
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn optimized and versatile synthesis to pyridinylimidazole-type p38α mitogen activated protein kinase inhibitors†
Ahmed
El-Gokha
ab,
Stefan A.
Laufer
c and
Pierre
Koch
*a
aInstitute of Pharmaceutical Sciences, Department of Medicinal and Pharmaceutical Chemistry, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 8, 72076 Tübingen, Germany. E-mail: pierre.koch@uni-tuebingen.de
bMenofia University, Chemistry Department, Faculty of Science, Menofia, Egypt
cInstitute of Pharmaceutical Sciences, Chair for Medicinal and Pharmaceutical Chemistry, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 8, 72076 Tübingen, Germany
First published on 3rd September 2015
Abstract
An optimized strategy for the synthesis of the potent p38α mitogen-activated protein kinase inhibitors 2-(2-hydroxyethylsulfanyl)-4-(4-fluorophenyl)-5-(2-aminopyridin-4-yl)imidazole (3) and 2-(2,3-dihydroxypropylsulfanyl)-4-(4-fluorophenyl)-5-(2-aminopyridin-4-yl)imidazole (4) starting from 2-fluoro-4-methylpyridine is reported. In contrast to a previously published synthesis starting from 2-bromo-4-methylpyridine, the overall yield could be increased from 3.6% to 29.4%. Moreover, this strategy avoids the use of palladium as a catalyst and is more diverse and versatile. Using this optimized protocol, both enantiomers of potent inhibitor 3 were synthesized. Biological data demonstrated that the (S)-enantiomer is the two times more potent eutomer.
Introduction
The p38α mitogen-activated protein (MAP) kinase is a serine/threonine kinase, which links extracellular signals to the intracellular machinery modulating a plethora of cellular processes, e.g., the release of pro-inflammatory cytokines like tumor necrosis factor-α (TNF-α) and interleukin-1β.1 Therefore, the inhibition of p38α MAP kinase was evaluated as a therapeutic strategy for the treatment of cytokine-driven diseases like rheumatoid arthritis or psoriasis. Other studies suggest an important role of p38α MAP kinase in the pathogenesis of neurodegenerative diseases like Parkinson's disease, Alzheimer's disease or multiple sclerosis.2–4 Recently, several clinical trials investigating inhibitors of p38α MAP kinase for the treatment of chronic obstructive pulmonary disease were terminated.5,6 Trisubstituted pyridinylimidazoles, like the prototypical inhibitor SB203580 (1) or ML3403 (2), are amongst the most prominent adenosine triphosphate (ATP) competitive inhibitors of p38α MAP kinase (Fig. 1).7–9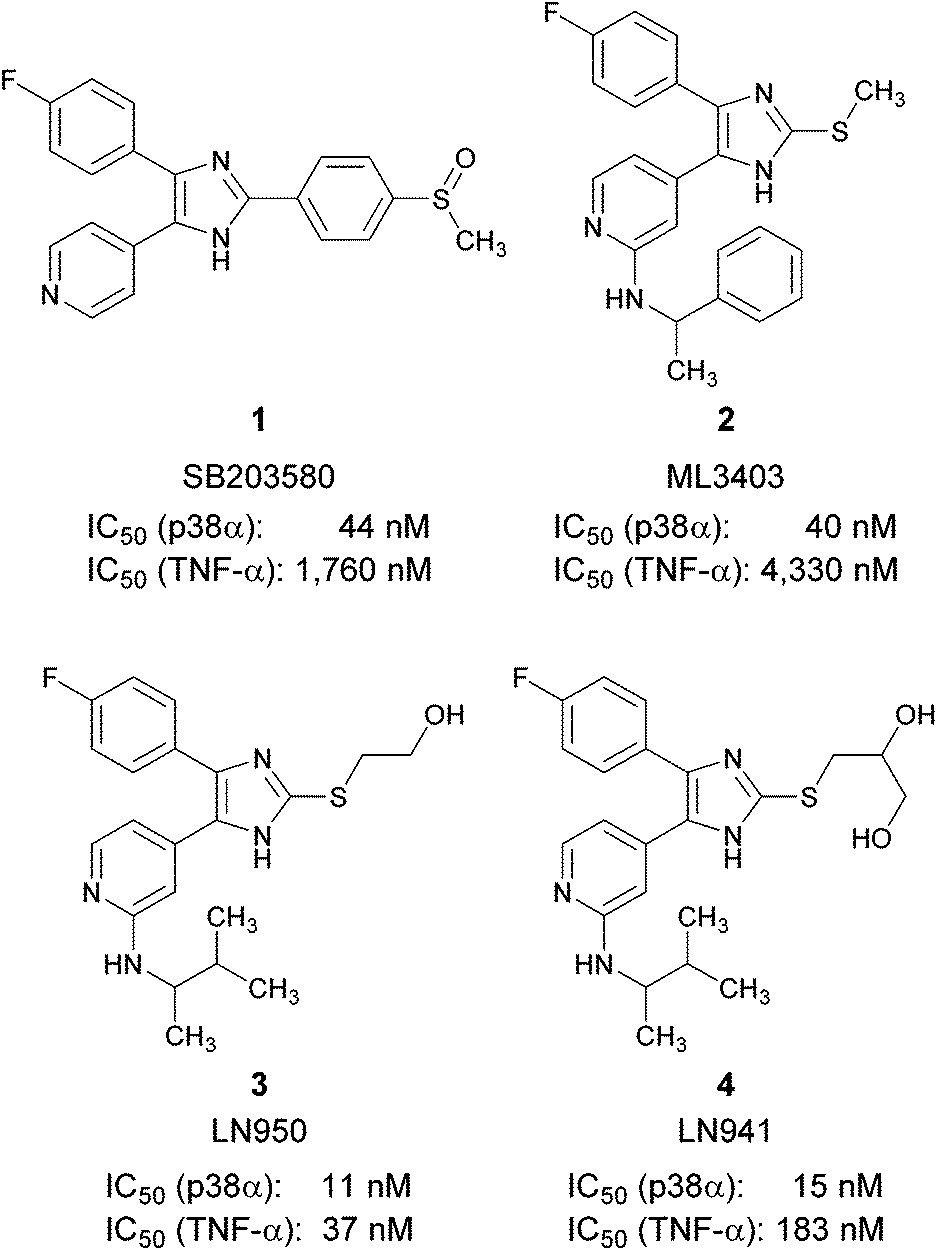 | ||
| Fig. 1 Pyridinylimidazole based p38α MAP kinase inhibitors. | ||
In 2008, we reported 2-(2-hydroxyethylsulfanyl)-4-(4-fluorophenyl)-5-(2-aminopyridin-4-yl)imidazole (3) and 2-(2,3-dihydroxypropylsulfanyl)-4-(4-fluorophenyl)-5-(2-aminopyridin-4-yl)imidazole (4) as optimized p38α MAP kinase inhibitors derived from ML3403 (Fig. 1).10 The hydroxyl containing moieties at the imidazole C2-position may interact with the ribose and the phosphate binding site of the enzyme. In contrast to the parent compound ML3403, compounds 3 and 4 show improved p38α MAP kinase inhibitory activity and a strong increase in inhibition of LPS-stimulated TNF-α release from human whole blood (in case of 3, >110-fold increase compared to 2). In order to profile these inhibitors in further in vitro and in vivo experiments, a high yielding and scalable synthesis is required.
The previously reported synthetic route toward inhibitors 3 and 4 starts from 2-bromo-4-methylpyridine (5) and is depicted in Scheme 1.10,11 In the first step of this synthesis, the amino moiety was introduced at the pyridine C2-position via Buchwald–Hartwig arylamination and the obtained secondary amine was directly protected with the Boc-group. Picoline 6 was converted into the ethanone 7 by reaction with ethyl 4-fluorobenzoate using NaHMDS as a base. Upon treatment with an excess of sodium nitrite in acetic acid, ethanone 7 was converted into the α-hydroxyiminoketone 8. Reduction of oxime 8 to a corresponding amine hydrochloride 9 accompanied by deprotection of the Boc-group was achieved by hydrogenation in methanolic hydrogen chloride using Pd/C as a catalyst. Cyclization was accomplished by treatment with potassium thiocyanate. Finally, the thione 10 was converted into the target compounds 3 and 4 by nucleophilic substitution reactions with 2-bromoethanol and 3-bromopropane-1,2-diol, respectively. The total yield of this synthetic strategy to pyridinylimidazoles 3 and 4 starting from 2-bromo-4-methylpyridine (5) is 3.6% and 3.9% (7 linear steps), respectively.
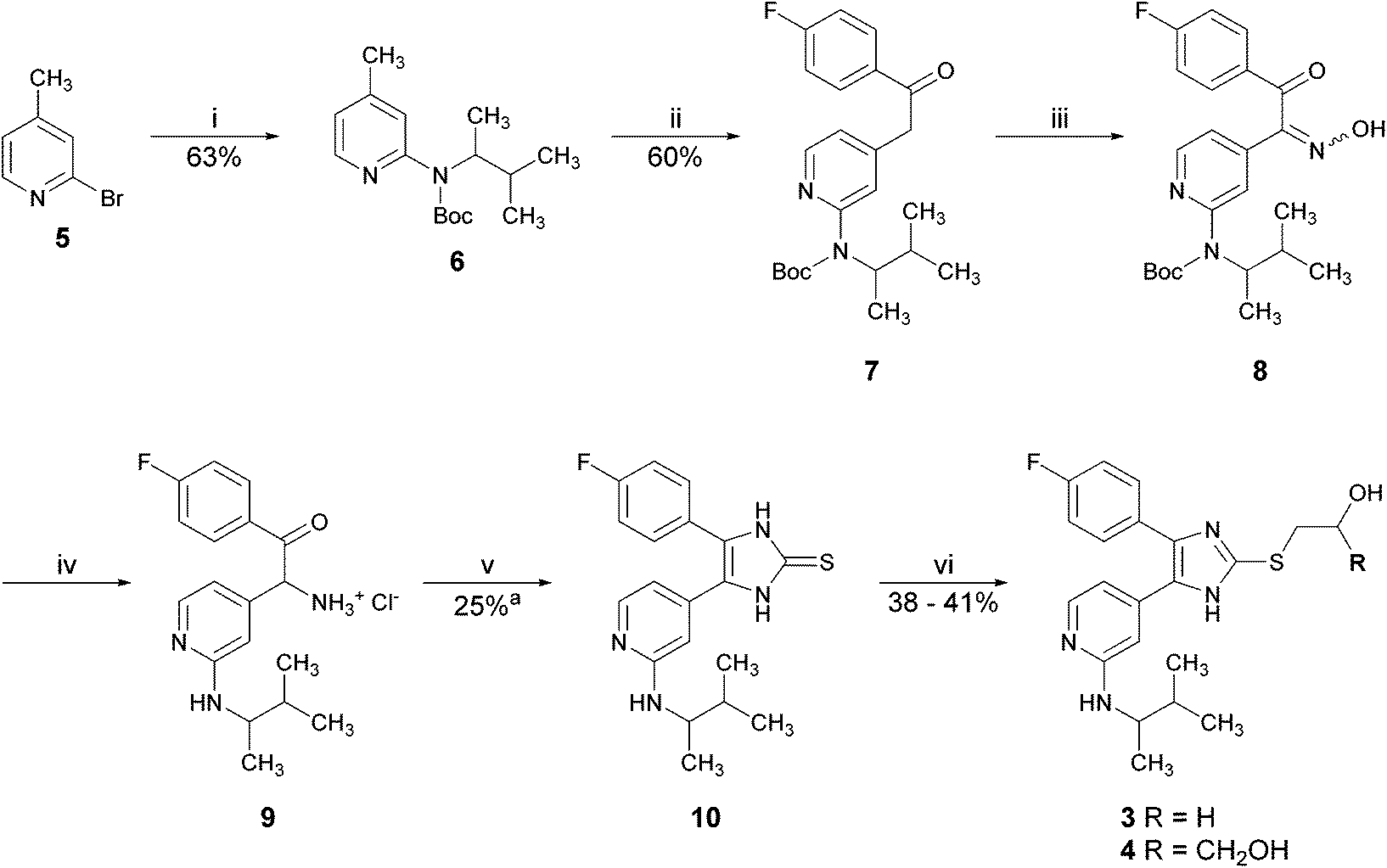 | ||
| Scheme 1 Reported synthesis of pyridinylimidazoles 3 and 4. Reagents and conditions: (i) 1. 3-methyl-2-butylamine (rac.), Pd2(dba)3, t-BuONa, BINAP, toluene, reflux temperature; 2. Boc2O, DMAP, CH2Cl2, r.t.; (ii) NaHMDS, ethyl 4-fluorobenzoate, THF, 0 °C – r.t.; (iii) NaNO2, acetic acid, 10 °C – r.t.; (iv) Pd/C 10%, MeOH/HCl, H2, atmospheric pressure, r.t.; (v) KSCN, DMF, reflux temperature, 3 h; (vi) 2-bromoethanol, t-BuOK, reflux temperature, 1.5 h in case of compound 3 or 3-bromopropane-1,2-diol, t-BuOK, reflux temperature, 3 h in case of compound 4; ayield over three steps, compounds 8 and 9 were not isolated. | ||
The major limitations of this synthetic strategy for scale-up are the introduction of the amino moiety in the first step of the synthesis, which requires the addition and removal of a protecting group and its low overall yield (>4%). Moreover, the required early stage use of the palladium catalyst is quite cost intensive due to the higher amount needed in scale-up.
Another attempted synthetic strategy to the target compounds 3 and 4 reported from our group is depicted in Scheme 2.12 The introduction of the 3-methyl-2-butylamino moiety at the pyridine C2-position as well as the moiety at the imidazole C2–S-position was envisaged in the last two steps of the synthesis. The key compound for this route 4-(4-fluorophenyl)-5-(2-fluoropyridin-4-yl)-1,3-dihydroimidazole-2-thione (12) was prepared according to a protocol from Laufer and co-workers8,13 in four steps starting from 2-fluoro-4-methylpyridine (11) in an overall yield of 57% (for details, see Scheme S1 in the ESI†). The alkylsulfanyl moiety was introduced by nucleophilic substitution of thione 12 and the appropriate alkyl halide. In the last step, the fluorine atom in 13a and 13b should be displaced by 3-methyl-2-butylamine (14) to obtain 3 and 4, respectively. By heating 13a or 13b with the amine 14, we observed an unexpected transformation of 2-alkylsulfanylimidazoles bearing a 2-hydroxyethyl or a 2,3-dihydroxypropyl moiety at the imidazole C2–S-position into the imidazol-2-one derivative 15.
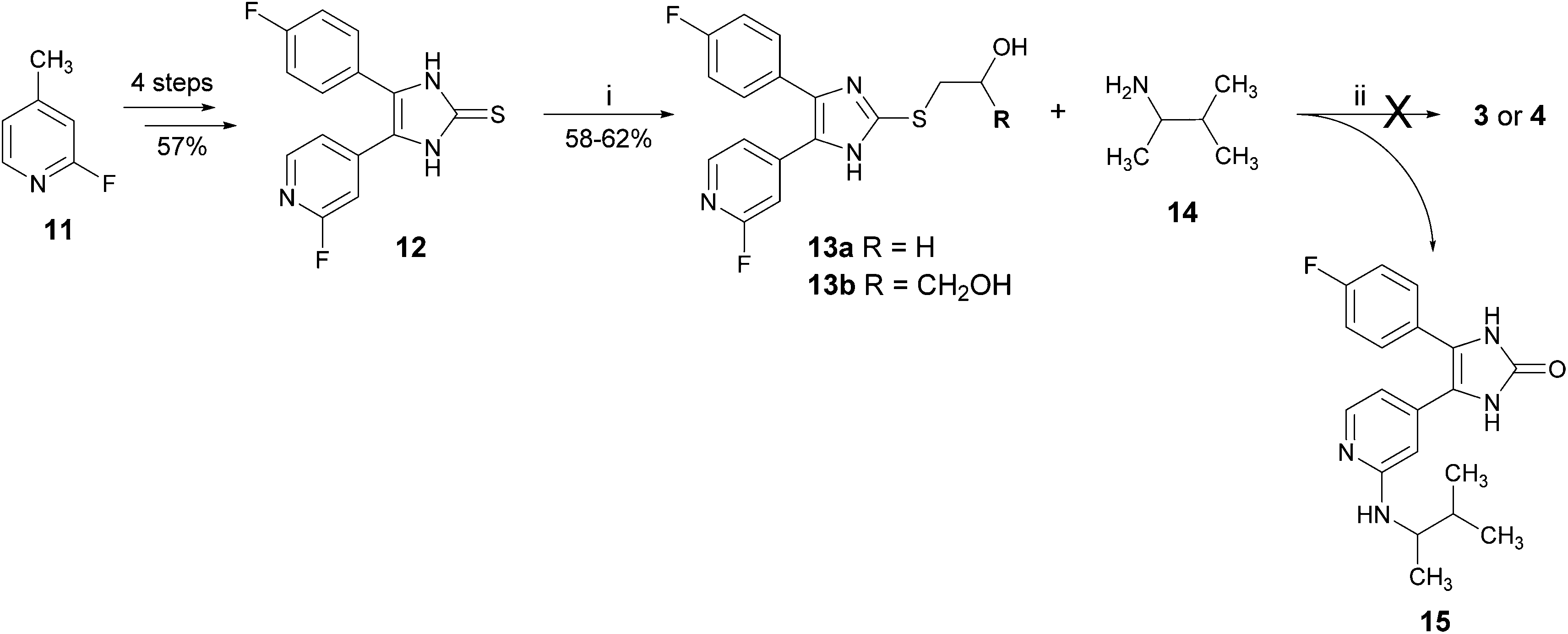 | ||
| Scheme 2 Unexpected reaction of 2-alkylimidazoles 13a and 13b into imidazol-2-ones. Reagents and conditions: (i) EtONa, methanol, 2-bromoethanol, r.t., 4 h (for synthesis of 13a) or EtONa, methanol, 3-bromopropane-1,2-diol, r.t., 16 h (for synthesis of 13b); (ii) 160 °C, 10 h, sealed tube. | ||
Herein, we report a distinct and optimized procedure to prepare pyridinylimidazoles 3 and 4 in higher yields and with increased versatility compared to the previously published protocols. Moreover, the described synthetic strategy avoids the use of palladium as a catalyst for introduction of the amino moiety at the pyridine C2-position.
Results & discussions
In order to avoid the aforementioned conversion of 2-alkylsulfanylimidazoles bearing a 2-hydroxyethyl or a 2,3-dihydroxypropyl moiety at the imidazole C2–S-position into the corresponding imidazol-2-one derivatives, we pursued the strategic introduction of a hydroxyl protecting group at the imidazole C2–S moiety prior to this step and its cleavage in the subsequent step (Scheme 3).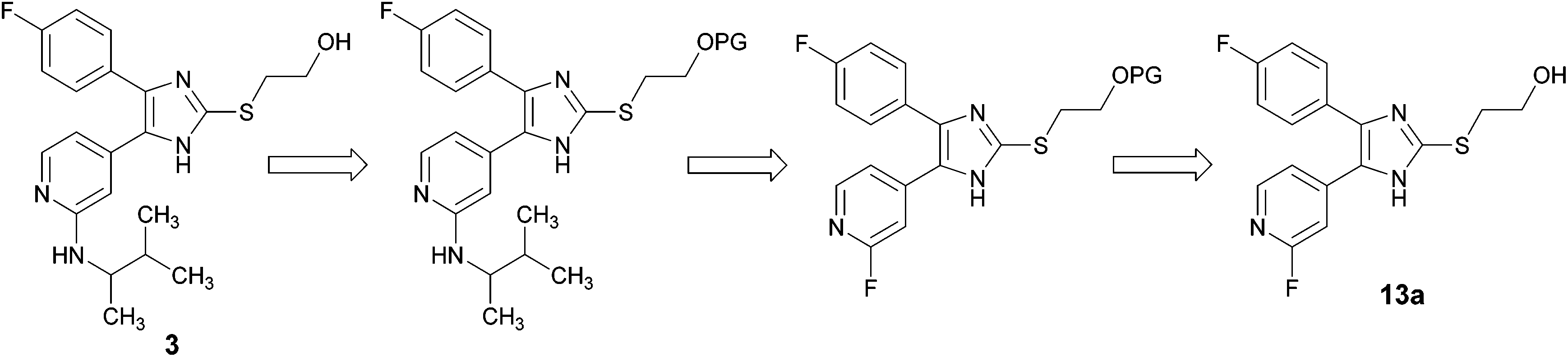 | ||
| Scheme 3 Retrosynthetic approach for synthesis of 3 (PG, protecting group). | ||
Initially, we attempted to make use of an acetyl ester to protect the hydroxy function (Scheme 4).14 Therefore, imidazole-2-thione 12 was treated with 2-bromoethyl acetate under mildly basic conditions in order to obtain ester 16, bearing the acetyl protected hydroxyethyl moiety at the imidazole C2–S-position. Compound 16 was reacted with 3-methyl-2-butylamine in a nucleophilic aromatic substitution reaction. Under these harsh and basic conditions (160 °C, excess of amine), the acetyl protecting group was cleaved and imidazol-2-one 15 was isolated as the major product of this reaction. Decrease of both, reaction temperature and excess of amine resulted in a slow or no conversion.
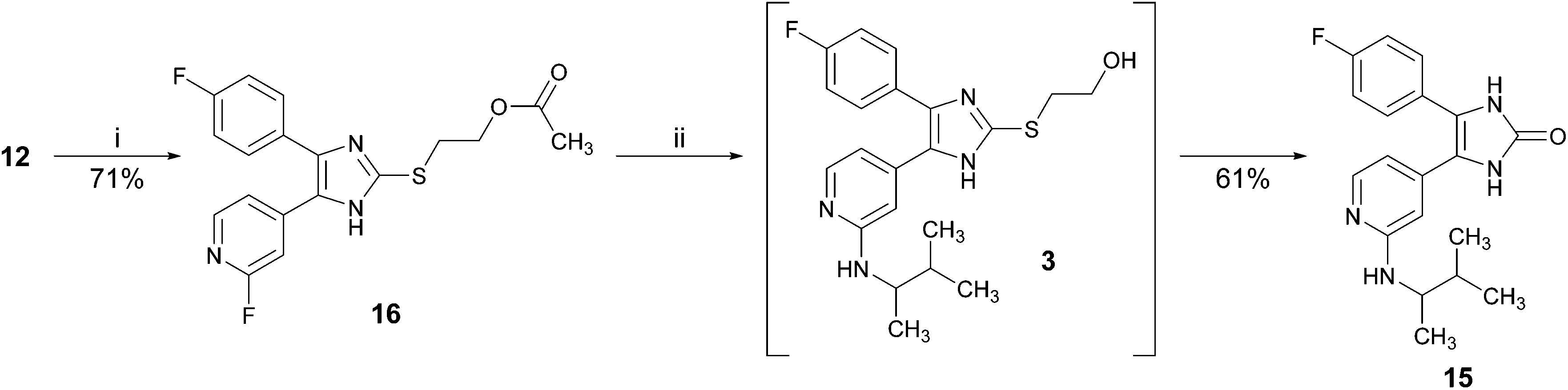 | ||
| Scheme 4 Conversion of ester 16 to imidazol-2-one 15. Reagents and conditions: (i) 2-bromoethyl acetate, Cs2CO3, DMF, 55 °C, 30 min; (ii) 3-methyl-2-butylamine (rac.), 160 °C, 16 h. | ||
As another protecting group for the hydroxy function, we used the acid labile but base stable tetrahydropyranyl (THP) group (Scheme 5).15 Imidazole-2-thione 12 was reacted with 2-(2-bromoethoxy)-tetrahydro-2H-pyran to give compound 17 in good to moderate yields. In the next step, the amino function at the pyridine C2-position was introduced via nucleophilic aromatic substitution under the above mentioned conditions. To our delight, we found the THP-group to be sufficiently stable and compound 18, the THP-protected derivative of 3, was obtained in good yields. Finally, treatment of 18 with ethanolic hydrochloric acid solution resulted in cleavage of the THP group and the target compound 3 was obtained in quantitative yields.
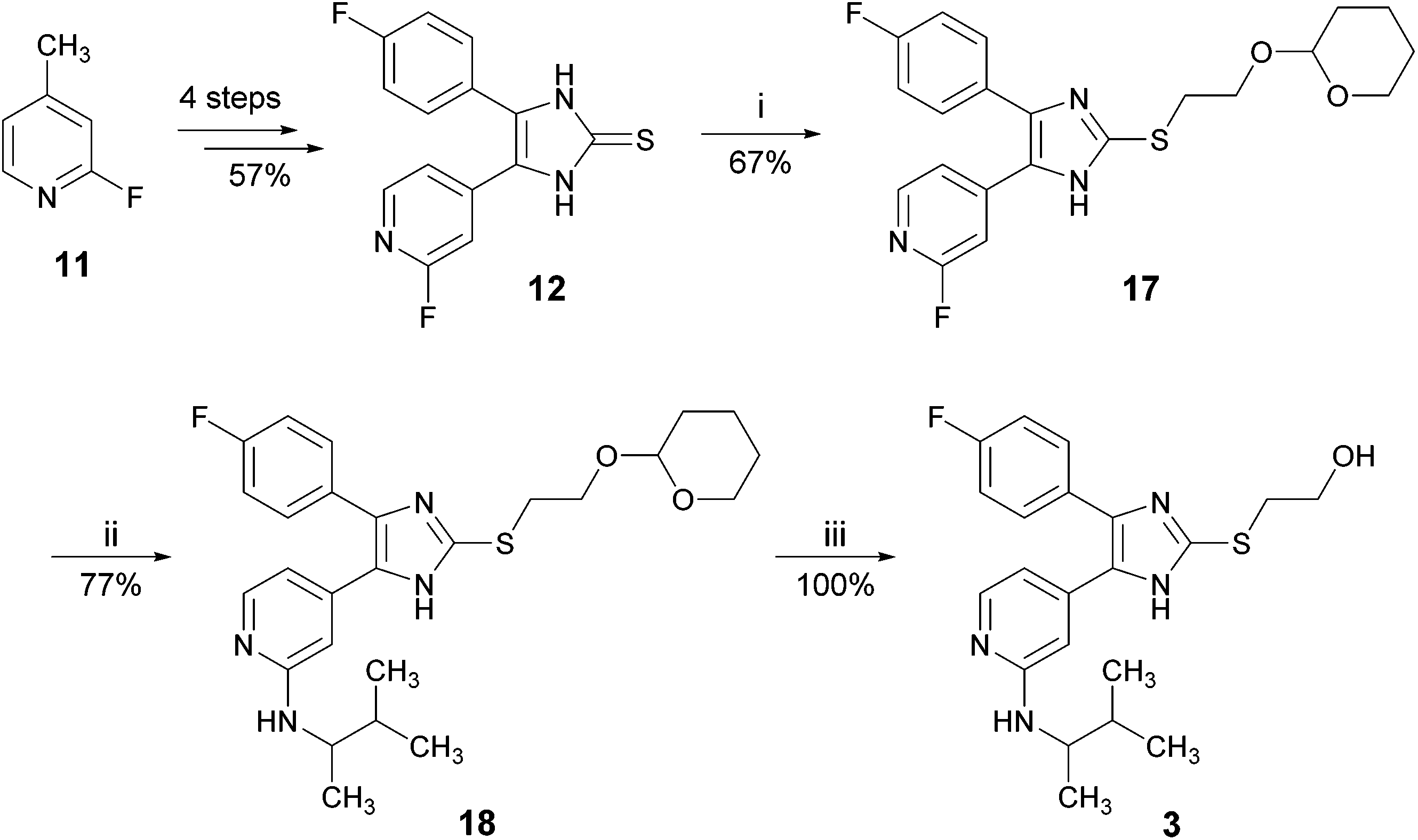 | ||
| Scheme 5 Optimized synthesis of 3 using THP as the hydroxyl protecting group. Reagents and conditions: (i) t-BuONa, methanol, 2-(2-bromoethoxy)-tetrahydro-2H-pyran, 30 min, 50 °C; (ii) 3-methyl-2-butylamine (rac.), 160 °C, 16 h, sealed tube; (iii) 1.25 M HCl/EtOH, 3 h, r.t. | ||
With a total yield of 29.4% in 7 linear steps, the optimized synthesis of compound 3 starting from 2-fluoro-4-methylpyridine (11) compares very well to the reported synthesis starting from 2-bromo-4-methylpyridine (5), giving more than eight times higher yields (compare Scheme 5vs.Scheme 1). Using this protocol, 2.3 g of inhibitor 3 were synthesized starting from 3.0 g imidazole-2-thione 12 in similar yields as reported in Scheme 5 (for details, see ESI†).
For optimizing the synthesis of inhibitor 4, we used an acetal group to protect the 1,2-dihydroxy function.16 Acetals are generally stable under basic conditions and liberate the diol under acidic conditions. The optimized route to 4 is depicted in Scheme 6. The dihydroxypropylsulfanylimidazole 13b was converted into the corresponding acetonide 19 by treatment with acetone under p-toluenesulfonic acid (p-TsOH) catalysis. In the next step, the amino function was introduced at the pyridine C2-position via nucleophilic aromatic substitution and finally the acetal protecting group was cleaved with diluted hydrochloric acid to yield inhibitor 4. The overall yield of this protocol starting from 11 is 20.2% (8 linear steps), which is five times higher than the previously reported synthesis starting from 5 (Scheme 1, total yield: 3.9%).
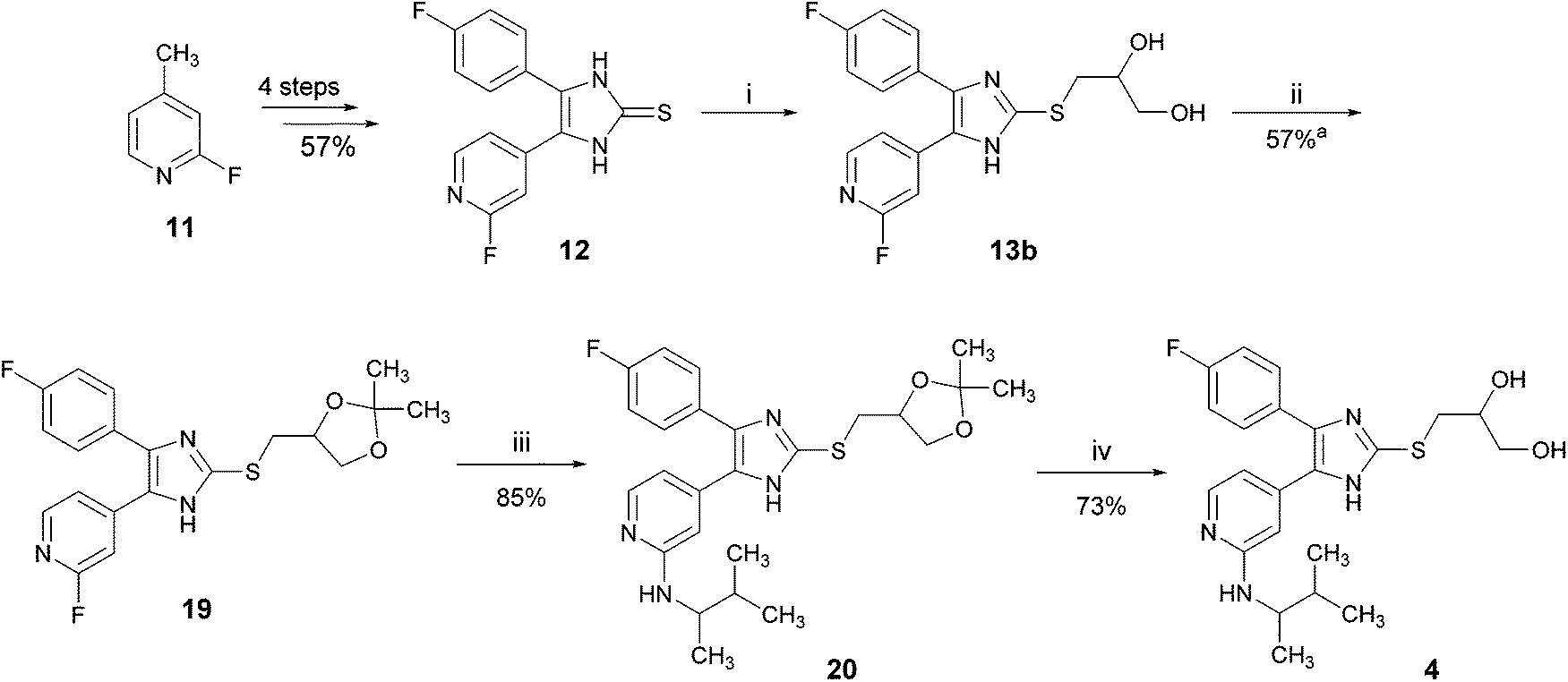 | ||
| Scheme 6 Optimized synthesis of 4 using acetal as the 1,2-dihydroxyl protecting group. Reagents and conditions: (i) t-BuONa, methanol, 3-bromopropane-1,2-diol, 70 °C, 1 h; (ii) acetone, p-TsOH, reflux temperature, 24 h; (iii) 3-methyl-2-butylamine (rac.), 160 °C, 16 h, sealed tube; (iv) 2 M aq. HCl, r.t., 3 h; ayield over two steps, compound 13b was not isolated. | ||
Both optimized synthetic strategies presented in Schemes 5 and 6 are versatile and diverse since the introduction of both, the imidazole C2–S moiety and the pyridine C2–amino function are in the last steps of the sequence. Therefore, these strategies allow a facile and high-yielding access to further pyridinylimidazole derivatives (Scheme 7, Schemes S2–S7, ESI†). Starting from intermediates 17 or 19, both enantiomers of 3 (compounds S2 and S4), the hydroxyethylsulfanyl and dihydroxypropylsulfanyl derivatives of ML3403 (S6 and S8) and both enantiomers of S6 (compounds S10 and S12) were prepared. The synthesized pyridinylimidazoles were tested in an ELISA-assay for their ability to inhibit the p38α MAP kinase (Table 1).17 Furthermore, the potential to inhibit the LPS-stimulated TNF-α release from human whole blood was determined for S2 and S4, the most promising inhibitors in this set, and compared to the racemic mixture, compound 3. In case of the chiral inhibitors S2, S4, S10 and S12, the biological data revealed the (S)-enantiomer as the eutomer. Compared to the racemic inhibitor 3, the LPS-stimulated TNF-α-release is inhibited two times more by the (S)-enantiomer S4, whereas, the (R)-enantiomer S2 is as expected two times less active in this assay.
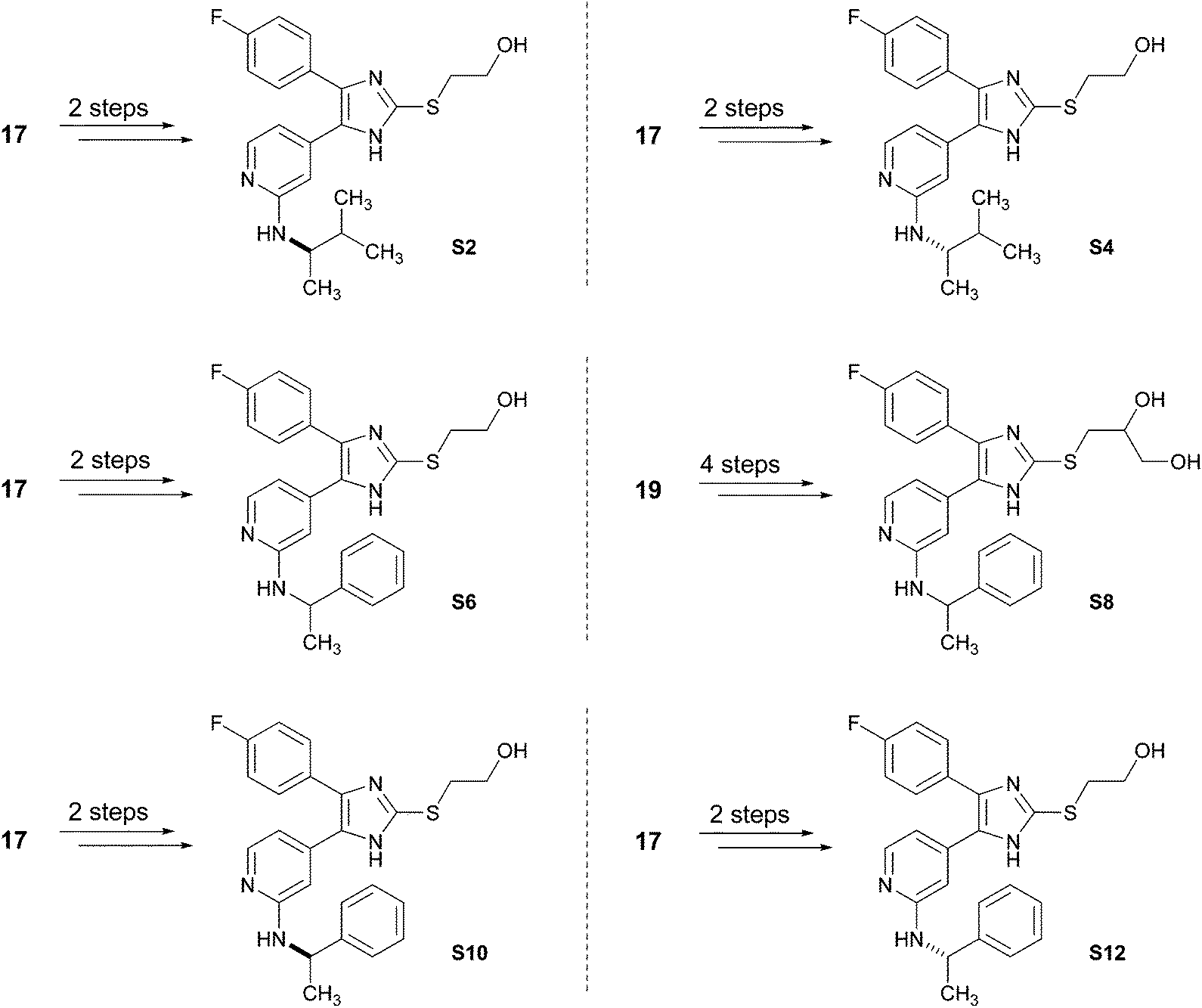 | ||
| Scheme 7 Synthesis of pyridinylimidazoles S2, S4, S6, S8, S10 and S12. For reagents and conditions, see the ESI.† | ||
| Cmpd | IC50 ± SEM | |
|---|---|---|
| p38α MAP kinasea | TNF-α releaseb | |
| a Results are from three experiments. b Tests were carried out in duplicate. c Test data are from Koch et al.10 d nd. not determined. | ||
| 3 (LN950) | 11 ± 1 nMc | 37 ± 4 nMc |
| 4 (LN941) | 15 ± 2 nMc | 183 ± 16 nMc |
| S2 [(R)-3] | 19 ± 2 nM | 84 ± 3 nM |
| S4 [(S)-3] | 14 ± 2 nM | 20 ± 3 nM |
| S6 | 32 ± 3 nM | ndd |
| S8 | 41 ± 7 nMc | 936 ± 15 nMc |
| S10 [(R)-6] | 42 ± 1 nM | nd |
| S12 [(S)-6] | 22 ± 0.2 nM | nd |
Conclusion
An optimized and diverse synthetic approach for the preparation of pyridinylimidazole-based p38α MAP kinase inhibitors is described. Compared to a previously reported method, an up to eight times higher overall yield was obtained.A series of eight pyridinylimidazoles was synthesized using these optimized conditions. In case of the chiral inhibitor 3, biological evaluation showed that the (S)-enantiomer is the two times more potent eutomer.
Acknowledgements
S. Bauer, M. Goettert, D. Müller, and K. Bauer are gratefully acknowledged for their assistance in biology testing.Notes and references
- Z. Chen, T. B. Gibson, F. Robinson, L. Silvestro, G. Pearson, B. E. Xu, A. Wright, C. Vanderbilt and M. H. Cobb, Chem. Rev., 2001, 101, 2449–2476 CrossRef CAS PubMed.
- L. Munoz and A. J. Ammit, Neuropharmacology, 2010, 58, 561–568 CrossRef CAS PubMed.
- K. Namiki, H. Matsunaga, K. Yoshioka, K. Tanaka, K. Murata, J. Ishida, A. Sakairi, J. Kim, N. Tokuhara, N. Shibakawa, M. Shimizu, Y. Wada, Y. Tokunaga, M. Shigetomi, M. Hagihara, S. Kimura, T. Sudo, A. Fukamizu and Y. Kasuya, J. Biol. Chem., 2012, 287, 24228–24238 CrossRef CAS PubMed.
- G. Wang, J. Pan and S. D. Chen, Prog. Neurobiol., 2012, 98, 207–221 CrossRef CAS PubMed.
- W. MacNee, R. J. Allan, I. Jones, M. C. De Salvo and L. F. Tan, Thorax, 2013, 68, 738–745 CrossRef PubMed.
- J. C. Betts, R. J. Mayer, R. Tal-Singer, L. Warnock, C. Clayton, S. Bates, B. E. Hoffman, C. Larminie and D. Singh, Pharmacol. Res. Perspect., 2015, 3, e00094 Search PubMed.
- C. Peifer, G. Wagner and S. Laufer, Curr. Top. Med. Chem., 2006, 6, 113–149 CrossRef CAS.
- S. A. Laufer, G. K. Wagner, D. A. Kotschenreuther and W. Albrecht, J. Med. Chem., 2003, 46, 3230–3244 CrossRef CAS PubMed.
- S. Laufer, D. Hauser, T. Stegmiller, C. Bracht, K. Ruff, V. Schattel, W. Albrecht and P. Koch, Bioorg. Med. Chem. Lett., 2010, 20, 6671–6675 CrossRef CAS PubMed.
- P. Koch, C. Bäuerlein, H. Jank and S. Laufer, J. Med. Chem., 2008, 51, 5630–5640 CrossRef CAS PubMed.
- S. Laufer and P. Koch, Org. Biomol. Chem., 2008, 6, 437–439 CAS.
- P. Koch and S. Laufer, J. Med. Chem., 2010, 53, 4798–4802 CrossRef CAS PubMed.
- S. A. Laufer and A. J. Liedtke, Tetrahedron Lett., 2006, 47, 7199–7203 CrossRef CAS PubMed.
- P. G. M. Green and T. W. Wuts, in Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, 3rd edn, 1999, pp. 150–160, 712–715 Search PubMed.
- P. G. M. Green and T. W. Wuts, in Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, 3rd edn, 1999, pp. 27–33, 708–711 Search PubMed.
- P. G. M. Green and T. W. Wuts, in Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, 3rd edn, 1999, pp. 207–215, 716–719 Search PubMed.
- M. Goettert, R. Graeser and S. A. Laufer, Anal. Biochem., 2010, 406, 233–234 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Schemes S1–S7, experimental procedures and analytical data, copies of 1H NMR and 13C NMR. See DOI: 10.1039/c5ob01505g |
| This journal is © The Royal Society of Chemistry 2015 |