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.45–7.38 (m, 1H), 7.35 (s, 1H), 7.21 (t, J = 7.4 Hz, 1H), 4.28 (t, J = 6.7 Hz, 2H), 1.94–1.78 (m, 2H), 1.06 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 157.9, 142.4, 133.8, 132.7, 128.9, 128.7, 122.0, 121.4, 119.5, 111.5, 99.1, 68.6, 22.7, 10.6; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C14H15N2O: 227.1184, found: 227.1174. A hydrochloride salt of 1 was prepared by the reported method31 to obtain 3-PBC·HCl (1·HCl): yellow solid; mp 194.5–195.5 °C (lit31 194.0–195.0 °C). The spectral data for this 1·HCl were in excellent agreement with the reported values (mp, 1H NMR).31
9a (145 mg, 32%) as a white solid; mp 125–126 °C: 1H NMR (300 MHz, CDCl3) δ 8.28 (t, J = 8.8 Hz, 1H), 8.20 (br, 1H), 7.60 (d, J = 8.7 Hz, 1H), 7.51–7.34 (m, 2H), 7.27 (t, J = 7.3 Hz, 1H), 6.83 (d, J = 8.7 Hz, 1H), 4.46 (t, J = 6.7 Hz, 2H), 1.99–1.80 (m, 2H), 1.10 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 159.5, 140.2, 138.2, 128.4, 126.8, 122.3, 121.6, 120.6, 119.7, 111.3, 108.6, 67.9, 22.6, 10.7; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C14H15N2O: 227.1184, found: 227.1180.
3-Isopropoxy-9H-pyrido[3,4-b]indole (3-ISOPBC, 2) and 3-isopropoxy-5H-pyrido[3,2-b]indole (9b)
Following the general procedure for the intramolecular Heck cyclization, 7b (526 mg, 2.0 mmol) was heated with Pd(OAc)2 (45 mg, 0.2 mmol), (t-Bu)3P·HBF4 (116 mg, 0.4 mmol) and K2CO3 (552 mg, 4.0 mmol) in DMA at 120 °C to afford a mixture of regioisomers 2 and 9b. After flash chromatography (silica gel, 5:1 hexanes/ethyl acetate), this procedure yielded pure 3-ISOPBC (2) and the byproduct 9b.
2 (239.5 mg, 53%): off-white solid; mp 134–136 °C: 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 8.19 (br, 1H), 8.04 (d, J = 7.8 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 7.34 (s, 1H), 7.21 (t, J = 7.4 Hz, 1H), 5.35–5.23 (m, 1H), 1.40 (d, J = 6.1 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 157.4, 142.1, 133.7, 132.5, 128.9, 128.8, 122.0, 121.6, 119.5, 111.3, 100.5, 68.6, 22.3; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C14H15N2O: 227.1184, found: 227.1184. A hydrochloride salt of 2 was prepared by the reported method37 to obtain 3-ISOPBC·HCl (2·HCl): light greenish yellow solid; mp 169–171 °C (lit.37 168–172 °C). The data for this compound matched in all respects (1H NMR, mp) with that reported in the literature.37
9b (163.1 mg, 36%): light brown solid; mp 110.4–111.5 °C: 1H NMR (300 MHz, CDCl3) δ 8.27 (d, J = 7.8 Hz, 1H), 7.99 (br, 1H), 7.67 (d, J = 8.7 Hz, 1H), 7.49–7.45 (m, 2H), 7.30–7.25 (m, 1H), 6.79 (d, J = 8.7 Hz, 1H), 5.60–5.48 (m, 1H), 1.45 (d, J = 6.1 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 158.9, 140.1, 138.4, 128.1, 126.7, 122.6, 121.3, 120.5, 119.7, 111.1, 109.4, 67.9, 22.2; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C14H15N2O: 227.1184, found: 227.1185.
tert-Butyl 9H-pyrido[3,4-b]indole-3-carboxylate (βCCt; 3) and tert-butyl 5H-pyrido[3,2-b]indole-3-carboxylate (9c)
Following the general procedure for the intramolecular Heck cyclization, 7c (2 g, 16.4 mmol), was heated with Pd(OAc)2 (147 mg, 0.656 mmol), (t-Bu)3P·HBF4 (380 mg, 0.4 mmol) and K2CO3 (1.8 g, 13.12 mmol) in DMA at 120 °C to afford crude 3 and 9c. After flash chromatography (silica gel, 1:1 hexanes/ethyl acetate), this afforded pure βCCt (3) and 9c.
3 (885 mg, 50%), white solid; mp 302.5–304.5 °C (lit33 301–303 °C): 1H NMR (300 MHz, CDCl3) δ 10.35 (br, 1H), 9.23 (s, 1H), 8.86 (s, 1H), 8.25 (d, J = 7.9 Hz, 1H), 7.80 (d, J = 8.3 Hz, 1H), 7.66–7.61 (m, 1H), 7.38 (t, J = 7.5 Hz, 1H), 1.75 (s, 9H); 13C NMR (75 MHz, CD3COCD3) δ 164.9, 141.2, 139.2, 137.7, 133.4, 128.6, 128.1, 121.8, 121.5, 120.3, 116.9, 112.2, 80.1, 27.6; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C16H17N2O2: 269.1290, found: 269.1286. The spectral data are in excellent agreement with the published values.33
9c (531 mg, 30%), fluffy white solid; mp 216.0–218.2 °C: 1H NMR (300 MHz, CDCl3) δ 9.46 (br, 1H), 8.38 (d, J = 7.8 Hz, 1H), 8.18 (d, J = 8.5 Hz, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.53–7.49 (m, 2H), 7.25–7.23 (m, 1H), 1.67 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 164.9, 142.4, 141.4, 141.1, 134.7, 128.6, 122.0, 121.9, 121.0, 120.8, 117.4, 111.5, 81.9, 28.2; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C16H17N2O2: 269.1290, found: 269.1289.
8-Propoxy-5H-pyrrolo[2,3-c:4,5-c′]dipyridine (6-aza-3-PBC, 4) and 2-propoxy-5H-pyrrolo[3,2-b:4,5-c′]dipyridine (9d)
Following the general procedure for the intramolecular Heck cyclization, the diaza compound 7d (3.0 g, 11.30 mmol) was heated with Pd(OAc)2 (255.0 mg, 1.13 mmol), (t-Bu)3P·HBF4 (657.0 mg, 2.26 mmol) and K2CO3 (3.2 g, 22.60 mmol) in DMA at 120 °C to afford crude 4 and 9d. After flash chromatography (silica gel, 1:24 methanol/dichloromethane) this process afforded the pure regioisomers 6-aza-3-PBC (4) and 9d as white solids.
4 (820 mg, 31.8%): mp 166–168 °C: 1H NMR (300 MHz, (CD3)2SO): δ 12. 13 (br, 1H), 9.51 (s, 1H), 8.57 (br, 2H), 7.68 (s, 1H), 7.61 (d, J = 5.7 Hz, 1H), 4.26 (t, J = 6.0, Hz, 2H), 1.83–1.71 (m, 2H), 1.01 (t, J = 6.0, Hz, 3H); 13C NMR (75 MHz, (CD3)2SO): δ 158.4, 147.0, 144.5, 143.4, 133.0, 131.5, 130.7, 118.2, 108.1, 100.4, 68.0, 22.5, 10.9; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H14N3O: 228.1137, found: 228.1144.
9d (1.62 g, 62.5%); mp 192–194 °C: 1H NMR (300 MHz, (CD3)2SO): 11.70 (s, 1H), 9.25 (s, 1H), 8.43 (d, J = 6.0 Hz, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.51 (d, J = 6.0 Hz, 1H), 6.93 (d, J = 9.0 Hz, 1H), 4.36 (t, J = 6.0 Hz, 2H), 1.84–1.77 (m, 2H), 1.03 (t, J = 6.0 Hz, 3H); 13C NMR (75 MHz, (CD3)2SO): δ 159.7, 145.2, 143.8, 142.8, 136.0, 128.8, 123.5, 118.5, 110.2, 107.6, 67.4, 22.4, 11.0; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H14N3O: 228.1137, found: 228.1140.
8-Isopropoxy-5H-pyrrolo[2,3-c:4,5-c′]dipyridine (6-aza-3-ISOPBC, 5) and 2-isopropoxy-5H-pyrrolo[3,2-b:4,5-c′]dipyridine (9e)
Following the general procedure for the intramolecular Heck cyclization, pyridine 7e (3.0 g, 11.30 mmol) was heated with Pd(OAc)2 (255.0 mg, 1.13 mmol), (t-Bu)3P·HBF4 (657.0 mg, 2.26 mmol) and K2CO3 (3.2 g, 22.60 mmol) in DMA at 120 °C to afford crude 5 and 9e. After flash chromatography (silica gel, 1:24 methanol/dichloromethane) this afforded the regioisomeric 6-aza-3-ISOPBC (5) and 9e as white solids.
5 (800 mg, 31.0%); mp 180.2–183.2 °C: 1H NMR (300 MHz, (CD3)2SO): δ 11.66 (s, 1H), 9.37 (s, 1H), 8.51 (s, 1H), 8.48 (d, J = 6.0 Hz, 1H), 7.56 (s, 1H), 7.46 (d, J = 6.0 Hz, 1H), 5.32–5.20 (m, 1H), 1.32 (d, J = 6.0 Hz, 6H); 13C NMR (125 MHz, (CD3)2SO): δ 157.4, 147.3, 146.3, 145.4, 132.7, 131.6, 130.1, 118.2, 107.4, 100.0, 68.0, 22.6; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H14N3O: 228.1137, found: 228.1150.
9e (1.6 g, 62.3%); mp 207.4–208.6 °C: 1H NMR (500 MHz, (CD3)2SO): δ 11.85 (s, 1H), 9.28 (s, 1H), 8.44 (d, J = 3.0 Hz, 1H), 7.94 (d, J = 6.0 Hz, 1H), 7.55 (d, J = 3.0 Hz, 1H), 6.89 (d, J = 6.0 Hz, 1H), 5.49–5.41 (m, 1H), 1.36 (d, J = 3.0 Hz, 6H); 13C NMR (125 MHz, (CD3)2SO): δ 159.2, 144.2, 144.0, 142.1, 136.0, 128.9, 123.7, 111.1, 107.8, 67.8, 22.4; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H14N3O: 228.1137, found: 228.1140.
tert-Butyl (2-chlorophenyl)(6-isopropoxypyridin-3-yl)carbamate (10)
To the amine 7b (275 mg, 1.05 mmol) in THF (6 mL) was added the di-tert-butyl dicarbonate (320 mg, 1.46 mmol) and 4-(dimethylamino)pyridine (DMAP) (51.1 mg, 0.42 mmol) and this mixture was stirred at rt for 24 h. The organic solvent was removed under reduced pressure and the crude product which resulted was purified by flash column chromatography (silica gel, 1:9 ethylacetate/hexane) to give the pure BOC protected amine 10 (323 mg, 85%).
1H NMR (300 MHz, CDCl3) δ 8.03 (d, J = 2.6 Hz, 1H), 7.60 (s, 1H), 7.44 (dd, J = 8.1, 5.9 Hz, 1H), 7.32–7.20 (m, 3H), 6.62 (d, J = 8.9 Hz, 1H), 5.30–5.16 (m, 1H), 1.43 (s, 9H), 1.31 (d, J = 6.2 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 160.9, 153.3, 143.9, 139.8, 136.7, 133.3, 132.2, 130.4, 130.3, 128.6, 127.7, 111.1, 81.6, 68.2, 28.1; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C19H24ClN2O3: 363.1475, found: 363.1469.
(9H-Fluoren-9-yl)methyl (2-chlorophenyl)(6-isopropoxypyridin-3-yl)carbamate (11)
The microwave tube was loaded with amine 7b (300 mg, 1.14 mmol) and Fmoc chloride (325 mg, 1.25 mmol). The tube was sealed and placed into a microwave apparatus (with a power of 100 W) at 80 °C for 1 h with stirring. At the end of this period, the reaction was directly purified, without quenching, by flash column chromatography(silica gel, 1:4 ethylacetate/hexane) to give pure Fmoc protected pyridine 11 (360 mg, 65%).
1H NMR (300 MHz, CDCl3) δ 8.08 (d, J = 2.7 Hz, 1H), 7.70 (d, J = 7.6 Hz, 3H), 7.50 (d, J = 3.8 Hz, 1H), 7.42–7.28 (m, 5H), 7.20–7.06 (m, 4H), 6.64 (d, J = 8.5 Hz, 1H), 5.33–5.15 (m, 1H), 4.49–4.41 (m, 2H), 4.16–4.09 (m, 1H), 1.34 (d, J = 6.1 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 154.3, 143.6, 141.3, 139.2, 139.1, 131.6, 130.60, 130.5, 129.2, 127.9, 127.7, 126.9, 125.0, 119.9, 111.4, 68.4, 68.2, 46.9, 22.1; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C29H25ClN2O3Na: 507.1451, found: 507.1448.
4-Chloro-6-isopropoxy-N-phenylpyridin-3-amine (16)
A heavy-wall pressure tube was equipped with a magnetic stir bar and fitted with a rubber septum that had been charged with 4-chloro-5-iodo-2-isopropoxypyridine 14 (75 mg, 0.252 mmol), aniline (27.6 μL, 0.256 mmol) and Cs2CO3 (410 mg, 1.26 mmol). The vessel was evacuated and backfilled with argon (this process was repeated a total of 3 times) and degassed toluene (1 mL) was injected into the tube with a degassed syringe under a positive pressure of argon. In another round bottom flask fitted with a rubber septum, Pd(OAc)2 (1.7 mg, 0.0076 mmol) and rac-BINAP (4.7 mg, 0.0076 mmol) was charged. This flask was evacuated and backfilled with argon (this process was repeated a total of 3 times) and then degassed toluene (0.5 mL) was added under a positive pressure of argon. This mixture was stirred for 10 min and then the mixture which resulted was added to the above pressure tube. The rubber septum was replaced with a screw-cap by quickly removing the rubber septum under the flow of argon and the sealed tube was introduced into a pre-heated oil bath at 110 °C. The reaction mixture was maintained at this temperature for 5 h. At the end of this time period the pressure tube was allowed to cool to rt. The reaction mixture was filtered through a short pad of celite, and the pad was washed with ethyl acetate (until no more product could be obtained; ≈50 mL). The combined organic eluents were washed with water (50 mL), brine (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography (silica gel, 20:1 hexanes/ethyl acetate) to afford only 16 (61 mg, 92%) as a pale yellow oil.
1H NMR (300 MHz, CDCl3) δ 8.18 (s, 1H), 7.28 (t, J = 7.9 Hz, 2H), 6.98–6.92 (m, 3H), 6.83 (s, 1H), 5.52 (s, 1H), 5.29–5.17 (m, 1H), 1.36 (d, J = 6.2 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 159.2, 143.5, 139.1, 138.2, 130.4, 129.5, 121.1, 116.8, 111.9, 68.8, 22.1; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C14H16ClN2O: 263.0951, found: 263.0958.
4-Chloro-6-propoxy-N-phenylpyridin-3-amine (15)
Following the above general procedure for 5 h at 110 °C, 4-chloro-5-iodo-2-propoxypyridine 13 (75 mg, 0.252 mmol), aniline (27.6 μL, 0.256 mmol), Pd(OAc)2 (1.7 mg, 0.0076 mmol), rac-BINAP (4.7 mg, 0.0076 mmol) and Cs2CO3 (410 mg, 1.26 mmol) were heated in toluene at reflux to afford a crude liquid which was purified by flash chromatography (silica gel, 20:1 hexanes/ethyl acetate) to furnish a pale yellow oil 15 (60.33 mg, 91%).
1H NMR (300 MHz, CDCl3) δ 8.20 (s, 1H), 7.28 (t, J = 7.9 Hz, 2H), 6.98–6.93 (m, 3H), 6.89 (s, 1H), 5.57 (s, 1H), 4.26 (t, J = 6.6 Hz, 2H), 1.89–1.77 (m, 2H), 1.06 (t, J = 7.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 159.9, 143.6, 139.3, 138.1, 130.6, 129.5, 121.1, 116.7, 111.4, 68.1, 22.4, 10.5; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C14H16ClN2O: 263.0951, found: 263.0946.
4-Chloro-6-propoxy-N-(pyridin-4-yl)pyridine-3-amine (17)
Following the above general procedure for 6 h at 140 °C, 4-chloro-5-iodo-2-propoxypyridine 13 (214 mg, 0.72 mmol), 4-aminopyridine (68.8 mg, 0.73 mmol), Pd(OAc)2 (4.8 mg, 0.0216 mmol) and rac-BINAP (13.4 mg, 0.0216 mmol) as well as Cs2CO3 (1.17 g, 3.6 mmol) were heated in toluene at reflux to afford a crude solid which was purified by flash chromatography (silica gel, ethyl acetate) to furnish a white solid 17 (137 mg, 72%); mp 119–120 °C, 1H NMR (300 MHz, CDCl3): δ 8.28 (d, J = 4.8 Hz, 2H), 8.19 (s, 1H), 6.92 (s, 1H), 6.65 (d, J = 5.4 Hz, 2H), 6.18 (br, 1H), 4.27 (t, J = 6.6 Hz, 2H), 1.88–1.76 (m, 2H), 1.04 (t, J = 7.5 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 162.3, 151.7, 150.0, 144.6, 142.0, 126.9, 111.9, 108.9, 68.4, 22.3, 10.5; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H15ClN3O 264.0904, found 264.0898.4-Chloro-6-isopropoxy-N-(pyridin-4-yl)pyridine-3-amine (18)
Following the above general procedure for 6 h at 140 °C, 4-chloro-5-iodo-2-isopropoxypyridine 13 (214 mg, 0.72 mmol), 4-aminopyridine (68.8 mg, 0.73 mmol), Pd(OAc)2 (4.8 mg, 0.0216 mmol) and rac-BINAP (13.4 mg, 0.0216 mmol) as well as Cs2CO3 (1.17 g, 3.6 mmol) were heated in toluene at reflux to afford a crude solid which was purified by flash chromatography (silica gel, ethyl acetate) to furnish a white solid 18 (135 mg, 71%); 1H NMR (300 MHz, CDCl3): δ 8.24 (d, J = 4.2 Hz, 2H), 8.18 (s, 1H), 6.87 (s, 1H), 6.74 (d, J = 5.7 Hz, 2H), 5.36–5.23 (m, 1H), 1.37 (d, J = 6.3 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 161.9, 152.4, 148.9, 144.8, 142.1, 126.4, 112.4, 108.9, 69.3, 22.0; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H15ClN3O 264.0904, found 264.0910.3-Propoxy-9H-pyrido[3,4-b]indole (3-PBC, 1)
Following the general procedure for the Heck cyclization for 16 h at 120 °C, 4-Chloro-6-propoxy-N-phenylpyridin-3-amine 15 (526 mg, 2.0 mmol), Pd(OAc)2 (44.8 mg, 0.2 mmol), (t-Bu)3P·HBF4 (116 mg, 0.4 mmol) and K2CO3 (552 mg, 4.0 mmol) were heated to give a solid which was purified by a wash column (silica gel, 5:1 hexanes/ethyl acetate) to yield 3-PBC 1 (416.80 mg, 92%).
3-Isopropoxy-9H-pyrido[3,4-b]indole (3-ISOPBC, 2)
Following the general procedure for the Heck cyclization for 16 h at 120 °C, 4-chloro-6-isopropoxy-N-phenylpyridin-3-amine 16 (526 mg, 2.0 mmol), Pd(OAc)2 (44.8 mg, 0.2 mmol), (t-Bu)3P·HBF4 (116 mg, 0.4 mmol) and K2CO3 (552 mg, 4.0 mmol) were heated to give a solid which was purified by a wash column (silica gel, 5:1 hexanes/ethyl acetate) to yield 3-ISOPBC 2 (412.30 mg, 91%).
8-Propoxy-5H-pyrrolo[2,3-c:4,5-c′]dipyridine (6-aza-3-PBC, 4)
Following the general procedure for the Heck cyclization for 16 h at 120 °C, 4-chloro-6-propoxy-N-(pyridin-4-yl)pyridine-3-amine 17 (125 mg, 0.475 mmol), Pd(OAc)2 (10.7 mg, 0.047 mmol), (t-Bu)3P·HBF4 (27.6 mg, 0.095 mmol) and K2CO3 (131.3 mg, 0.95 mmol) were heated to give a solid which was purified by a wash column (silica gel, 1:24 methanol/dichloromethane) to yield 6-aza-3-PBC 4 (97.15 mg, 90%).
8-Isopropoxy-5H-pyrrolo[2,3-c:4,5-c′]dipyridine (6-aza-3-ISOPBC, 5)
Following the general procedure for the Heck cyclization for 16 h at 120 °C, 4-chloro-6-isopropoxy-N-(pyridin-4-yl)pyridine-3-amine 18 (125 mg, 0.475 mmol), Pd(OAc)2 (10.7 mg, 0.047 mmol), (t-Bu)3P·HBF4 (27.6 mg, 0.095 mmol) and K2CO3 (131.3 mg, 0.95 mmol) were heated to give a solid which was purified by a wash column (silica gel, 1:24 methanol/dichloromethane) to yield 6-aza-3-ISOPBC 5 (99.31 mg, 92%).
Large-scale synthesis of 3-ISOPBC (2)
:1 hexanes/ethyl acetate) to afford only 16 (19.86 g, 90%) as a pale yellow oil.
:1 hexanes/ethyl acetate) to afford 3-ISOPBC (2) (15.74 g, 92%) as an off white solid.
Acknowledgements
We thank the NIMH (1R01MH096463-01A1), NIAAA (7R01AA016179) and NINDS (5R01NS076517-03) for generous financial support. The X-ray crystallographic work was supported by NIDA through Interagency Agreement #ADA 12003 with the Naval Research Laboratory (NRL). We also wish to acknowledge the Bradley-Herzfeld Foundation and The Milwaukee Institute for Drug Discovery for support.References
- R. Cao, W. Peng, Z. Wang and A. Xu, Curr. Med. Chem., 2007, 14, 479–500 CrossRef CAS.
- P. Venault and G. Chapouthier, Sci. World J., 2007, 7, 204–223 CrossRef CAS PubMed.
- M. Stahre, J. Roeber, D. Kanny, R. D. Brewer and X. Zhang, Prev. Chronic Dis., 2014, 11, E109 Search PubMed.
- A. R. Yang, J. Liu, H. S. Yi, K. T. Warnock, M. Wang, H. L. June Jr., A. C. Puche, A. Elnabawi, W. Sieghart, L. Aurelian and H. L. June Sr., Front. Neurosci., 2011, 5, 123 CAS.
- M. C. Moffett, A. Vicentic, M. Kozel, P. Plotsky, D. D. Francis and M. J. Kuhar, Biochem. Pharmacol., 2007, 73, 321–330 CrossRef CAS PubMed.
- I. Nylander and E. Roman, Psychopharmacology, 2013, 229, 555–569 CrossRef CAS PubMed.
- J. N. Jaworski, D. D. Francis, C. L. Brommer, E. T. Morgan and M. J. Kuhar, Psychopharmacology, 2005, 181, 8–15 CrossRef CAS PubMed.
- B. A. Johnson and N. Ait-Daoud, Psychopharmacology, 2000, 149, 327–344 CrossRef CAS.
- H. R. Kranzler, Alcohol Alcohol., 2000, 35, 537–547 CrossRef CAS.
- M. Davies, J. Psychiatry Neurosci., 2003, 28, 263–274 Search PubMed.
- G. Kalsi, C. A. Prescott, K. S. Kendler and B. P. Riley, Trends Genet., 2009, 25, 49–55 CrossRef CAS PubMed.
- G. F. Koob, Biochem. Pharmacol., 2004, 68, 1515–1525 CrossRef CAS PubMed.
- W. Sieghart and M. Ernst, Curr. Med. Chem.: Cent. Nerv. Syst. Agents, 2005, 5, 217–242 CrossRef CAS.
- M. M. Silveri, J. T. Sneider, D. J. Crowley, M. J. Covell, D. Acharya, I. M. Rosso and J. E. Jensen, Biol. Psychiatry, 2013, 74, 296–304 CrossRef CAS PubMed.
- S. Kumar, P. Porcu, D. F. Werner, D. B. Matthews, J. L. Diaz-Granados, R. S. Helfand and A. L. Morrow, Psychopharmacology, 2009, 205, 529–564 CrossRef CAS PubMed.
- L. Churchill, A. Bourdelais, M. C. Austin, S. J. Lolait, L. C. Mahan, A. M. O'Carroll and P. W. Kalivas, Synapse, 1991, 8, 75–85 CrossRef CAS PubMed.
- G. E. Duncan, G. R. Breese, H. E. Criswell, T. J. McCown, J. S. Herbert, L. L. Devaud and A. L. Morrow, Neuroscience, 1995, 64, 1113–1128 CrossRef CAS.
- S. C. Harvey, K. L. Foster, P. F. McKay, M. R. Carroll, R. Seyoum, J. E. Woods 2nd, C. Grey, C. M. Jones, S. McCane, R. Cummings, D. Mason, C. Ma, J. M. Cook and H. L. June, J. Neurosci., 2002, 22, 3765–3775 CAS.
- W. Wisden, D. J. Laurie, H. Monyer and P. H. Seeburg, J. Neurosci., 1992, 12, 1040–1062 CAS.
- J. Liu, A. R. Yang, T. Kelly, A. Puche, C. Esoga, H. L. June Jr., A. Elnabawi, I. Merchenthaler, W. Sieghart, H. L. June Sr. and L. Aurelian, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4465–4470 CrossRef CAS PubMed.
- G. F. Koob and M. Le Moal, Nat. Neurosci., 2005, 8, 1442–1444 CrossRef CAS PubMed.
- G. F. Koob, A. J. Roberts, G. Schulteis, L. H. Parsons, C. J. Heyser, P. Hyytia, E. Merlo-Pich and F. Weiss, Alcohol. Clin. Exp. Res., 1998, 22, 3–9 CrossRef CAS PubMed.
- W. J. McBride and T. K. Li, Crit. Rev. Neurobiol., 1998, 12, 339–369 CrossRef CAS.
- H. L. June, K. L. Foster, P. F. McKay, R. Seyoum, J. E. Woods II, S. C. Harvey, W. J. A. Eiler, II, C. Grey, M. R. Carroll, S. McCane, C. M. Jones, W. Yin, D. Mason, R. Cummings, M. Garcia, C. Ma, P. V. V. S. Sarma, J. M. Cook and P. Skolnick, Neuropsychopharmacology, 2003, 28, 2124–2137 CAS.
- E. D. Cox, T. J. Hagen, R. M. Mckernan and J. M. Cook, Med. Chem. Res., 1995, 5, 710 CAS.
- E. K. Sawyer, C. Moran, M. H. Sirbu, M. Szafir, M. Van Linn, O. Namjoshi, V. V. N. P. B. Tiruveedhula, J. M. Cook and D. M. Platt, Alcohol. Clin. Exp. Res., 2014, 38, 1108–1117 CrossRef CAS PubMed.
- D. S. O'Tousa, K. T. Warnock, L. M. Matson, O. A. Namjoshi, M. V. Linn, V. V. Tiruveedhula, M. E. Halcomb, J. Cook, N. J. Grahame and H. L. June, Addict. Biol., 2015, 20, 236–247 CrossRef PubMed.
- J. K. Rowlett, R. D. Spealman, S. Lelas, J. M. Cook and W. Yin, Psychopharmacology, 2003, 165, 209–215 CAS.
- J. Kovacevic, T. Timic, V. V. Tiruveedhula, B. Batinic, O. A. Namjoshi, M. Milic, S. Joksimovic, J. M. Cook and M. M. Savic, Brain Res. Bull., 2014, 104, 1–6 CrossRef CAS PubMed.
- J. Divljakovic, M. Milic, O. A. Namjoshi, V. V. Tiruveedhula, T. Timic, J. M. Cook and M. M. Savic, Brain Res. Bull., 2013, 91, 1–7 CrossRef CAS PubMed.
- M. S. Allen, T. J. Hagen, M. L. Trudell, P. W. Codding, P. Skolnick and J. M. Cook, J. Med. Chem., 1988, 31, 1854–1861 CrossRef CAS.
- T. J. Hagen, F. Guzman, C. Schultz, J. M. Cook, P. Skolnick and H. E. Shannon, Heterocycles, 1986, 24, 2845–2855 CrossRef CAS.
- W. Yin, S. Majumder, T. Clayton, S. Petrou, M. L. VanLinn, O. A. Namjoshi, C. Ma, B. A. Cromer, B. L. Roth, D. M. Platt and J. M. Cook, Bioorg. Med. Chem., 2010, 18, 7548–7564 CrossRef CAS PubMed.
- W. Yin, P. V. V. S. Sarma, J. Ma, D. Han, J. L. Chen and J. M. Cook, Tetrahedron Lett., 2005, 46, 6363–6368 CrossRef CAS PubMed.
- O. A. Namjoshi, A. Gryboski, G. O. Fonseca, M. L. Van Linn, Z.-j. Wang, J. R. Deschamps and J. M. Cook, J. Org. Chem., 2011, 76, 4721–4727 CrossRef CAS PubMed.
- M. S. Allen, A. J. LaLoggia, L. J. Dorn, M. J. Martin, G. Costantino, T. J. Hagen, K. F. Koehler, P. Skolnick and J. M. Cook, J. Med. Chem., 1992, 35, 4001–4010 CrossRef CAS.
- M. S. Allen, Y. C. Tan, M. L. Trudell, K. Narayanan, L. R. Schindler, M. J. Martin, C. Schultz, T. J. Hagen, K. F. Koehler, P. W. Codding, P. Skolnick and J. M. Cook, J. Med. Chem., 1990, 33, 2343–2357 CrossRef CAS.
- J. M. Cook, M. L. Van Linn and W. Yin, U. S. Patent, 8268854, 2012 Search PubMed.
- J. M. Bailey, G. Bruton, A. Huxley, P. H. Milner and B. S. Orlek, PCT Int. Patent, 2005014571, 2005 Search PubMed.
- J.-R. Jansen, M. Fuesslein, W. Hallenbach, O. Ort, C. Arnold, E.-M. Franken, O. Malsam, U. Reckmann, E. Sanwald and U. Goergens, PCT Int. Patent, 2009068194, 2009 Search PubMed.
- M. Godoi, G. V. Botteselle, J. Rafique, M. S. T. Rocha, J. M. Pena and A. L. Braga, Asian J. Org. Chem., 2013, 2, 746–749 CrossRef CAS PubMed.
- T. Heffron, B. Safina, S. Staben, D. P. Sutherlin, B. Wei, R. Elliott, R. Heald, E. M. Seward, E. Gancia and B. Waskowycs, US Patent, 20120245144, 2012 Search PubMed.
- M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC.
- K. T. J. Loones, B. U. W. Maes, G. Rombouts, S. Hostyn and G. Diels, Tetrahedron, 2005, 61, 10338–10348 CrossRef CAS PubMed.
- S. Hostyn, B. U. W. Maes, G. Van Baelen, A. Gulevskaya, C. Meyers and K. Smits, Tetrahedron, 2006, 62, 4676–4684 CrossRef CAS PubMed.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- N. C. Bruno and S. L. Buchwald, Org. Lett., 2013, 15, 2876–2879 CrossRef CAS PubMed.
- B. P. Fors, N. R. Davis and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 5766–5768 CrossRef CAS PubMed.
- B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552–13554 CrossRef CAS PubMed.
- J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 1997, 62, 6066–6068 CrossRef CAS.
- B. U. W. Maes, K. T. J. Loones, T. H. M. Jonckers, G. L. F. Lemière, R. A. Dommisse and A. Haemers, Synlett, 2002, 1995–1998 CrossRef CAS.
- C. Meyers, B. U. W. Maes, K. T. J. Loones, G. Bal, G. L. F. Lemière and R. A. Dommisse, J. Org. Chem., 2004, 69, 6010–6017 CrossRef CAS PubMed.
- Y. Sunesson, E. Limé, S. O. Nilsson Lill, R. E. Meadows and P.-O. Norrby, J. Org. Chem., 2014, 79, 11961–11969 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of spectra and crystallographic information files in CIF format. CCDC 1040831–1040833 and 1044936. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob01572c |
| This journal is © The Royal Society of Chemistry 2015 |