
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted synthesis of 3-aminobenzo[b]thiophene scaffolds for the preparation of kinase inhibitors†
Mark C.
Bagley
*a,
Jessica E.
Dwyer
a,
Maria D. Beltran
Molina
b,
Alexander W.
Rand
a,
Hayley L.
Rand
a and
Nicholas C. O.
Tomkinson
b
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Falmer, Brighton, East Sussex, BN1 9QJ, UK. E-mail: m.c.bagley@sussex.ac.uk
bWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK
First published on 12th May 2015
Abstract
Microwave irradiation of 2-halobenzonitriles and methyl thioglycolate in the presence of triethylamine in DMSO at 130 °C provides rapid access to 3-aminobenzo[b]thiophenes in 58–96% yield. This transformation has been applied in the synthesis of the thieno[2,3-b]pyridine core motif of LIMK1 inhibitors, the benzo[4,5]thieno[3,2-e][1,4]diazepin-5(2H)-one scaffold of MK2 inhibitors and a benzo[4,5]thieno[3,2-d]pyrimidin-4-one inhibitor of the PIM kinases.
Introduction
Benzothiophenes are naturally-occurring heterocycles, found in petroleum deposits in their simplest form but also discovered recently as a motif in more complex glycosides isolated from the roots of E. grijissii.1 Benzothiophenes are important components of organic semiconductors due to their potential for elongated and highly delocalised electronic structures.2,3 Substituted benzothiophenes have also found application in drug discovery as highly-privileged structures and valuable building blocks in medicinal chemistry, being incorporated into tubulin polymerisation inhibitors,4,5 acetyl-CoA carboxylase inhibitors,6 antidepressants,7 and as estrogen receptor modulators.8,9 Benzothiophenes are present in a number of clinical agents, including Raloxifene,10 a selective estrogen receptor modulator, Zileuton,11 an inhibitor of 5-lipoxygenase and leukotriene biosynthesis used for the treatment of asthma, and the antifungal agent Sertaconazole, which inhibits the synthesis of ergosterol.12Scaffolds based upon 2- or 3-aminobenzo[b]thiophenes have enormous potential for further derivatization and have shown great promise in fragment-based drug discovery and in hit identification or lead development, including approaches towards antimitotic agents5,13 and in the development of inhibitors of kinase targets, such as the LIMK protein family,14 PIM-kinases15 and MAPK-2 kinase (MK2) (Fig. 1).16,17 A number of 3-aminothieno[2,3-b]pyridine-2-carboxamide hits, such as 1, were identified from high throughput screening (HTS) as inhibitors of LIMK1, leading to the development of tricyclic derivatives such as 2a and the benzothieno[3,2-d]pyrimidine 2b as a LIMK1 inhibitor lead candidate, to disrupt actin polymerisation and thus prevent the metastatic potential of tumour cells where LIMK is over-expressed.14
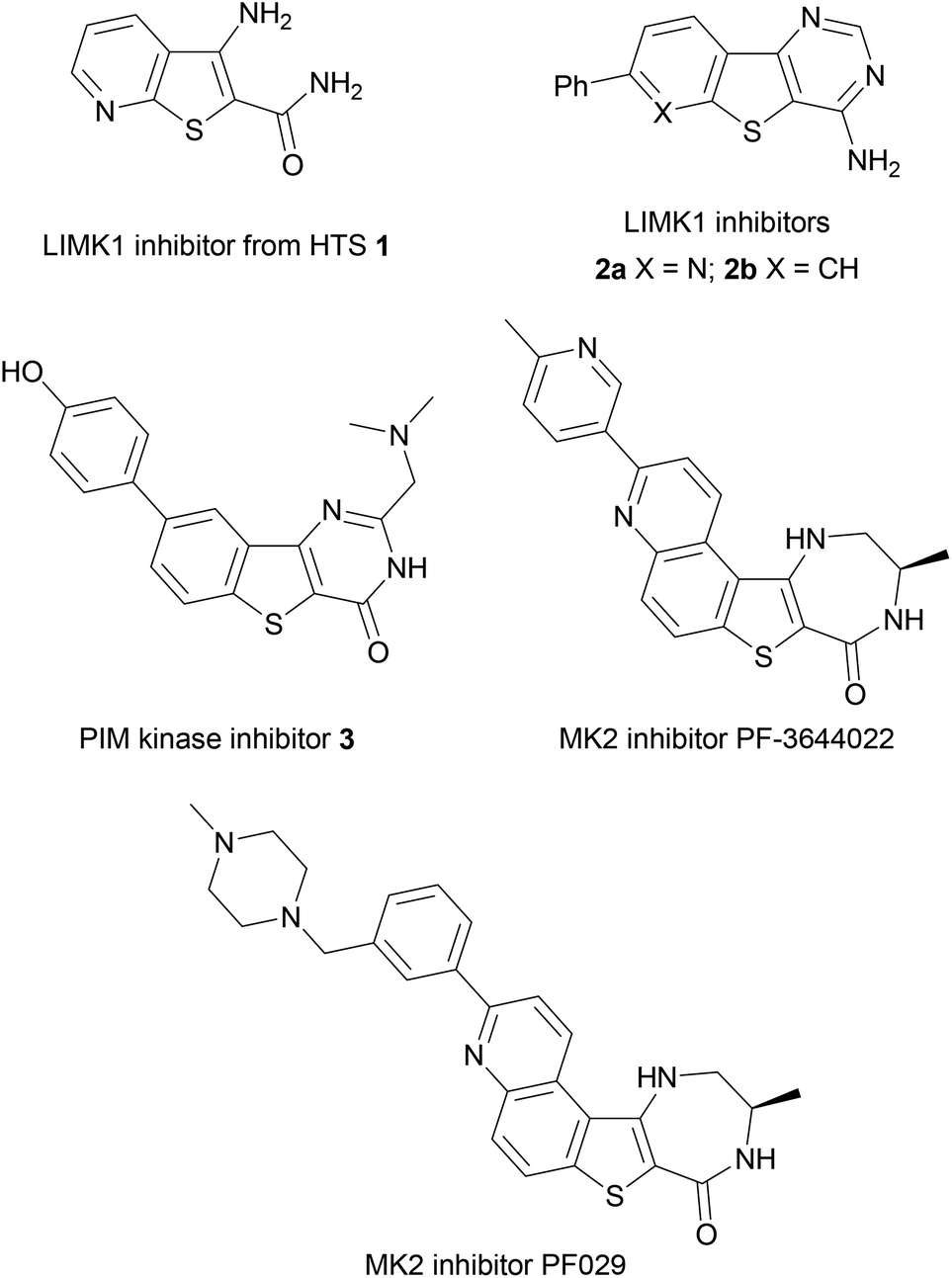 | ||
| Fig. 1 Aminobenzothiophene scaffolds in drug discovery. | ||
Benzothienopyrimidinones have been investigated as PIM kinase inhibitors.15 The PIM kinases (PIM1, PIM2 and PIM3) have been implicated in tumourigenesis and simultaneous targeting of all three isoforms has presented itself as a promising approach in cancer therapy, with PIM triple knockout mice found to be viable and fertile.18 The benzothiophene scaffold was again identified from an initial HTS hit,15 leading to the development of a range of potent and selective benzo[b]thiophene-derived inhibitors such as 3 with nM activity (Ki values of 2, 3 and 0.5 nM against PIM1, PIM2 and PIM3, respectively) with oral bioavailability in mouse models.
Examples of aminobenzothiophene derivatives are also found amongst inhibitors of the mitogen activated protein kinase (MAPK) family of enzymes. These enzymes are essential for inflammatory cell signalling events and contain historically popular drug targets for inflammatory diseases, including rheumatoid arthritis and Crohn's disease because of their involvement in the production of pro-inflammatory cytokines,19 as well as being implicated in accelerated cellular ageing in Werner syndrome (WS) via p38.20–23 MAPK-activated protein kinase (MK2) is a rate-limiting kinase downstream of p38 in the MAPK pathway and has been the subject of many studies in recent years,24 as MK2 knock-out mice possess normal healthy phenotypes whereas p38 knock-out mice are lethal.25 The aminobenzo[b]thiophene derivative PF-3644022 shows excellent kinase selectivity for MK2, in vivo potency on a nanomolar scale and projected ADME characteristics that suggested it was suitable for oral human dosing.17,26,27 However, PF-3644022 was found to result in hepatotoxicity in dogs27 and so the analogue PF029 was developed and exhibited an improved toxicological profile with no loss of cellular potency. This was rationalized through installation of a metabolic shunt onto the reactive diazepinone ring and extension of the biaryl ring section to increase the compound's cationic character, thus reducing its molecular affinity for transporter proteins.
As part of our interest in the synthesis of MAPK inhibitors for the study of cellular ageing in Werner syndrome,20,28–33 the benzothiophene scaffold, and its selectivity and cellular activity profile for MK2 exemplified by PF-3644022, made it an attractive target for synthesis. We have shown that treating young WS cell cultures with p38 MAPK inhibitors can bring about a complete reversal of the ageing phenotype, giving increased replicative life-span, growth rates comparable to normal young cells and a reduction in levels of F-actin stress fibres.20,23 These findings suggested that WS could be amenable to therapeutic intervention, but with high toxicity and poor kinase selectivity exhibited in vivo by many p38 inhibitors,34–36 an inhibitor scaffold that targeted the downstream kinase MK2 would offer a promising alternative target.20,37
With such a range of biological properties, there is a continuing interest in the search for new methods to access substituted benzothiophenes.38 One approach, with the potential to incorporate diversity into a target library, would be to employ transition metal-mediated processes from the corresponding 3-halobenzo[b]thiophenes.39 However, methods for the synthesis of 3-halobenzo[b]thiophenes are currently fairly limited. In particular, routes can be problematic when using ring halogenation due to the low reactivity of the heteroaryl unit and its functional group compatibility.40–43 The 5-endo-dig halocyclisation of ortho-alkynylaryl thiophenol derivatives offers an alternative approach,44–48 but this requires installation of an alkyne by metal-catalyzed cross-coupling followed by cyclisation, mediated by a halogen-containing electrophile, so can exhibit a number of inherent disadvantages.
Herein, we present an annulation-based method for the rapid preparation of 3-halo and 3-amino-2-substituted benzo[b]thiophenes suitable for elaboration to a range of kinase inhibitors.49 It employs microwave irradiation as a convenient platform for fast reaction kinetics, and to improve reaction efficiency, and avoids the need for metal-catalyzed processes to establish the parent heterocycle. This method is shown to be suitable to access the pharmacophore of a range of biologically-active scaffolds for application in medicinal chemistry and drug discovery.
Results and discussion
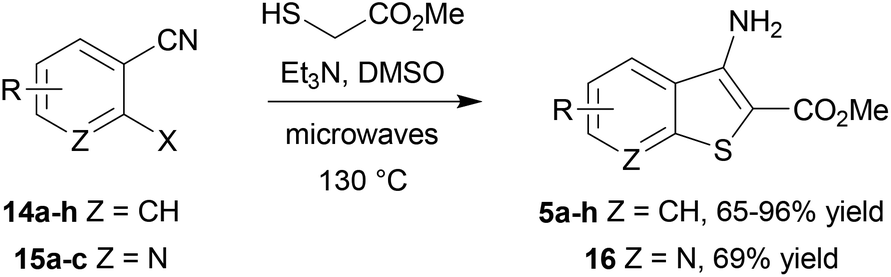
The benzothiophene-containing chemotypes appearing in recent drug discovery programmes (Fig. 1) feature, or could in principle be derived from, electron-poor aminobenzothiophene intermediates or their 7-aza analogues (Scheme 1). For example, the PIM kinase inhibitor scaffold 4 has been accessed from 5-bromobenzothiophene 5a, using the halogen as a handle for library diversification in a late-stage Suzuki coupling.15 Similarly, it could be hypothesized that inhibitors of MK2 for study in WS cells, such as PF-3644022 (Fig. 1), could be prepared from the same core motif 5, using 5-nitrobenzothiophene 5b, rather than by the functionalization of 6-nitroquinoline as reported by Anderson et al.17 This approach would enable the synthesis of a range of diverse chemical tools from a single common template. The original route to MK2 hit compound 6a, prior to the development of PF-3644022,16 employed cinnamic acid 8a in reaction with thionyl chloride in chlorobenzene at 120 °C to establish the 3-chlorobenzothiophene scaffold 7a (Scheme 1).50 Unfortunately this route would be wholly inappropriate for the synthesis of benzothiophene 7b for elaboration to the desired intermediate 6b on route to PF-3644022, as altering the substituent-directing effects to a nitro group results in poor yields and inseparable mixtures of 7b and 7c in the benzothiophene synthesis, as well as giving other side products, as reported by Higa.51 Hence an alternative route had to be sought.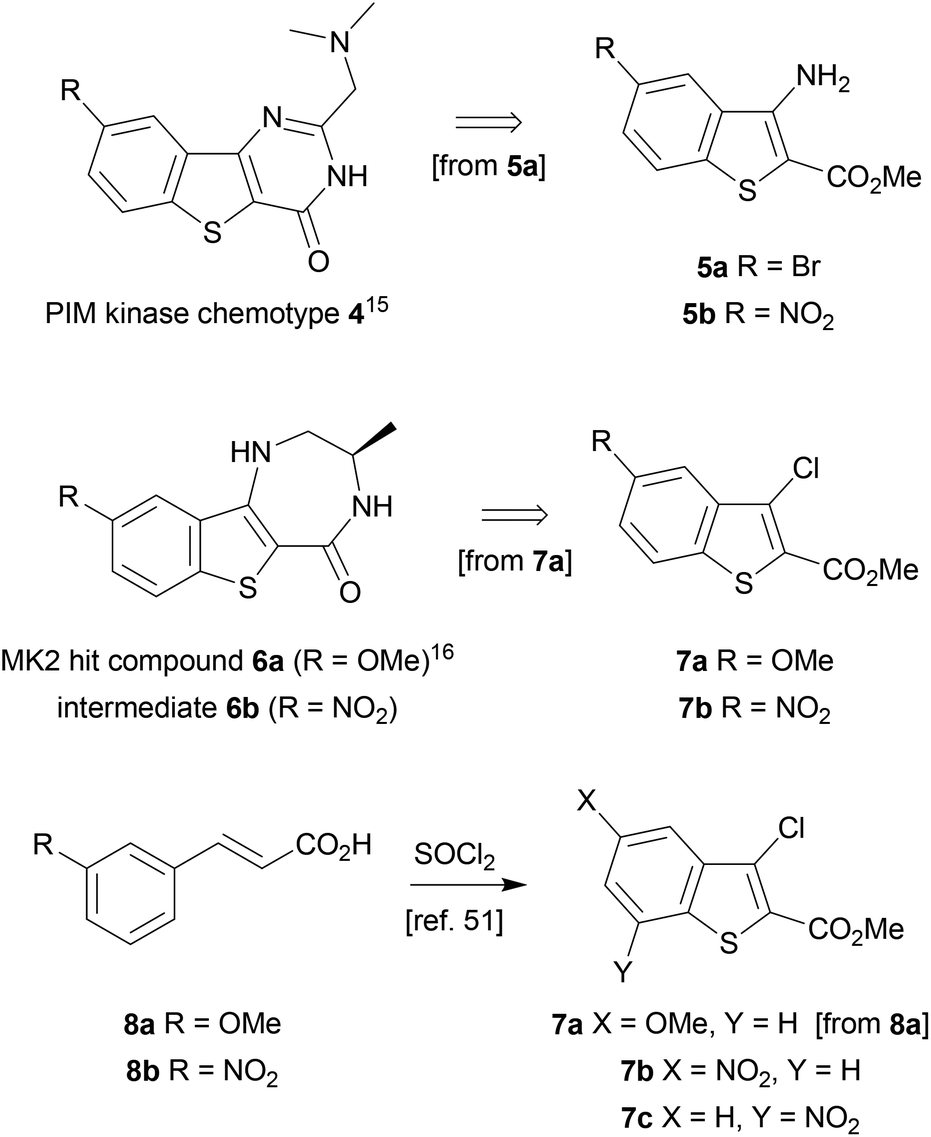 | ||
| Scheme 1 Benzothiophene precursors to kinase inhibitors. | ||
Our first approach towards scaffold 6b used an alternative and established method to access 3-halobenzothiophenes by halogenation of the corresponding benzothiophene.40 The condensation of methyl thioglycolate with 2-chloro-5-nitrobenzaldehyde (9) under basic conditions gave methyl 5-nitrobenzo[b]thiophene-2-carboxylate (10) in high yield (Scheme 2). We have shown in previous work how microwave dielectric heating can be used to dramatically reduce reaction times in the synthesis of inhibitor scaffolds.30,32,33,37,52 Given that elevated temperature has promoted this,53,54 and a closely-related process for the synthesis of 2-acetylbenzothiophenes,55 we carried out this transformation under microwave irradiation at 90 °C to give the benzothiophene 10 in good yield, whilst shortening the reaction time from 17 h to 15 min. Selective halogenation at C-3 of 10 was facilitated in a convoluted sequence of reactions via the carboxylic acid 11 to overcome poor heterocycle reactivity.40 Saponification, again conducted by microwave dielectric heating, under basic conditions was complete in 3 min at 100 °C and gave the carboxylic acid 11 in excellent yield. Subsequent heating with excess bromine and sodium acetate in glacial acetic acid did give 3-bromobenzothiophene 13 in good yield after esterification using methyl iodide on a number of occasions. However, the bromination was found to be highly variable and efforts to develop an alternative process using microwave heating were constantly frustrated56 and so a more reliable and efficient route was sought. The poor yield of this bromination reaction is catalogued in a recent report.57
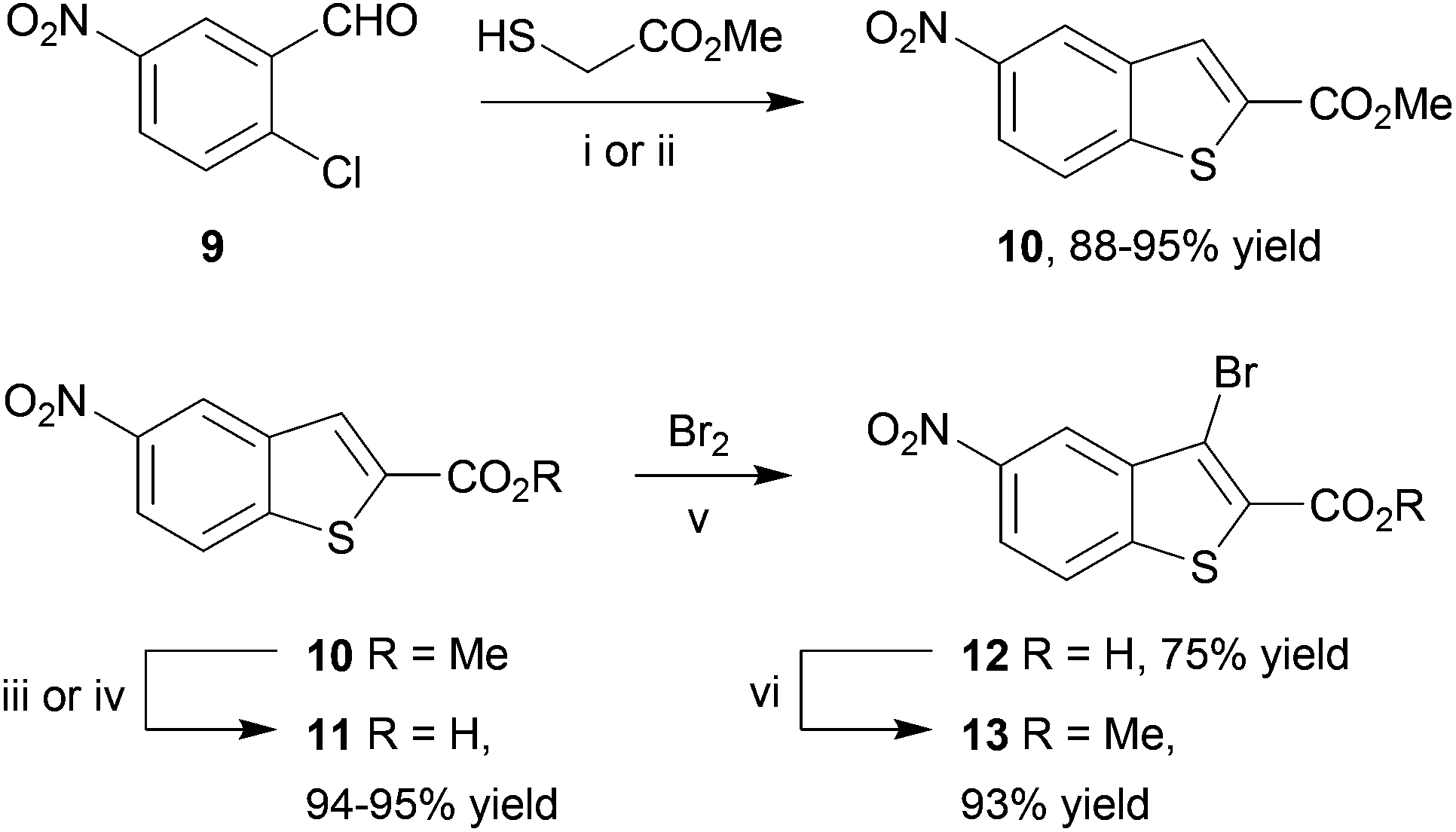 | ||
| Scheme 2 Reagents and conditions: (i) K2CO3, DMF, RT, 17 h; (ii) K2CO3, DMF, microwaves, 90 °C, 15 min; (iii) NaOH, MeOH–H2O, reflux, 3 h; (iv) NaOH, MeOH–H2O, microwaves, 100 °C, 3 min; (v) Br2, AcOH, NaOAc, 55 °C, 48 h; (vi) MeI, K2CO3, DMF, RT, 3 h. | ||
In an alternative approach, our success in the microwave-assisted synthesis of 10 was adapted to incorporate an amino group at C-3, amenable by diazotization chemistry to provide efficient access to 3-bromobenzothiophene 13. The cyclocondensation of methyl thioglycolate with 2-nitrobenzonitriles under basic conditions has been established by Beck,58–60 and adapted methods with halide displacement have also been reported.61–64 By switching the base from NaOMe61 to Et3N63 and heating either 5-bromo-2-fluorobenzonitrile (14a) or 2-fluoro-5-nitrobenzonitrile (14b) and methyl thioglycolate gave the corresponding 3-aminobenzothiophene 5a,b in very high yield (Scheme 3), e.g. for 5b either at 100 °C in DMSO for 2 h using conductive heating (95% yield) or under microwave irradiation at 130 °C for 11 min (94% yield), after simply pouring the reaction mixture into ice-water and collecting the product by filtration. Subsequent deaminative bromination60 of aminobenzo[b]thiophene 5b using tert-butyl nitrite in acetonitrile in the presence of copper(II) bromide gave the target bromobenzothiophene 13 in excellent yield by this much more direct route.
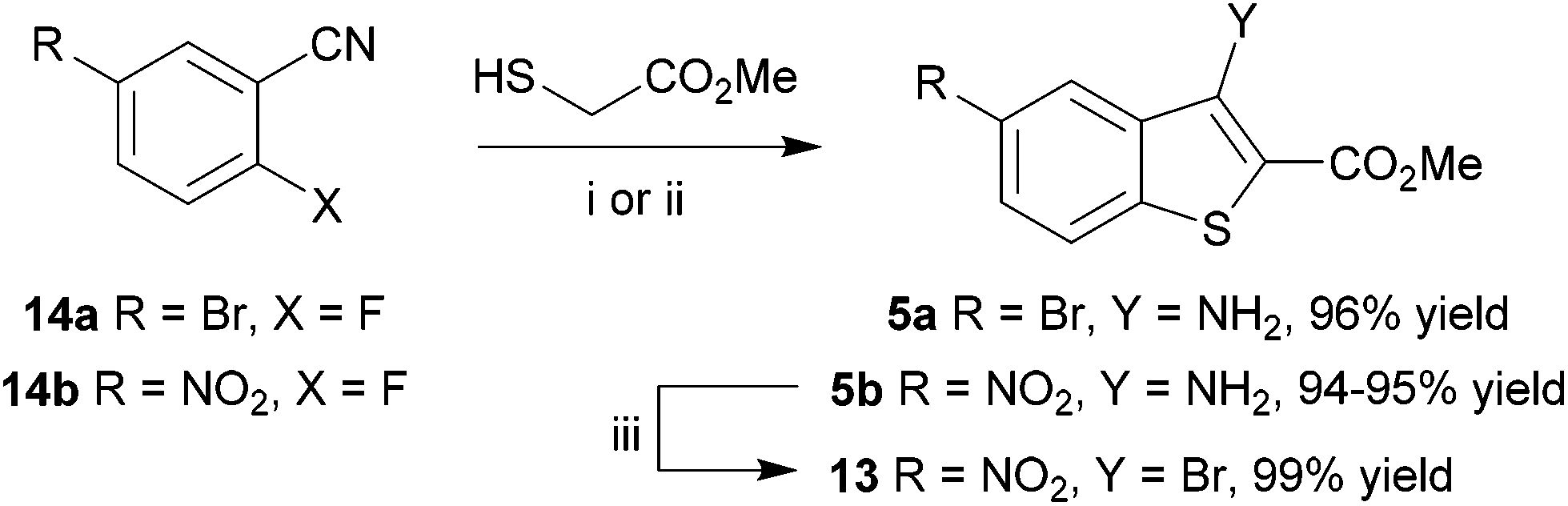 | ||
| Scheme 3 Reagents and conditions: (i) Et3N, DMSO, 100 °C, 2 h; H2O; (ii) Et3N, DMSO, microwaves, 130 °C, 11 min; H2O; (iii) tert-BuONO, CuBr2, MeCN, 0 °C; RT, 2 h; HCl (aq.). | ||
The scope of this method was further explored by investigating a number of substrates (Scheme 4, Table 1), suitable for elaboration to a range of benzothiophene-containing scaffolds found in drug discovery (Fig. 1).
 | ||
| Scheme 4 Synthesis of benzothiophenes 5a–h and 7-aza–16. | ||
| Entry | Substrate | R | X | Z | Time/mina | Product | Yieldb (%) |
|---|---|---|---|---|---|---|---|
| a Hold time at the given temperature, as measured by the in-built IR sensor, by modulation of the initial microwave power. b Isolated yield of product 5 or 16 after reaction according to Scheme 4, cooling in a stream of compressed air and pouring the reaction mixture into iced water. c Product was isolated by aqueous work up, followed by purification by column chromatography on silica gel. | |||||||
| 1 | 14a | 5-Br | F | CH | 11 | 5a | 96 |
| 2 | 14b | 5-NO2 | F | CH | 11 | 5b | 94 |
| 3 | 14c | 5-Cl | F | CH | 11 | 5c | 92 |
| 4 | 14d | 4-CF3 | F | CH | 18 | 5d (R = 6-CF3) | 80 |
| 5 | 14e | 4-NO2 | F | CH | 35 | 5e (R = 6-NO2) | 67 |
| 6c | 14f | 5-Ph | F | CH | 15 | 5f | 85 |
| 7 | 14g | H | F | CH | 15 | 5g | 65 |
| 8 | 14g | H | Br | CH | 15 | 5g | 23 |
| 9 | 14g | H | I | CH | 15 | 5g | 47 |
| 10 | 14h | 5-CF3 | F | CH | 20 | 5h | 88 |
| 11 | 15a | H | F | N | 15 | 16 | 66 |
| 12 | 15b | H | Cl | N | 15 | 16 | 69 |
| 13 | 15c | H | Br | N | 15 | 16 | 51 |
It was found that the process was most efficient for highly electron-poor precursors, such as 14a–d (entries 1–4), and was generally most effective with 2-fluorides, but could also accommodate bromides and iodides albeit with reduced efficiency (Table 1, entries 8 and 9). In most cases a very simple work up procedure was effective, providing benzothiophenes 5a–h in 65–96% yield in reaction times varying between 11 and 35 min, depending upon substrate. The efficiency compared well to other available methods (cf.Table 1, entry 7, with Beck synthesis of 5g,58 52% yield after 20 h), and so this method was adopted as the route of choice to access the benzothiophene scaffold. Furthermore, it was possible to apply the procedure to the synthesis of 7-azabenzothiophene 16 using 2-halonicotinonitriles 15a–c. Interestingly, the choice of halogen as substrate did not cause much variation in the yield of azabenzothiophene product 16 (entries 11–13), which represents the core heterocyclic motif of the 3-aminothieno[2,3-b]pyridine-2-carboxamide inhibitors14 of LIMK1.
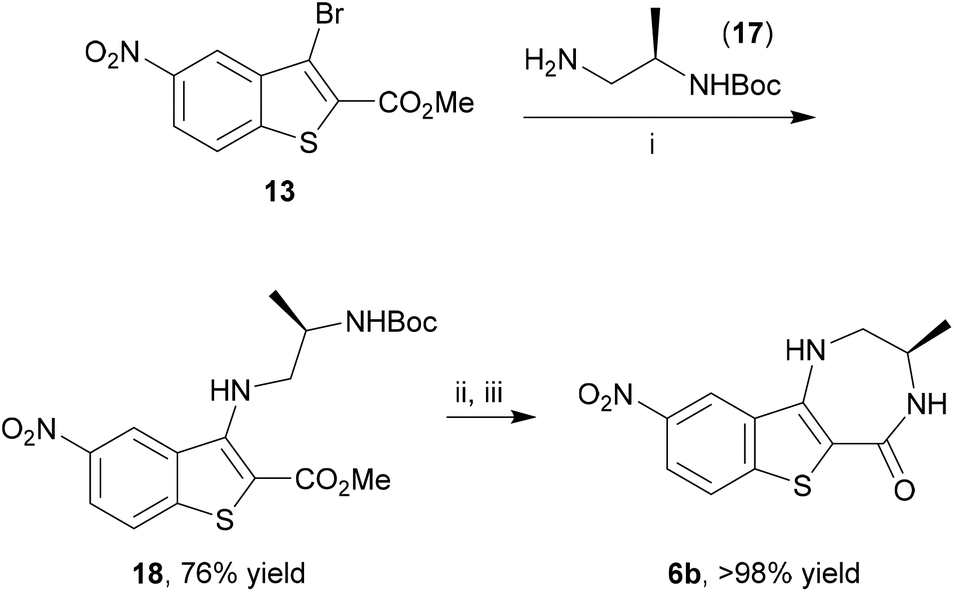
Having developed this rapid microwave-assisted method to prepare benzothiophenes, the 5-nitro analogue 13 was further elaborated to the benzo[4,5]thieno[3,2-e][1,4]diazepin-5(2H)-one MK2 inhibitor scaffold 6b. Buchwald–Hartwig coupling of (R)-tert-butyl (1-aminopropan-2-yl)carbamate (17) and bromobenzothiophene 13 gave a good yield of the N-arylated product 18 under microwave irradiation at 150 °C after 75 min (Scheme 5). Subsequent Boc-deprotection using TFA and lactamization by treatment with NaOMe using a modified procedure of Boschelli65 under conductive heating gave the MK2 inhibitor scaffold 6b, bearing suitable functionality for further elaboration, in essentially quantitative yield.
 | ||
| Scheme 5 Reagents and conditions: (i) Pd(OAc)2 (5 mol%), Cs2CO3, (±)-BINAP (13 mol%), PhMe, microwaves, 150 °C, 75 min; (ii) TFA, CH2Cl2, RT, 4.5 h; (iii) NaOMe, MeOH, 50 °C, 2 h; reflux, 2 h; HCl (aq.). | ||
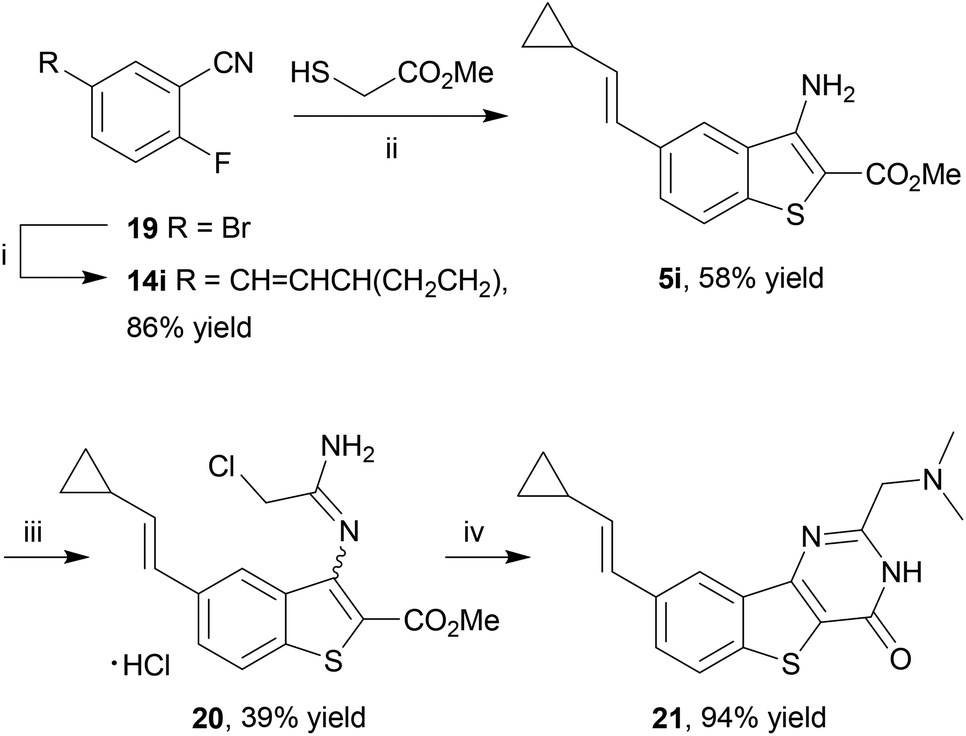
Finally, an application of our microwave-assisted method to access a pre-functionalized benzothiophene 5i for direct transformation to a benzo[4,5]thieno[3,2-d]pyrimidin-4-one scaffold 21 as a chemical tool for PIM kinase inhibition15,66 was investigated. This inhibitor exhibits subnanomolar to single-digit nanomolar Ki values against all three PIM kinases and has been co-crystallised with PIM1, guiding subsequent SAR studies. It was postulated that rather than introducing the cyclopropylvinyl group by a late-stage Suzuki coupling, in accordance with the original diversification study, this group could be incorporated from the start in order to access 20 directly. To that end, 2-fluorobenzonitrile 14i was prepared by the Pd-catalyzed Suzuki–Miyaura coupling of 5-bromo-2-fluorobenzonitrile (14a) and the corresponding boronate ester at 80 °C,15 and heated with methyl thioglycolate in the presence of Et3N in DMSO at 130 °C for 35 min under microwave irradiation to give the corresponding benzothiophene 5i in reasonable yield after purification by column chromatography (Scheme 6). The cyclopropylvinyl group was found to be compatible with the subsequent chemistry: reaction of 5i with chloroacetonitrile in 4 N HCl in dioxane gave chloromethyl derivative 19 which, on reaction with dimethylamine, underwent further cyclization to give the thienopyrimidinone scaffold 20 after purification by immobilization on an acidic resin. Thus the method delivered this known PIM kinase inhibitor by an extremely rapid route, suitable for biological study.
 | ||
| Scheme 6 Reagents and conditions: (i) trans-2-cyclopropylvinylboronic acid pinacol ester, Pd(Ph3)2Cl2 (10 mol%), 1 M Na2CO3, DME/EtOH/H2O, 80 °C, 16 h; (ii) Et3N, DMSO, microwaves, 130 °C, 35 min; (iii) 2-chloroacetonitrile, HCl, dioxane, RT, 16 h; (iv) Me2NH, EtOH, RT, 16 h; SCX column, MeOH/CH2Cl2; NH3, MeOH. | ||
Experimental
Materials and methods
Commercially available reagents were used without further purification; solvents were dried by standard procedures. Light petroleum refers to the fraction with bp 40–60 °C and ether refers to diethyl ether. Unless otherwise stated, reactions were performed under an atmosphere of air. Flash chromatography was carried out using Merck Kieselgel 60 H silica or Matrex silica 60. Analytical thin layer chromatography was carried out using aluminium-backed plates coated with Merck Kieselgel 60 GF254 that were visualised under UV light (at 254 and/or 360 nm). Microwave irradiation experiments were performed in a sealed Pyrex tube using a self-tunable CEM Discover, CEM Explorer or Biotage Initiator 2.5 EXP EU focused monomodal microwave synthesizer at the given temperature using the instrument's in-built IR temperature measuring device, by varying the irradiation power (initial power given in parentheses).Fully characterized compounds were chromatographically homogeneous. Melting points were determined on a Kofler hot stage apparatus or Stanford Research Systems Optimelt and are uncorrected. Specific rotations were measured at the indicated temperature (in °C) using a ADP440 polarimeter (Bellingham + Stanley) at the sodium D line and are given in deg cm−3 g−1 dm−1 with concentration c in 10−2 g cm−3. Infra-red spectra were recorded in the range 4000–600 cm−1 on a Perkin-Elmer 1600 series FTIR spectrometer using an ATR probe or a Shimadzu IRAffinity-1 equipped with an ATR accessory and are reported in cm−1. NMR spectra were recorded using a Varian VNMRS instrument operating at 400 or 500 MHz or a Bruker Avance III 400 MHz or Bruker Avance DRX 500 MHz for 1H spectra and 100 or 126 MHz for 13C spectra; J values were recorded in Hz and multiplicities were expressed by the usual conventions. Low resolution mass spectra were determined using a Waters Q-TOF Ultima using electrospray positive ionization, A Waters LCT premier XE using atmospheric pressure chemical ionization (APcI), an Agilent 6130 single quadrupole with an APcI/electrospray dual source, a Fisons Instrument VG Autospec using electron ionization at 70 eV (low resolution) or a ThermoQuest Finnigan LCQ DUO electrospray, unless otherwise stated. TOFMS refers to time-of-flight mass spectrometry, ES refers to electrospray ionization, CI refers to chemical ionization (ammonia), FTMS refers to Fourier transform mass spectrometry, NSI refers to nano-electrospray ionization and EI refers to electron ionization. A number of high resolution mass spectra were obtained courtesy of the EPSRC Mass Spectrometry Service at University College of Wales, Swansea, UK using the ionization methods specified.
Synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.44 (1H, d, J = 1.9 Hz, 4-CH), 7.82 (1H, d, J = 8.6 Hz, 7-CH), 7.64 (1H, dd, J = 8.6, 1.9 Hz, 6-CH), 7.17 (2H, bs, NH2), 3.79 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.6 (C), 148.6 (C), 137.7 (C), 133.2 (C), 131.0 (C), 125.7 (CH), 125.2 (CH), 117.1 (CH), 96.1 (C), 51.4 (Me); m/z (ES) 286 (M[79Br]H+, 100%).
O), 1572 (NO2), 1432 (C–C), 1328 (NO2), 1276 (C–O), 1093 (C–O); 1H NMR (500 MHz, d6-DMSO) δH/ppm 9.24 (1H, d, J = 2 Hz, 4-CH), 8.29 (1H, dd, J = 9, 2 Hz, 6-CH), 8.12 (1H, d, J = 9 Hz, 7-CH), 7.47 (2H, bs, NH2), 3.82 (3H, s, Me); 13C NMR (100 MHz, d6-DMSO) δC/ppm 164.2 (C), 149.6 (C), 144.9 (C), 144.5 (C), 131.4 (C), 124.3 (CH), 122.0 (CH), 119.4 (CH), 96.8 (C), 51.4 (Me); m/z (EI) 252 (M˙+, 100%), 219 (72). 1H and 13C NMR spectroscopic analyses were in good agreement with literature data.67
O), 1278 (C–O); 1H NMR (500 MHz, d6-DMSO) δH/ppm 8.30 (1H, d, J = 2.1 Hz, 4-CH), 7.88 (1H, d, J = 8.6 Hz, 7-CH), 7.53 (1H, dd, J = 8.6, 2.1 Hz, 6-CH), 7.17 (2H, bs, NH2), 3.79 (3H, s, Me); 13C NMR (126 MHz, d6-DMSO) δC/ppm 164.6 (C), 148.7 (C), 137.3 (C), 132.7 (C), 129.1 (C), 128.4 (CH), 125.0 (CH), 122.7 (CH), 96.2 (C), 51.4 (Me); m/z (ES) 242 (M[35Cl]H+, 100%). 1H NMR spectroscopic analyses were in good agreement with literature data.15
O); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.38–8.32 (2H, 5-CH and 7-CH), 7.72 (1H, d, J = 8.7 Hz, 4-CH), 7.26 (2H, bs, NH2), 3.82 (3H, s, Me); 13C NMR (126 MHz, d6-DMSO) δC/ppm 164.5 (C), 148.8 (C), 138.9 (C), 134.1 (C), 128.4 (q, 2JC–F = 31.8 Hz, C), 124.2 (q, 1JC–F = 272.7 Hz, C), 124.1 (CH), 121.0–120.7 (m, CH), 120.3–120.0 (m, CH), 97.4 (C), 51.5 (Me); m/z (ES) 276 (MH+, 100%).
O), 1624 (C–O); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.90 (1H, d, J = 2.1 Hz, 7-CH), 8.37 (1H, d, J = 8.7 Hz, 4-CH), 8.19 (1H, dd, J = 8.7, 2.1 Hz, 5-CH), 7.30 (2H, bs, NH2), 3.83 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.3 (C), 148.4 (C), 147.0 (C), 138.8 (C), 135.6 (C), 124.2 (CH), 119.8 (CH), 118.5 (CH), 99.8 (C), 51.7 (Me); m/z (ES) 253 (MH+, 20%), 252 (M˙+, 100). 1H NMR spectroscopic analyses were in good agreement with literature data.63
O); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.44 (1H, d, J = 1.9 Hz, 4-CH), 7.82 (1H, d, J = 8.6 Hz, 7-CH), 7.64 (1H, dd, J = 8.6, 1.9 Hz, 6-CH), 7.17 (2H, bs, NH2), 3.79 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.6 (C), 148.6 (C), 137.7 (C), 133.2 (C), 131.0 (C), 125.7 (CH), 125.2 (CH), 117.1 (CH), 96.1 (C), 51.4 (Me); m/z (ES) 286 (M[79Br]H+, 100%).
O), 1572 (NO2), 1432 (C–C), 1328 (NO2), 1276 (C–O), 1093 (C–O); 1H NMR (500 MHz, d6-DMSO) δH/ppm 9.24 (1H, d, J = 2 Hz, 4-CH), 8.29 (1H, dd, J = 9, 2 Hz, 6-CH), 8.12 (1H, d, J = 9 Hz, 7-CH), 7.47 (2H, bs, NH2), 3.82 (3H, s, Me); 13C NMR (100 MHz, d6-DMSO) δC/ppm 164.2 (C), 149.6 (C), 144.9 (C), 144.5 (C), 131.4 (C), 124.3 (CH), 122.0 (CH), 119.4 (CH), 96.8 (C), 51.4 (Me); m/z (EI) 252 (M˙+, 100%), 219 (72). 1H and 13C NMR spectroscopic analyses were in good agreement with literature data.67
O), 1278 (C–O); 1H NMR (500 MHz, d6-DMSO) δH/ppm 8.30 (1H, d, J = 2.1 Hz, 4-CH), 7.88 (1H, d, J = 8.6 Hz, 7-CH), 7.53 (1H, dd, J = 8.6, 2.1 Hz, 6-CH), 7.17 (2H, bs, NH2), 3.79 (3H, s, Me); 13C NMR (126 MHz, d6-DMSO) δC/ppm 164.6 (C), 148.7 (C), 137.3 (C), 132.7 (C), 129.1 (C), 128.4 (CH), 125.0 (CH), 122.7 (CH), 96.2 (C), 51.4 (Me); m/z (ES) 242 (M[35Cl]H+, 100%). 1H NMR spectroscopic analyses were in good agreement with literature data.15
O); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.38–8.32 (2H, 5-CH and 7-CH), 7.72 (1H, d, J = 8.7 Hz, 4-CH), 7.26 (2H, bs, NH2), 3.82 (3H, s, Me); 13C NMR (126 MHz, d6-DMSO) δC/ppm 164.5 (C), 148.8 (C), 138.9 (C), 134.1 (C), 128.4 (q, 2JC–F = 31.8 Hz, C), 124.2 (q, 1JC–F = 272.7 Hz, C), 124.1 (CH), 121.0–120.7 (m, CH), 120.3–120.0 (m, CH), 97.4 (C), 51.5 (Me); m/z (ES) 276 (MH+, 100%).
O), 1624 (C–O); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.90 (1H, d, J = 2.1 Hz, 7-CH), 8.37 (1H, d, J = 8.7 Hz, 4-CH), 8.19 (1H, dd, J = 8.7, 2.1 Hz, 5-CH), 7.30 (2H, bs, NH2), 3.83 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.3 (C), 148.4 (C), 147.0 (C), 138.8 (C), 135.6 (C), 124.2 (CH), 119.8 (CH), 118.5 (CH), 99.8 (C), 51.7 (Me); m/z (ES) 253 (MH+, 20%), 252 (M˙+, 100). 1H NMR spectroscopic analyses were in good agreement with literature data.63
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), gave the title compound (380 mg, 85%) as a colourless solid, mp 96–97 °C (Found [ES+]: 284.0741. C16H14NO2S [MH] requires 284.0740); IR (neat) νmax/cm−1 3439 (N–H), 3338 (N–H), 2949 (C–H), 1658 (CO); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.54 (1H, s, 4-CH), 7.91 (1H, d, J = 8.5 Hz, 7-CH), 7.84 (1H, d, J = 8.5 Hz, 6-CH), 7.78 (2H, d, J = 7.5 Hz, 2′,6′-PhH), 7.51 (2H, app t, J = 7.5 Hz, 3′,5′-PhH), 7.39 (1H, t, J = 7.5 Hz, 4′-PhH), 7.26 (2H, bs, NH2), 3.80 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.8 (C), 149.9 (C), 139.6 (C), 138.0 (C), 136.2 (C), 132.2 (C), 129.0 (CH), 127.5 (CH), 127.2 (CH), 126.7 (CH), 123.6 (CH), 121.0 (CH), 95.0 (C), 51.2 (Me); m/z (ES) 284 (MH+, 100%).
O), 1520, 1289 (C–O); 1H NMR (500 MHz, d6-DMSO) δH/ppm 8.13 (1H, d, J = 8.1 Hz, 4-CH), 7.82 (1H, d, J = 8.1 Hz, 7-CH), 7.50 (1H, m, 6-CH), 7.39 (1H, m, 5-CH), 7.15 (2H, bs, NH2), 3.78 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.8 (CO), 149.8 (3-C), 138.8 (7α-C), 131.4 (3α-C), 128.5 (6-CH), 123.8 (5-CH), 123.1 (4-CH), 123.1 (7-CH), 94.4 (2-C), 51.2 (Me); m/z (ES) 207 (M˙+, 93%), 176 (30), 175 (100), 147 (34), 146 (37). NMR spectroscopic analyses were in good agreement with literature data.59,68
O); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.67 (1H, bs, 4-CH), 8.09 (1H, d, J = 8.5 Hz, 7-CH), 7.79 (1H, dd, J = 8.5, 1.5 Hz, 6-CH), 7.34 (2H, s, NH2), 3.82 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.5 (C), 149.3 (C), 142.5 (C), 131.3 (C), 124.9 (q, 2JC–F = 32.7 Hz, C), 124.5 (q, 1JC–F = 272.1 Hz, C), 124.4 (C–H), 124.1 (m, C–H), 120.7 (m, C–H), 96.2 (C), 51.4 (Me); m/z (ES) 276 (MH+, 100%).
:1), gave the title compound (210 mg, 58%) as a colourless solid, mp 131–132 °C (Found [ES+]: 274.0899. C15H16NO2S [M] requires 274.0896); IR (neat) νmax/cm−1 3481 (N–H), 3365 (N–H), 1672 (CO); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.12 (1H, d, J = 1.6 Hz, 4-CH), 7.71 (1H, d, J = 8.5 Hz, 7-CH), 7.50 (1H, dd, J = 8.5, 1.6 Hz, 6-CH), 7.12 (2H, bs, NH2), 6.53 (1H, d, J = 15.8 Hz, CH), 5.93 (1H, dd, J = 15.8, 9.1 Hz, CH), 3.78 (3H, s, Me), 1.69–1.55 (1H, m, CH), 0.86–0.80 (2H, m, CHH), 0.56–0.49 (2H, m, CHH); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.8 (C), 149.7 (C), 136.9 (C), 135.2 (C), 133.7 (CH), 131.9 (C), 126.4 (CH), 126.4 (CH), 123.1 (CH), 119.4 (CH), 94.9 (C), 51.2 (Me), 14.4 (CH), 7.1 (CH2); m/z (ES) 274 (MH+, 100%).
:9)] the mixture was evaporated in vacuo. The residue was dissolved in a mixture of MeOH (5 mL) and NaOMe (25 wt% in MeOH; 1 mL, 4.4 mmol) and warmed to 50 °C for 2 h before heating at reflux for 2 h. The reaction mixture was then allowed to cool to room temperature, cooled in an ice bath, neutralized by the addition of hydrochloric acid (1 M) and stirred at 0 °C for 30 min. The resulting solid was isolated by filtration under reduced pressure, washed with water and dried in air to give the title compound (0.18 g, 100%) as a red powder, mp 337.4–342.4 °C (dec.) (Found [ES+]: 278.0590. C12H1sN3O3S [MH] requires 278.0594); [α]24D +110.6 (c 0.04, MeOH); IR (neat) νmax/cm−1 3307 (N–H), 1598 (CO), 1501 (NO2), 1438 (C–C), 1318 (NO2), 1102 (C–N), 736 (N–H); 1H NMR (500 MHz, d6-DMSO) δH/ppm 9.03 (1H, d, J = 2 Hz, 10-CH), 8.22 (1H, dd, J = 9, 2 Hz, 8-CH), 8.05 (1H, d, J = 9 Hz, 7-CH), 8.02 (1H, m, 1-NH), 7.95 (1H, d, J = 3 Hz, 4-NH), 3.60 (1H, m, 3-CH), 3.39 (2H, m, 2-CH2), 1.16 (3H, d, J = 7 Hz, Me); 13C NMR (125 MHz, d6-DMSO) δC/ppm 164.2 (5-C), 145.3 (C), 144.2 (C), 141.8 (C), 132.9 (C), 123.8 (7-CH), 120.8 (8-CH), 118.6 (10-CH), 108.6 (C), 50.6 (2-CH2), 47.6 (3-CH), 18.6 (Me); m/z (ES) 277 (M˙+, 87%), 262 (24), 234 (29), 206 (30).
O), 1528 (NO2), 1439 (C–C), 1342 (NO2), 1302 (C–O); 1H NMR (500 MHz, CDCl3) δH/ppm 8.80 (1H, d, J = 2 Hz, 4-CH), 8.32 (1H, dd, J = 9, 2 Hz, 6-CH), 8.20 (1H, s, 3-CH), 8.01 (1H, d, J = 9 Hz, 7-CH), 4.00 (3H, s, Me); 13C NMR (125 MHz, CDCl3) δC/ppm 162.2 (C), 147.4 (C), 145.9 (C), 138.3 (C), 137.2 (C), 130.7 (3-CH), 123.6 (7-CH), 121.2 (9-CH), 120.9 (7-CH), 52.9 (Me); m/z (EI) 237 (M˙+, 100%), 206 (63), 160 (25). 1H NMR spectroscopic analyses were in good agreement with literature data.53,70
O), 1600 (C–C), 1511 (NO2), 1344 (NO2), 1313 (C–O); 1H NMR (500 MHz, CD3OD) δH/ppm 8.87 (1H, d, J = 2 Hz, 4-CH), 8.30 (1H, dd, J = 9, 2 Hz, 6-CH), 8.24 (1H, s, 3-CH), 8.15 (1H, d, J = 9 Hz, 7-CH); 13C NMR (125 MHz, CD3OD) δC/ppm 164.8 (C), 148.9 (5-C), 147.5 (C), 140.3 (C), 140.0 (C), 132.0 (3-CH), 125.1 (7-CH), 122.3 (4-CH), 121.9 (6-CH); m/z (EI) 223 (M˙+, 100%), 195 (38), 149 (37). 1H NMR spectroscopic analyses were in good agreement with literature data.53
O), 1600 (C–C), 1511 (NO2), 1347 (NO2), 1270 (C–O), 620 (C–Br); 1H NMR (500 MHz, CD3OD) δH/ppm 8.83 (1H, d, J = 2 Hz, 4-CH), 8.39 (1H, dd, J = 9, 2 Hz, 6-CH), 8.21 (1H, d, J = 9 Hz, 7-CH); m/z (EI) 303 (M[81Br]˙+, 98%), 301 (M[79Br]˙+, 96), 293 (17), 291 (13).
O), 1600 (C–C), 1514 (NO2), 1347 (NO2), 1089 (C–O), 1052 (C–O), 617 (C–Br); 1H NMR (500 MHz, CDCl3) δH/ppm 8.90 (1H, d, J = 2 Hz, 4-CH), 8.38 (1H, dd, J = 9, 2 Hz, 6-CH), 8.00 (1H, d, J = 9 Hz, 7-CH), 4.02 (3H, s, Me); 13C NMR (125 MHz, CDCl3) δc/ppm 161.0 (C), 146.5 (5-C), 144.6 (C), 138.9 (C), 131.0 (3α-C), 123.8 (7-CH), 122.2 (6-CH), 121.3 (4-CH), 115.6 (3-CH), 53.0 (Me); m/z (EI) 317 (M[81Br]˙+, 100%), 315 (M[79Br]˙+, 94), 286 (58), 284 (55).
:2:3) (50 mL) was added aqueous Na2CO3 solution (1 M; 27 mL). The resulting mixture was placed under an atmosphere of nitrogen and heated at 80 °C for 16 h after which time the reaction was cooled to room temperature and filtered through Celite. Purification by flash column chromatography on silica gel, eluting with light petroleum–EtOAc (9:1), gave the title compound (2.4 g, 86%) as a yellow oil (Found [ES+]: 188.0868. C12H11NF [MH] requires 188.0870); IR (neat) νmax/cm−1 3007 (C–H), 2496 (CN), 1672 (CC); 1H NMR (400 MHz, CDCl3) δH/ppm 7.53–7.46 (2H, m), 7.16–7.08 (1H, m), 6.39 (1H, d, J = 15.7 Hz), 5.71 (1H, dd, J = 15.7, 9.0 Hz), 1.65–1.53 (1H, m), 0.92–0.84 (2H, m), 0.59–0.52 (2H, m); 13C NMR (101 MHz, CDCl3) δC/ppm 161.7 (d, J = 258.2 Hz), 137.9, 135.3 (d, J = 3.5 Hz), 131.8 (d, J = 7.9 Hz), 130.0, 124.2, 116.5 (d, J = 20.3 Hz), 101.5 (d, J = 15.7 Hz), 77.2, 14.7, 7.6; m/z (ES) 188 (MH+, 100%).
O), 1291 (C–O), 1127 (C–O); 1H NMR (500 MHz, d6-DMSO) δH/ppm 8.68 (1H, dd, J = 4.6, 1.6 Hz, 6-CH), 8.54 (1H, dd, J = 8.1, 1.6 Hz, 4-CH), 7.46 (1H, dd, J = 8.1, 4.6 Hz, 5-CH), 7.30 (2H, bs, NH2), 3.80 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.7 (CO), 159.7 (7α-C), 150.7 (6-CH), 147.7 (3-C), 131.4 (4-CH), 125.5 (3α-C), 119.4 (5-CH), 93.2 (2-C), 51.4 (Me); m/z (EI) 208 (M˙+, 100%), 177 (28), 176 (93), 148 (60). 1H NMR spectroscopic analyses were in good agreement with literature data.13
:9)] (Found [ES+]: 175.1443. C8H19N2O2 [MH] requires 175.1441); [α]24D −5.7 (c 1.1, MeOH); IR (neat) νmax/cm−1 2973 (C–H), 1685 (CO), 1521 (N–H), 1364 (CH3 deformation), 1247 (C–O), 1165 (C–N), 1045 (C–O), 872 (N–H); 1H NMR (500 MHz, CDCl3) δH/ppm 4.78 (1H, m, NHBoc), 3.60 (1H, m, 2-CH), 2.70 (1H, m, 1-CHH), 2.59 (1H, m, 1-CHH), 1.40 (9H, s, CMe3), 1.08 (3H, d, J = 7 Hz, 3-Me); 13C NMR (125 MHz, CDCl3) δC/ppm 155.7 (C), 79.1 (C), 48.6 (2-CH), 47.4 (4-CH2), 28.4 (CMe3), 18.5 (Me). 1H NMR spectroscopic analyses were in good agreement with literature data.75
:1), gave the title compound (50 mg, 76%) as a red solid, mp 182.9–184.0 °C (Found [ES+]: 410.1378. C18H24N3O6S [MH] requires 410.1380); [α]24D +55.3 (c 0.1, MeOH); IR (neat) νmax/cm−1 3355 (N–H), 2954 (C–H), 1672 (CO), 1588 (NO2) 1508 (C–C), 1325 (NO2), 1230 (C–O), 1060 (C–N); 1H NMR (500 MHz, d6-DMSO) δH/ppm 8.94 (1H, m, 4-CH), 8.27 (1H, d, J = 9 Hz, 6-CH), 8.15 (1H, d, J = 9 Hz, 7-CH), 7.54 (1H, m, 3-NH), 6.87 (1H, d, J = 6 Hz, 2′-NH), 3.81 (4H, m, 1′-CHH and OMe), 3.76 (1H, m, 2′-CH), 3.57 (1H, m, 1′-CHH), 1.32 (9H, s, CMe3), 1.12 (1H, d, J = 6 Hz, 3′-Me); 13C NMR (125 MHz, d6-DMSO) δC/ppm 164.0 (C), 155.1 (C), 150.3 (3-C), 145.2 (C), 144.3 (C), 131.1 (3α-C), 124.7 (7-CH), 121.6 (6-CH), 120.5 (4-CH), 99.5 (2-C), 77.6 (CMe3), 51.7 (OMe), 50.5 (1′-CH2), 46.2 (2′-CH), 28.1 (CMe3), 18.3 (3′-Me); m/z (EI) 410 (MH+, 100%), 409 (M˙+, 70).
O), 1620 (CC); 1H NMR (400 MHz, CD3OD) δH/ppm 7.91 (1H, d, J = 8.6 Hz, 7-CH), 7.72 (1H, d, J = 1.5 Hz, 4-CH), 7.67 (1H, dd, J = 8.6, 1.5 Hz, 6-CH), 6.60 (1H, d, J = 15.8 Hz, CH), 5.96 (1H, dd, J = 15.8, 9.1 Hz, CH), 4.73 (2H, s, CH2), 3.94 (3H, s, Me), 1.65–1.55 (1H, m, CH), 0.87–0.79 (2H, m, CHH), 0.59–0.48 (2H, m, CHH); 13C NMR (101 MHz, CD3OD) δC/ppm 166.3 (C), 162.4 (C), 138.9 (C), 138.3 (CH), 138.0 (C), 135.8 (C), 130.5 (C), 130.4 (C), 127.7 (CH), 127.3 (CH), 124.6 (CH), 119.6 (CH), 53.4 (Me), 40.1 (CH), 15.5 (CH), 7.8 (CH2, CH2); m/z (ES) 349 (M[35Cl]H+, 100%).
:1; 5 mL) and loaded onto an SCX cartridge (1 g). The cartridge was flushed with MeOH (3 × 5 mL) and then eluted with a solution of NH3 in MeOH (2 M; 8 mL) to give the title compound as a colourless solid (55 mg, 94%), mp 202–203 °C (dec.) (Found [ES+]: 326.1324 C18H20N3OS [MH] requires 326.1322); IR (neat) νmax/cm−1 2873 (br, C–H), 1672 (CO), 1587 (CC); 1H NMR (400 MHz, d6-DMSO) δH/ppm 12.41 (1H, bs, NH), 8.09 (1H, d, J = 1.5 Hz, 4-CH), 8.03 (1H, d, J = 8.5 Hz, 7-CH), 7.69 (1H, dd, J = 8.5, 1.5 Hz, 6-CH), 6.68 (1H, d, J = 15.8 Hz, CH), 6.01 (1H, dd, J = 15.8, 9.2 Hz, CH), 3.52 (2H, s, CH2), 2.29 (6H, s, Me), 1.66–1.56 (1H, m, CH), 0.85–0.78 (2H, m, CHH), 0.61–0.54 (2H, m, CHH); 13C NMR (101 MHz, d6-DMSO) δC/ppm 158.5 (C), 157.2 (C), 152.7 (C), 138.3 (C), 136.2 (CH), 135.2 (CH), 134.6 (C), 126.5 (C), 126.2 (CH), 123.9 (CH), 122.0 (C), 119.7 (CH), 61.3 (CH2), 45.1 (Me, Me), 14.7 (CH), 7.2 (CH2, CH2); m/z (ES) 326 (MH+, 100%). 1H NMR spectroscopic analyses were in good agreement with literature data for the corresponding HCl salt.15
:1), gave the title compound (380 mg, 85%) as a colourless solid, mp 96–97 °C (Found [ES+]: 284.0741. C16H14NO2S [MH] requires 284.0740); IR (neat) νmax/cm−1 3439 (N–H), 3338 (N–H), 2949 (C–H), 1658 (CO); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.54 (1H, s, 4-CH), 7.91 (1H, d, J = 8.5 Hz, 7-CH), 7.84 (1H, d, J = 8.5 Hz, 6-CH), 7.78 (2H, d, J = 7.5 Hz, 2′,6′-PhH), 7.51 (2H, app t, J = 7.5 Hz, 3′,5′-PhH), 7.39 (1H, t, J = 7.5 Hz, 4′-PhH), 7.26 (2H, bs, NH2), 3.80 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.8 (C), 149.9 (C), 139.6 (C), 138.0 (C), 136.2 (C), 132.2 (C), 129.0 (CH), 127.5 (CH), 127.2 (CH), 126.7 (CH), 123.6 (CH), 121.0 (CH), 95.0 (C), 51.2 (Me); m/z (ES) 284 (MH+, 100%).
O), 1520, 1289 (C–O); 1H NMR (500 MHz, d6-DMSO) δH/ppm 8.13 (1H, d, J = 8.1 Hz, 4-CH), 7.82 (1H, d, J = 8.1 Hz, 7-CH), 7.50 (1H, m, 6-CH), 7.39 (1H, m, 5-CH), 7.15 (2H, bs, NH2), 3.78 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.8 (CO), 149.8 (3-C), 138.8 (7α-C), 131.4 (3α-C), 128.5 (6-CH), 123.8 (5-CH), 123.1 (4-CH), 123.1 (7-CH), 94.4 (2-C), 51.2 (Me); m/z (ES) 207 (M˙+, 93%), 176 (30), 175 (100), 147 (34), 146 (37). NMR spectroscopic analyses were in good agreement with literature data.59,68
O); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.67 (1H, bs, 4-CH), 8.09 (1H, d, J = 8.5 Hz, 7-CH), 7.79 (1H, dd, J = 8.5, 1.5 Hz, 6-CH), 7.34 (2H, s, NH2), 3.82 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.5 (C), 149.3 (C), 142.5 (C), 131.3 (C), 124.9 (q, 2JC–F = 32.7 Hz, C), 124.5 (q, 1JC–F = 272.1 Hz, C), 124.4 (C–H), 124.1 (m, C–H), 120.7 (m, C–H), 96.2 (C), 51.4 (Me); m/z (ES) 276 (MH+, 100%).
:1), gave the title compound (210 mg, 58%) as a colourless solid, mp 131–132 °C (Found [ES+]: 274.0899. C15H16NO2S [M] requires 274.0896); IR (neat) νmax/cm−1 3481 (N–H), 3365 (N–H), 1672 (CO); 1H NMR (400 MHz, d6-DMSO) δH/ppm 8.12 (1H, d, J = 1.6 Hz, 4-CH), 7.71 (1H, d, J = 8.5 Hz, 7-CH), 7.50 (1H, dd, J = 8.5, 1.6 Hz, 6-CH), 7.12 (2H, bs, NH2), 6.53 (1H, d, J = 15.8 Hz, CH), 5.93 (1H, dd, J = 15.8, 9.1 Hz, CH), 3.78 (3H, s, Me), 1.69–1.55 (1H, m, CH), 0.86–0.80 (2H, m, CHH), 0.56–0.49 (2H, m, CHH); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.8 (C), 149.7 (C), 136.9 (C), 135.2 (C), 133.7 (CH), 131.9 (C), 126.4 (CH), 126.4 (CH), 123.1 (CH), 119.4 (CH), 94.9 (C), 51.2 (Me), 14.4 (CH), 7.1 (CH2); m/z (ES) 274 (MH+, 100%).
:9)] the mixture was evaporated in vacuo. The residue was dissolved in a mixture of MeOH (5 mL) and NaOMe (25 wt% in MeOH; 1 mL, 4.4 mmol) and warmed to 50 °C for 2 h before heating at reflux for 2 h. The reaction mixture was then allowed to cool to room temperature, cooled in an ice bath, neutralized by the addition of hydrochloric acid (1 M) and stirred at 0 °C for 30 min. The resulting solid was isolated by filtration under reduced pressure, washed with water and dried in air to give the title compound (0.18 g, 100%) as a red powder, mp 337.4–342.4 °C (dec.) (Found [ES+]: 278.0590. C12H1sN3O3S [MH] requires 278.0594); [α]24D +110.6 (c 0.04, MeOH); IR (neat) νmax/cm−1 3307 (N–H), 1598 (CO), 1501 (NO2), 1438 (C–C), 1318 (NO2), 1102 (C–N), 736 (N–H); 1H NMR (500 MHz, d6-DMSO) δH/ppm 9.03 (1H, d, J = 2 Hz, 10-CH), 8.22 (1H, dd, J = 9, 2 Hz, 8-CH), 8.05 (1H, d, J = 9 Hz, 7-CH), 8.02 (1H, m, 1-NH), 7.95 (1H, d, J = 3 Hz, 4-NH), 3.60 (1H, m, 3-CH), 3.39 (2H, m, 2-CH2), 1.16 (3H, d, J = 7 Hz, Me); 13C NMR (125 MHz, d6-DMSO) δC/ppm 164.2 (5-C), 145.3 (C), 144.2 (C), 141.8 (C), 132.9 (C), 123.8 (7-CH), 120.8 (8-CH), 118.6 (10-CH), 108.6 (C), 50.6 (2-CH2), 47.6 (3-CH), 18.6 (Me); m/z (ES) 277 (M˙+, 87%), 262 (24), 234 (29), 206 (30).
O), 1528 (NO2), 1439 (C–C), 1342 (NO2), 1302 (C–O); 1H NMR (500 MHz, CDCl3) δH/ppm 8.80 (1H, d, J = 2 Hz, 4-CH), 8.32 (1H, dd, J = 9, 2 Hz, 6-CH), 8.20 (1H, s, 3-CH), 8.01 (1H, d, J = 9 Hz, 7-CH), 4.00 (3H, s, Me); 13C NMR (125 MHz, CDCl3) δC/ppm 162.2 (C), 147.4 (C), 145.9 (C), 138.3 (C), 137.2 (C), 130.7 (3-CH), 123.6 (7-CH), 121.2 (9-CH), 120.9 (7-CH), 52.9 (Me); m/z (EI) 237 (M˙+, 100%), 206 (63), 160 (25). 1H NMR spectroscopic analyses were in good agreement with literature data.53,70
O), 1600 (C–C), 1511 (NO2), 1344 (NO2), 1313 (C–O); 1H NMR (500 MHz, CD3OD) δH/ppm 8.87 (1H, d, J = 2 Hz, 4-CH), 8.30 (1H, dd, J = 9, 2 Hz, 6-CH), 8.24 (1H, s, 3-CH), 8.15 (1H, d, J = 9 Hz, 7-CH); 13C NMR (125 MHz, CD3OD) δC/ppm 164.8 (C), 148.9 (5-C), 147.5 (C), 140.3 (C), 140.0 (C), 132.0 (3-CH), 125.1 (7-CH), 122.3 (4-CH), 121.9 (6-CH); m/z (EI) 223 (M˙+, 100%), 195 (38), 149 (37). 1H NMR spectroscopic analyses were in good agreement with literature data.53
O), 1600 (C–C), 1511 (NO2), 1347 (NO2), 1270 (C–O), 620 (C–Br); 1H NMR (500 MHz, CD3OD) δH/ppm 8.83 (1H, d, J = 2 Hz, 4-CH), 8.39 (1H, dd, J = 9, 2 Hz, 6-CH), 8.21 (1H, d, J = 9 Hz, 7-CH); m/z (EI) 303 (M[81Br]˙+, 98%), 301 (M[79Br]˙+, 96), 293 (17), 291 (13).
O), 1600 (C–C), 1514 (NO2), 1347 (NO2), 1089 (C–O), 1052 (C–O), 617 (C–Br); 1H NMR (500 MHz, CDCl3) δH/ppm 8.90 (1H, d, J = 2 Hz, 4-CH), 8.38 (1H, dd, J = 9, 2 Hz, 6-CH), 8.00 (1H, d, J = 9 Hz, 7-CH), 4.02 (3H, s, Me); 13C NMR (125 MHz, CDCl3) δc/ppm 161.0 (C), 146.5 (5-C), 144.6 (C), 138.9 (C), 131.0 (3α-C), 123.8 (7-CH), 122.2 (6-CH), 121.3 (4-CH), 115.6 (3-CH), 53.0 (Me); m/z (EI) 317 (M[81Br]˙+, 100%), 315 (M[79Br]˙+, 94), 286 (58), 284 (55).
:2:3) (50 mL) was added aqueous Na2CO3 solution (1 M; 27 mL). The resulting mixture was placed under an atmosphere of nitrogen and heated at 80 °C for 16 h after which time the reaction was cooled to room temperature and filtered through Celite. Purification by flash column chromatography on silica gel, eluting with light petroleum–EtOAc (9:1), gave the title compound (2.4 g, 86%) as a yellow oil (Found [ES+]: 188.0868. C12H11NF [MH] requires 188.0870); IR (neat) νmax/cm−1 3007 (C–H), 2496 (CN), 1672 (CC); 1H NMR (400 MHz, CDCl3) δH/ppm 7.53–7.46 (2H, m), 7.16–7.08 (1H, m), 6.39 (1H, d, J = 15.7 Hz), 5.71 (1H, dd, J = 15.7, 9.0 Hz), 1.65–1.53 (1H, m), 0.92–0.84 (2H, m), 0.59–0.52 (2H, m); 13C NMR (101 MHz, CDCl3) δC/ppm 161.7 (d, J = 258.2 Hz), 137.9, 135.3 (d, J = 3.5 Hz), 131.8 (d, J = 7.9 Hz), 130.0, 124.2, 116.5 (d, J = 20.3 Hz), 101.5 (d, J = 15.7 Hz), 77.2, 14.7, 7.6; m/z (ES) 188 (MH+, 100%).
O), 1291 (C–O), 1127 (C–O); 1H NMR (500 MHz, d6-DMSO) δH/ppm 8.68 (1H, dd, J = 4.6, 1.6 Hz, 6-CH), 8.54 (1H, dd, J = 8.1, 1.6 Hz, 4-CH), 7.46 (1H, dd, J = 8.1, 4.6 Hz, 5-CH), 7.30 (2H, bs, NH2), 3.80 (3H, s, Me); 13C NMR (101 MHz, d6-DMSO) δC/ppm 164.7 (CO), 159.7 (7α-C), 150.7 (6-CH), 147.7 (3-C), 131.4 (4-CH), 125.5 (3α-C), 119.4 (5-CH), 93.2 (2-C), 51.4 (Me); m/z (EI) 208 (M˙+, 100%), 177 (28), 176 (93), 148 (60). 1H NMR spectroscopic analyses were in good agreement with literature data.13
:9)] (Found [ES+]: 175.1443. C8H19N2O2 [MH] requires 175.1441); [α]24D −5.7 (c 1.1, MeOH); IR (neat) νmax/cm−1 2973 (C–H), 1685 (CO), 1521 (N–H), 1364 (CH3 deformation), 1247 (C–O), 1165 (C–N), 1045 (C–O), 872 (N–H); 1H NMR (500 MHz, CDCl3) δH/ppm 4.78 (1H, m, NHBoc), 3.60 (1H, m, 2-CH), 2.70 (1H, m, 1-CHH), 2.59 (1H, m, 1-CHH), 1.40 (9H, s, CMe3), 1.08 (3H, d, J = 7 Hz, 3-Me); 13C NMR (125 MHz, CDCl3) δC/ppm 155.7 (C), 79.1 (C), 48.6 (2-CH), 47.4 (4-CH2), 28.4 (CMe3), 18.5 (Me). 1H NMR spectroscopic analyses were in good agreement with literature data.75
:1), gave the title compound (50 mg, 76%) as a red solid, mp 182.9–184.0 °C (Found [ES+]: 410.1378. C18H24N3O6S [MH] requires 410.1380); [α]24D +55.3 (c 0.1, MeOH); IR (neat) νmax/cm−1 3355 (N–H), 2954 (C–H), 1672 (CO), 1588 (NO2) 1508 (C–C), 1325 (NO2), 1230 (C–O), 1060 (C–N); 1H NMR (500 MHz, d6-DMSO) δH/ppm 8.94 (1H, m, 4-CH), 8.27 (1H, d, J = 9 Hz, 6-CH), 8.15 (1H, d, J = 9 Hz, 7-CH), 7.54 (1H, m, 3-NH), 6.87 (1H, d, J = 6 Hz, 2′-NH), 3.81 (4H, m, 1′-CHH and OMe), 3.76 (1H, m, 2′-CH), 3.57 (1H, m, 1′-CHH), 1.32 (9H, s, CMe3), 1.12 (1H, d, J = 6 Hz, 3′-Me); 13C NMR (125 MHz, d6-DMSO) δC/ppm 164.0 (C), 155.1 (C), 150.3 (3-C), 145.2 (C), 144.3 (C), 131.1 (3α-C), 124.7 (7-CH), 121.6 (6-CH), 120.5 (4-CH), 99.5 (2-C), 77.6 (CMe3), 51.7 (OMe), 50.5 (1′-CH2), 46.2 (2′-CH), 28.1 (CMe3), 18.3 (3′-Me); m/z (EI) 410 (MH+, 100%), 409 (M˙+, 70).
O), 1620 (CC); 1H NMR (400 MHz, CD3OD) δH/ppm 7.91 (1H, d, J = 8.6 Hz, 7-CH), 7.72 (1H, d, J = 1.5 Hz, 4-CH), 7.67 (1H, dd, J = 8.6, 1.5 Hz, 6-CH), 6.60 (1H, d, J = 15.8 Hz, CH), 5.96 (1H, dd, J = 15.8, 9.1 Hz, CH), 4.73 (2H, s, CH2), 3.94 (3H, s, Me), 1.65–1.55 (1H, m, CH), 0.87–0.79 (2H, m, CHH), 0.59–0.48 (2H, m, CHH); 13C NMR (101 MHz, CD3OD) δC/ppm 166.3 (C), 162.4 (C), 138.9 (C), 138.3 (CH), 138.0 (C), 135.8 (C), 130.5 (C), 130.4 (C), 127.7 (CH), 127.3 (CH), 124.6 (CH), 119.6 (CH), 53.4 (Me), 40.1 (CH), 15.5 (CH), 7.8 (CH2, CH2); m/z (ES) 349 (M[35Cl]H+, 100%).
:1; 5 mL) and loaded onto an SCX cartridge (1 g). The cartridge was flushed with MeOH (3 × 5 mL) and then eluted with a solution of NH3 in MeOH (2 M; 8 mL) to give the title compound as a colourless solid (55 mg, 94%), mp 202–203 °C (dec.) (Found [ES+]: 326.1324 C18H20N3OS [MH] requires 326.1322); IR (neat) νmax/cm−1 2873 (br, C–H), 1672 (CO), 1587 (CC); 1H NMR (400 MHz, d6-DMSO) δH/ppm 12.41 (1H, bs, NH), 8.09 (1H, d, J = 1.5 Hz, 4-CH), 8.03 (1H, d, J = 8.5 Hz, 7-CH), 7.69 (1H, dd, J = 8.5, 1.5 Hz, 6-CH), 6.68 (1H, d, J = 15.8 Hz, CH), 6.01 (1H, dd, J = 15.8, 9.2 Hz, CH), 3.52 (2H, s, CH2), 2.29 (6H, s, Me), 1.66–1.56 (1H, m, CH), 0.85–0.78 (2H, m, CHH), 0.61–0.54 (2H, m, CHH); 13C NMR (101 MHz, d6-DMSO) δC/ppm 158.5 (C), 157.2 (C), 152.7 (C), 138.3 (C), 136.2 (CH), 135.2 (CH), 134.6 (C), 126.5 (C), 126.2 (CH), 123.9 (CH), 122.0 (C), 119.7 (CH), 61.3 (CH2), 45.1 (Me, Me), 14.7 (CH), 7.2 (CH2, CH2); m/z (ES) 326 (MH+, 100%). 1H NMR spectroscopic analyses were in good agreement with literature data for the corresponding HCl salt.15
Conclusions
This method for the microwave-assisted synthesis of 3-aminobenzo[b]thiophenes is rapid, simple to carry out and generally high yielding. It has been applied in the synthesis of a range of functional benzothiophenes and their 7-aza analogues and constitutes a very efficient route to the corresponding 3-halobenzothiophenes using diazonium chemistry. Applications of this process have been shown in the synthesis of the thieno[2,3-b]pyridine core motif of LIMK1 inhibitors using 2-halopyridine-3-carbonitriles, the benzo[4,5]thieno[3,2-e][1,4]diazepin-5(2H)-one scaffold of MK2 inhibitors with functionality suitable for subsequent modification using Buchwald–Hartwig chemistry, and a benzo[4,5]thieno[3,2-d]pyrimidin-4-one target as a chemical tool for inhibition of PIM kinases. In the case of inhibitors of MK2, the use of this approach has been shown to be superior to traditional bromination chemistry using the parent benzothiophene heterocycle. Given the speed, efficiency and reliability of these methods, and their ability to incorporate a wide range of functionality, these approaches are likely to find application in providing chemical tools, rapidly, reliably and efficiently, for advancing studies in chemical biology, as well as to access targets in medicinal chemistry.Acknowledgements
We thank the EPSRC-BBSRC-MRC sponsored network SMS-Drug (EP/I037229/1; award to MCB and NCOT), EPSRC (studentship awards to JED and HLR) and the R M Phillips Trust (award to MCB) for support of this work and Dr Alaa Abdul-Sada at the University of Sussex and the EPSRC Mass Spectrometry Service at the University of Wales, Swansea UK for mass spectra.Notes and references
- K. Koike, Z. Jia, T. Nikaido, Y. Liu, Y. Zhao and D. Guo, Org. Lett., 1999, 1, 197 CrossRef CAS.
- J. Huang, H. Luo, L. Wang, Y. Guo, W. Zhang, H. Chen, M. Zhu, Y. Liu and G. Yu, Org. Lett., 2012, 14, 3300 CrossRef CAS PubMed.
- Y. Ni, K. Nakajima, K. Kanno and T. Takahashi, Org. Lett., 2009, 11, 3702 CrossRef CAS PubMed.
- K. G. Pinney, a. D. Bounds, K. M. Dingeman, V. P. Mocharla, G. R. Pettit, R. Bai and E. Hamel, Bioorg. Med. Chem. Lett., 1999, 9, 1081 CrossRef CAS.
- R. Romagnoli, P. G. Baraldi, M. D. Carrion, C. L. Cara, D. Preti, F. Fruttarolo, M. G. Pavani, M. A. Tabrizi, M. Tolomeo, S. Grimaudo, A. Di Cristina, J. Balzarini, J. A. Hadfield, A. Brancale and E. Hamel, J. Med. Chem., 2007, 50, 2273 CrossRef CAS PubMed.
- T. Chonan, D. Wakasugi, D. Yamamoto, M. Yashiro, T. Oi, H. Tanaka, A. Ohoka-Sugita, F. Io, H. Koretsune and A. Hiratate, Bioorg. Med. Chem., 2011, 19, 1580 CrossRef CAS PubMed.
- L. Berrade, B. Aisa, M. J. Ramirez, S. Galiano, S. Guccione, L. R. Moltzau, F. O. Levy, F. Nicoletti, G. Battaglia, G. Molinaro, I. Aldana, A. Monge and S. Perez-Silanes, J. Med. Chem., 2011, 54, 3086 CrossRef CAS PubMed.
- K. C. Lee, B. S. Moon, J. H. Lee, K.-H. Chung, J. A. Katzenellenbogen and D. Y. Chi, Bioorg. Med. Chem., 2003, 11, 3649 CrossRef CAS.
- C. D. Jones, M. G. Jevnikar, A. J. Pike, M. K. Peters, L. J. Black, A. R. Thompson, J. F. Falcone and J. A. Clemens, J. Med. Chem., 1984, 27, 1057 CrossRef CAS.
- Editorial, Lancet Oncol., 2006, 7, 443 CrossRef CAS PubMed; V. G. Vogel, J. P. Costantino, D. L. Wickerham, W. M. Cronin, R. S. Cecchini, J. N. Atkins, T. B. Bevers, L. Fehrenbacher, E. R. Pajon, J. L. Wade, A. Robidoux, R. G. Margolese, J. James, S. M. Lippman, C. D. Runowicz, P. A. Ganz, S. E. Reis, W. McCaskill-Stevens, L. G. Ford, V. C. Jordan and N. Wolmark, JAMA, 2006, 295, 2727 CrossRef CAS PubMed.
- P. Lu, M. L. Schrag, D. E. Slaughter, C. E. Raab, M. Shou and A. D. Rodrigues, Drug Metab. Dispos., 2003, 31, 1352 CrossRef CAS PubMed.
- J. D. Croxtall and G. L. Plosker, Drugs, 2009, 69, 339 CrossRef CAS PubMed.
- R. Romagnoli, P. G. Baraldi, M. K. Salvador, D. Preti, M. A. Tabrizi, M. Bassetto, A. Brancale, E. Hamel, I. Castagliuolo, R. Bortolozzi, G. Basso and G. Viola, J. Med. Chem., 2013, 56, 2606 CrossRef CAS PubMed.
- B. E. Sleebs, A. Levit, I. P. Street, H. Falk, T. Hammonds, A. C. Wong, M. D. Charles, M. F. Olson and J. B. Baell, Med. Chem. Commun., 2011, 2, 977 RSC.
- Z.-F. Tao, L. A. Hasvold, J. D. Leverson, E. K. Han, R. Guan, E. F. Johnson, V. S. Stoll, K. D. Stewart, G. Stamper, N. Soni, J. J. Bouska, Y. Luo, T. J. Sowin, N.-H. Lin, V. S. Giranda, S. H. Rosenberg and T. D. Penning, J. Med. Chem., 2009, 52, 6621 CrossRef CAS PubMed.
- D. R. Anderson, M. J. Meyers, R. G. Kurumbail, N. Caspers, G. I. Poda, S. A. Long, B. S. Pierce, M. W. Mahoney and R. J. Mourey, Bioorg. Med. Chem. Lett., 2009, 19, 4878 CrossRef CAS PubMed.
- D. R. Anderson, M. J. Meyers, R. G. Kurumbail, N. Caspers, G. I. Poda, S. A. Long, B. S. Pierce, M. W. Mahoney, R. J. Mourey and M. D. Parikh, Bioorg. Med. Chem. Lett., 2009, 19, 4882 CrossRef CAS PubMed.
- H. Mikkers, M. Nawijn, J. Allen, C. Brouwers, E. Verhoeven, J. Jonkers and A. Berns, Mol. Cell. Biol., 2004, 24, 6104 CrossRef CAS PubMed.
- S. Kumar, J. Boehm and J. C. Lee, Nat. Rev. Drug Discovery, 2003, 2, 717 CrossRef CAS PubMed.
- M. C. Bagley, T. Davis, P. G. S. Murziani, C. S. Widdowson and D. Kipling, Pharmaceuticals, 2010, 3, 1842 CrossRef CAS PubMed.
- D. Kipling, T. Davis, E. L. Ostler and R. G. Faragher, Science, 2004, 305, 1426 CrossRef CAS PubMed.
- T. Davis, M. F. Haughton, C. J. Jones and D. Kipling, Ann. N. Y. Acad. Sci., 2006, 1067, 243 CrossRef CAS PubMed.
- T. Davis, D. M. Baird, M. F. Haughton, C. J. Jones and D. Kipling, J. Gerontol., Ser. A, 2005, 60, 1386 CrossRef PubMed.
- A. Schlapbach and C. Huppertz, Future Med. Chem., 2009, 1, 1243 CrossRef CAS PubMed.
- N. Ronkina, A. Kotlyarov and M. Gaestel, Front. Biosci., 2008, 13, 5511 CrossRef CAS PubMed.
- R. J. Mourey, B. L. Burnette, S. J. Brustkern, J. S. Daniels, J. L. Hirsch, W. F. Hood, M. J. Meyers, S. J. Mnich, B. S. Pierce, M. J. Saabye, J. F. Schindler, S. A. South, E. G. Webb, J. Zhang and D. R. Anderson, J. Pharmacol. Exp. Ther., 2010, 333, 797 CrossRef CAS PubMed.
- J. S. Daniels, Y. Lai, S. South, P.-C. Chiang, D. Walker, B. Feng, R. Mireles, L. O. Whiteley, J. W. McKenzie, J. Stevens, R. Mourey, D. Anderson and J. W. Davis II, Drug Metab. Lett., 2013, 7, 15 CrossRef CAS.
- T. Davis, M. J. Rokicki, M. C. Bagley and D. Kipling, Chem. Cent. J., 2013, 7, 18 CrossRef CAS PubMed.
- M. C. Bagley, M. Baashen, J. Dwyer, P. Milbeo, D. Kipling and T. Davis, in Microwaves in Drug Discovery and Development: Recent Advances, ed. J. Spencer and M. C. Bagley, Future Science Ltd., 2014, ch. 5, pp. 86–104 Search PubMed.
- M. C. Bagley, M. Baashen, V. L. Paddock, D. Kipling and T. Davis, Tetrahedron, 2013, 69, 8429 CrossRef CAS PubMed.
- T. Davis, M. C. Dix, M. J. Rokicki, C. S. Widdowson, A. J. C. Brook, D. Kipling and M. C. Bagley, Chem. Cent. J., 2011, 5, 83 CrossRef CAS PubMed.
- M. C. Bagley, T. Davis, M. C. Dix, V. Fusillo, M. Pigeaux, M. J. Rokicki and D. Kipling, J. Org. Chem., 2009, 74, 8336 CrossRef CAS PubMed.
- M. C. Bagley, T. Davis, M. C. Dix, C. S. Widdowson and D. Kipling, Org. Biomol. Chem., 2006, 4, 4158 CAS.
- T. Force, K. Kuida, M. Namchuk, K. Parang and J. M. Kyriakis, Circulation, 2004, 109, 1196 CrossRef CAS PubMed.
- D. M. Goldstein, A. Kuglstatter, Y. Lou and M. J. Soth, J. Med. Chem., 2010, 53, 2345 CrossRef CAS PubMed.
- M. C. Genovese, Arthritis Rheum., 2009, 60, 317 CrossRef PubMed.
- T. Davis, M. C. Bagley, M. C. Dix, P. G. S. Murziani, M. J. Rokicki, C. S. Widdowson, J. M. Zayed, M. A. Bachler and D. Kipling, Bioorg. Med. Chem. Lett., 2007, 17, 6832 CrossRef CAS PubMed.
- C. Hou, Q. He and C. Yang, Org. Lett., 2014, 16, 5040 CrossRef CAS PubMed.
- E. David, J. Perrin, S. Pellet-Rostaing, J. Fournier dit Chabert and M. Lemaire, J. Org. Chem., 2005, 70, 3569 CrossRef CAS PubMed.
- M. Martin-Smith and S. Reid, J. Chem. Soc., 1960, 938 RSC.
- M. Martin-Smith and M. Gates, J. Am. Chem. Soc., 1956, 78, 5351 CrossRef CAS.
- M. Martin-Smith and M. Gates, J. Am. Chem. Soc., 1956, 78, 6177 CrossRef CAS.
- J. Fournier Dit Chabert, L. Joucla, E. David and M. Lemaire, Tetrahedron, 2004, 60, 3221 CrossRef CAS PubMed.
- K. O. Hessian and B. L. Flynn, Org. Lett., 2003, 5, 4377 CrossRef CAS PubMed.
- B. L. Flynn, P. Verdier-Pinard and E. Hamel, Org. Lett., 2001, 3, 651 CrossRef CAS PubMed.
- D. Yue and R. C. Larock, J. Org. Chem., 2002, 67, 1905 CrossRef CAS PubMed.
- S. Kim, N. Dahal and T. Kesharwani, Tetrahedron Lett., 2013, 54, 4373 CrossRef CAS PubMed.
- V. Guilarte, M. A. Fernández-Rodriquez, P. García-García, E. Hernando and R. Sanz, Org. Lett., 2011, 13, 5100 CrossRef CAS PubMed.
- S. Knapp, P. Arruda, J. Blagg, S. Burley, D. H. Drewry, A. Edwards, D. Fabbro, P. Gillespie, N. S. Gray, B. Kuster, K. E. Lackey, P. Mazzafera, N. C. O. Tomkinson, T. M. Willson, P. Workman and W. J. Zuercher, Nat. Chem. Biol., 2013, 9, 3 CrossRef CAS PubMed.
- T. Higa and A. J. Krubsack, J. Org. Chem., 1976, 41, 3399 CrossRef CAS.
- T. Higa and A. J. Krubsack, J. Org. Chem., 1975, 40, 3037 CrossRef CAS.
- M. C. Bagley, T. Davis, M. C. Dix, P. G. S. Murziani, M. J. Rokicki and D. Kipling, Bioorg. Med. Chem. Lett., 2008, 18, 3745 CrossRef CAS PubMed.
- R. A. Zambias and M. L. Hammond, Synth. Commun., 1991, 21, 959 CrossRef CAS PubMed.
- J. Cai, S. Zhang, M. Zheng, X. Wu, J. Chen and M. Ji, Bioorg. Med. Chem. Lett., 2012, 22, 806 CrossRef CAS PubMed.
- J. Debray, M. Lemaire and F. Popowycz, Synlett, 2013, 37 CAS.
- M. C. Bagley, J. E. Dwyer and P. Milbeo, unpublished work.
- J. M. Mbere, J. B. Bremner, B. W. Skelton and A. H. White, Tetrahedron, 2011, 67, 6895 CrossRef CAS PubMed.
- J. R. Beck, J. Org. Chem., 1972, 37, 3224 CrossRef CAS.
- M. H. Norman, F. Navas III, J. B. Thompson and G. C. Rigdon, J. Med. Chem., 1996, 39, 4692 CrossRef CAS PubMed.
- V. O. Iaroshenko, S. Ali, S. Mkrtchyan, A. Gevorgyan, T. M. Babar, V. Semeniuchenko, Z. Hassan, A. Villinger and P. Langer, Tetrahedron Lett., 2012, 53, 7135 CrossRef CAS PubMed.
- I. Abdillahi and G. Kirsch, Synthesis, 2011, 1314 Search PubMed.
- I. Abdillahi and G. Kirsch, Synthesis, 2010, 1428 CAS.
- A. J. Bridges and H. Zhou, J. Heterocycl. Chem., 1997, 34, 1163 CrossRef CAS PubMed.
- M. D. Meyer, R. J. Altenbach, F. Z. Basha, W. A. Carroll, S. Condon, S. W. Elmore, J. F. Kerwin Jr., K. B. Sippy, K. Tietje, M. D. Wendt, A. A. Hancock, M. E. Brune, S. A. Buckner and I. Drizin, J. Med. Chem., 2000, 43, 1586 CrossRef CAS PubMed.
- S. S. Khatana, D. H. Boschelli, J. B. Kramer, D. T. Connor, H. Barth and P. Stoss, J. Org. Chem., 1996, 61, 6060 CrossRef CAS.
- R. Ekambaram, E. Enkvist, A. Vaasa, M. Kasari, G. Raidaru, S. Knapp and A. Uri, ChemMedChem, 2013, 8, 909 CrossRef CAS PubMed.
- V. Bertolasi, K. Dudová, P. Šimůnek, J. Černý and V. Macháček, J. Mol. Struct., 2003, 658, 33 CrossRef CAS.
- L. N. Tumey, Y. Bennani and D. C. Bom, WO Pat., 014 647 A2, 2006 Search PubMed.
- V. G. Matassa, F. J. Brown, P. R. Bernstein, H. S. Shapiro, T. P. Maduskuie Jr., L. A. Cronk, E. P. Vacek, Y. K. Yee, D. W. Snyder, R. D. Krell, C. L. Lerman and J. J. Maloney, J. Med. Chem., 1990, 61, 2621 CrossRef.
- D. L. Boger, B. E. Fink and M. P. Hedrick, J. Am. Chem. Soc., 2000, 122, 6382 CrossRef CAS.
- L. Fieser and R. Kennelly, J. Am. Chem. Soc., 1935, 57, 1611 CrossRef CAS.
- W. Wu, Z. Li, G. Zhou and S. Jiang, Tetrahedron Lett., 2011, 52, 2488 CrossRef CAS PubMed.
- Z. Xia and C. D. Smith, J. Org. Chem., 2001, 66, 3459 CrossRef CAS PubMed.
- A. Marquart and B. Podlogar, J. Org. Chem., 1994, 59, 2092 CrossRef CAS.
- Z. Huang, J. Jin, T. D. Machajewski, W. R. Antonios-McCrea, M. McKenna, D. Poon, P. A. Renhowe, M. Sendzik, C. M. Shafer, A. Smith, Y. Xu and Q. Zhang, WO Pat., 115 572 A2, 2009 Search PubMed.
- M.-J. R. P. Queiroz, A. Begouin, I. C. F. R. Ferreira, G. Kirsch, R. C. Calhelha, S. Barbosa and L. M. Estevinho, Eur. J. Org. Chem., 2004, 3679 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00819k |
| This journal is © The Royal Society of Chemistry 2015 |