
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigations of possible prodrug structures for 2-(2-mercaptophenyl)tetrahydropyrimidines: reductive conversion from anti-HIV agents with pyrimidobenzothiazine and isothiazolopyrimidine scaffolds†
Shiho
Okazaki
a,
Shinya
Oishi
*a,
Tsukasa
Mizuhara
a,
Kazuya
Shimura
b,
Hiroto
Murayama
b,
Hiroaki
Ohno
a,
Masao
Matsuoka
b and
Nobutaka
Fujii
*a
aGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: soishi@pharm.kyoto-u.ac.jp; nfujii@pharm.kyoto-u.ac.jp; Fax: +81-75-753-4570; Tel: +81-75-753-4561
bInstitute for Virus Research, Kyoto University, Sakyo-ku, Kyoto, 606-8507, Japan
First published on 10th March 2015
Abstract
3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine (PD 404182) and 3,4-dihydro-2H-benzo[4,5]isothiazolo[2,3-a]pyrimidine are the heterocyclic antiretroviral agents against human immunodeficiency virus type 1 (HIV-1) infection. On the basis of similar structure–activity relationships of anti-HIV activities toward the early-stage of viral infection between these unique scaffolds, the transformations under the bioassay conditions were investigated. The distinctive S–N bond in the isothiazolopyrimidine scaffold was immediately cleaved under reductive conditions in the presence of GSH to generate a thiophenol derivative. A similar rapid conversion of PD 404182 into the same thiophenol derivative was observed, suggesting that pyrimidobenzothiazine and isothiazolopyrimidine scaffolds may work as prodrug forms of the common bioactive thiophenol derivatives.
Introduction
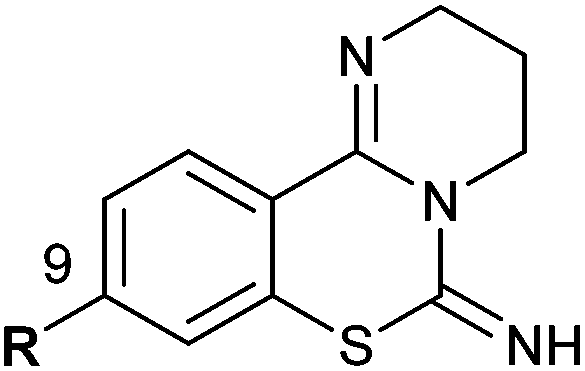
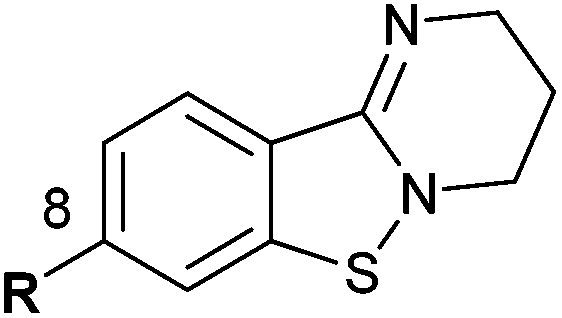
PD 404182 (1a, 3,4-dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine)1–4 is an antiviral agent against human hepatitis C virus (HCV),5,6 HIV,6–10 simian immunodeficiency virus (SIV),6 and herpes simplex virus (HSV) (Fig. 1).10 3,4-Dihydro-2H-benzo[4,5]isothiazolo[2,3-a]pyrimidine 2a with a unique heterocyclic scaffold also exhibits potent anti-HIV activity (EC50 = 0.29 ± 0.09 μM) (Fig. 1).11 In our recent studies, we revealed that modification of the 9-position of PD 404182 derivatives with hydrophobic aryl groups improved the anti-HIV activity.7–9 For example, the introduction of an aryl (1b, 1c and 1f) or bromo (1d) substituent at the 9-position led to a two- to three-fold increase in the anti-HIV activity compared with compound 1a (Table 1). This improvement was duplicated by the modification of benzo[4,5]isothiazolo[2,3-a]pyrimidine derivatives at the 8-position (2b, 2c, 2d and 2f), which is the equivalent position of the 9-position in pyrimido[1,2-c][1,3]benzothiazin-6-imine. Similarly, introduction of acetamido group at the equivalent positions resulted in a complete loss of the anti-HIV activity of both the PD 404182 derivative (1e) and isothiazolopyrimidine derivative (2e). Furthermore, a complete loss of anti-HIV activity was observed by the introduction of the 8-bromo and 7-bromo groups to the parent compounds (1g and 2g, respectively). These similar structure–activity relationships implied that the same target molecule(s) or mechanism(s) of action could possibly be involved in the anti-HIV activities of PD 404182 and the isothiazolopyrimidine derivatives.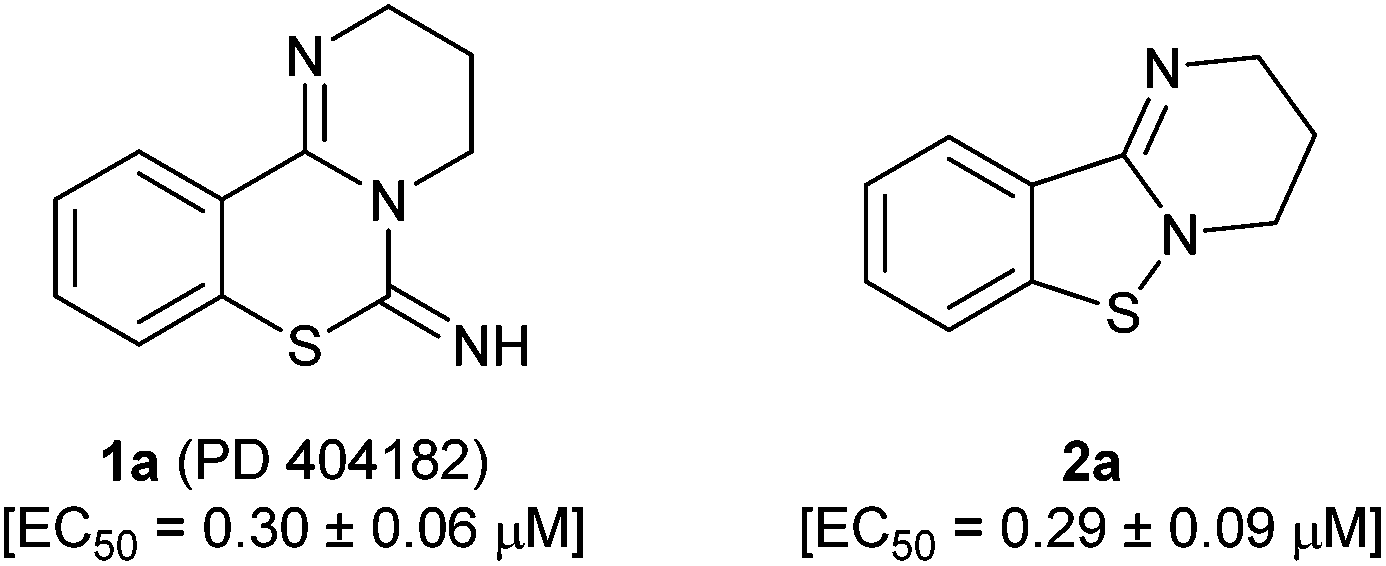 | ||
| Fig. 1 Structures of PD 404182 (1a) and benzo[4,5]isothiazolo[2,3-a]pyrimidine (2a). | ||
| Cmpd | EC50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a,7,8 (μM) a,7,8 (μM) |
Cmpd | EC50b,11 (μM) |
||
|---|---|---|---|---|---|
| a EC50 values represent the concentration of the compound required to inhibit the HIV-1 infection by 50%. The data were obtained from three independent experiments by the MAGI assay. b EC50 values were obtained by the NCK assay. c EC50 values were obtained by the NCK assay, compound 1a: 0.30 ± 0.06 μM; compound 1d: 0.14 ± 0.02 μM; compound 1g: >10 μM. d Cytotoxicity was observed at 10 μM. | |||||
|
|
||||
| 1a (PD 404182) | R = H | 0.44 ± 0.08c | 2a | R = H | 0.29 ± 0.09 |
| 1b | R = Ph | 0.24 ± 0.04 | 2b | R = Ph | 0.30 ± 0.10 |
| 1c | R = m-anisyl | 0.15 ± 0.05 | 2c | R = m-anisyl | 0.10 ± 0.03 |
| 1d | R = Br | 0.25 ± 0.09c | 2d | R = Br | 0.22 ± 0.07 |
| 1e | R = NHAc | >10 | 2e | R = NHAc | >10 |
| 1f |
|
0.15 ± 0.03 | 2f |
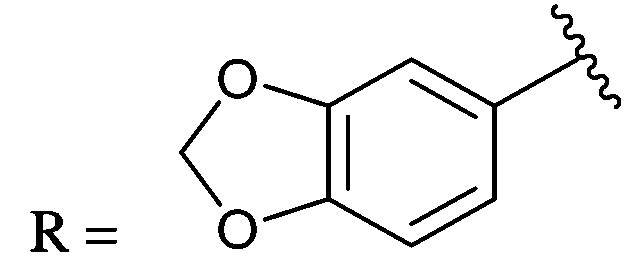
|
0.16 ± 0.02 |
| 1g |
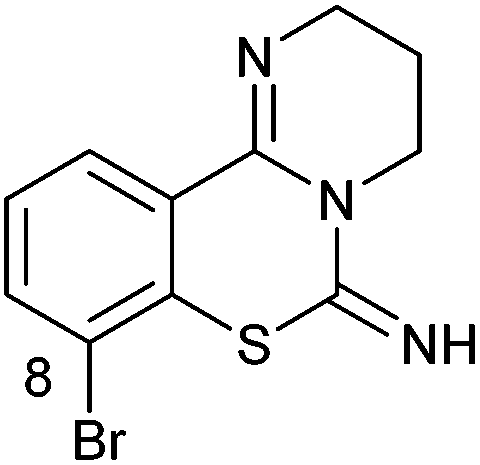
|
>10c | 2g |
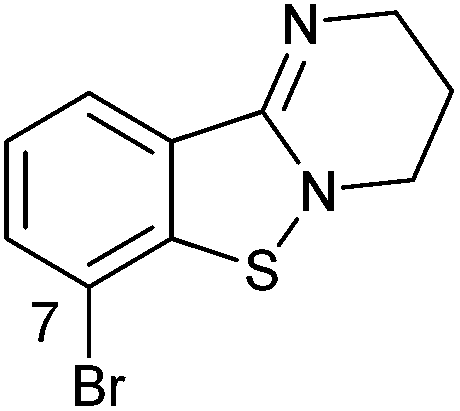
|
>10d |
In addition, heterocyclic compounds 1a and 2a both inhibited early-stage HIV infection including virus attachment and membrane fusion to host cells as exemplified in comparison with DS 5000 (adsorption inhibitor)12 and enfuvirtide (fusion inhibitor).13 Although it was suggested that the antiviral effects of compound 1a are derived from its virucidal effect against viral particles,14 its antiviral mechanism of action has not yet been sufficiently detailed. In the current study, we investigated the chemical transformations of pyrimido[1,2-c][1,3]benzothiazin-6-imine derivatives 1 and benzo[4,5]isothiazolo[2,3-a]pyrimidine derivatives 2 under the bioassay conditions.
Results and discussion
Synthesis
PD 404182 derivatives 1 and benzo[4,5]isothiazolo[2,3-a]pyrimidine derivatives 2 were synthesized according to the protocols in our previous studies (Scheme 1).7,8,11 2-(2-Mercaptophenyl)tetrahydropyrimidine derivatives 4 were obtained by trifluoroacetic acid (TFA)-mediated ethanolysis of the imino group in pyrimido[1,2-c][1,3]benzothiazin-6-imines 1. The alternative route via alkaline hydrolysis of pyrimido[1,2-c][1,3]benzothiazin-6-thione 3 also generated thiophenol derivatives 4. Some thiophenol derivatives 4 could be isolated as stable crystals, such as 2-(2-mercaptophenyl)-, 2-(4-bromo-2-mercaptophenyl)- or 2-(3-bromo-2-mercaptophenyl)-tetrahydropyrimidine (4a, d, g). Thioanisole derivative 7 was obtained by the oxidative amidination of the corresponding benzaldehyde 5.15,16 Sulfuric acid-mediated deprotection of N-tosylated tetrahydropyrimidine 8, derived from benzaldehyde 6 by the identical protocol,7 afforded aniline derivative 9.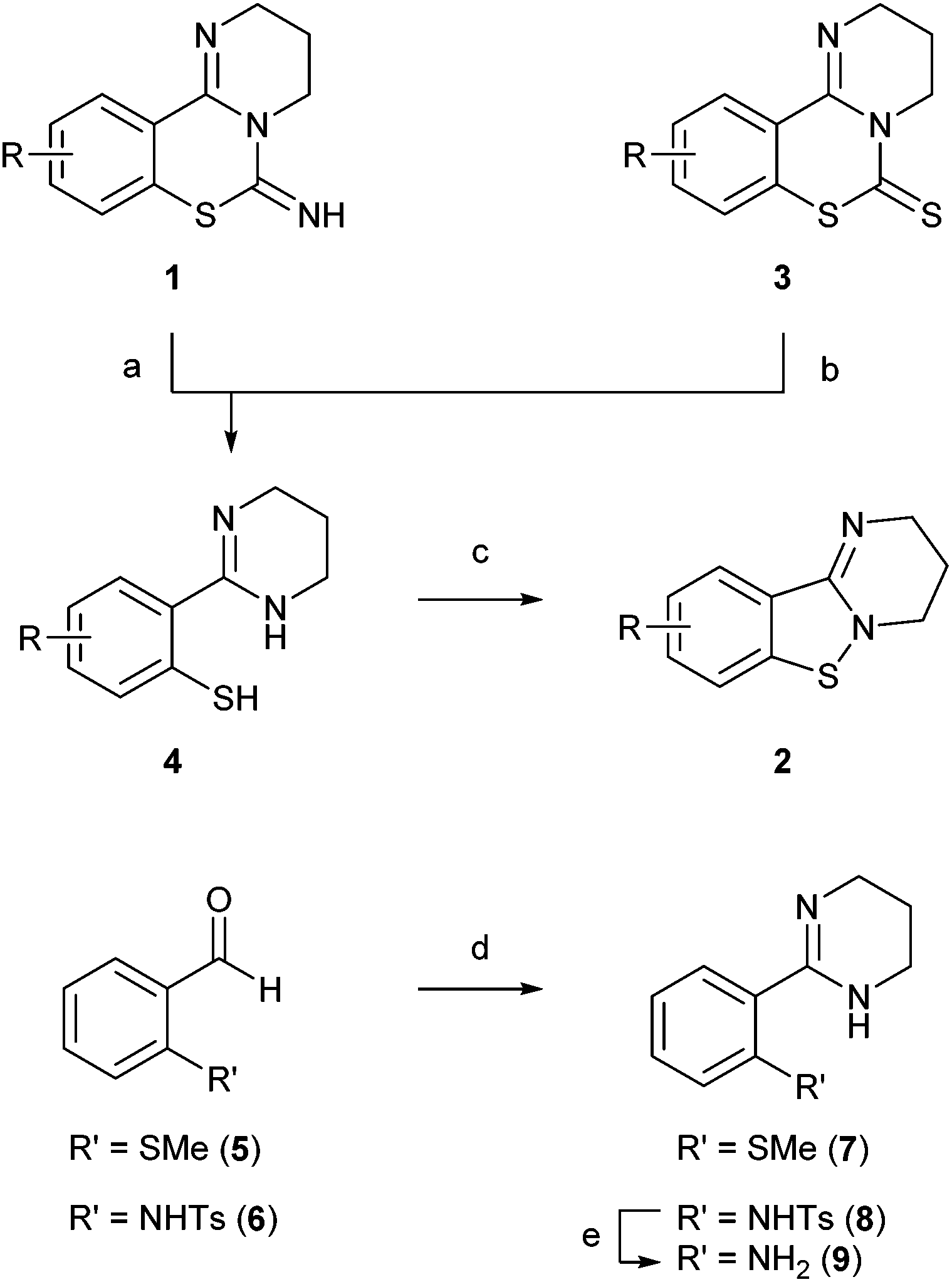 | ||
| Scheme 1 Synthesis of benzo[4,5]isothiazolo[2,3-a]pyrimidine and 2-phenyl-1,4,5,6-tetrahydropyrimidine derivatives. Reagents and conditions: (a) TFA, CHCl3–EtOH or EtOH, rt; (b) NaOH, MeOH–H2O, reflux; (c) PIDA, CHCl3 and/or EtOH, rt; (d) 1,3-propanediamine, I2, K2CO3, t-BuOH, 70 °C; (e) conc. H2SO4, 100 °C. | ||
Identification of the inactive ingredient by transformation from PD 404182 in aqueous media
Recently, Chamoun-Emanuelli et al. reported that the antiviral activity of PD 404182 (1a) disappeared during storage under some conditions (for example, in Dulbecco's phosphate-buffered saline (pH 7) at 42 °C).10 During our studies on PD 404182 derivatives, we also found that the anti-HIV activity from the stock solution of 1a in DMSO was often attenuated. X-ray crystallography of the predominant component from 1a revealed that the resulting product was 3,4-dihydro-2H,6H-benzo[e]pyrimido[2,1-b][1,3]thiazin-6-one 10 (Fig. 2). An apparent rearrangement of the cyclic amidine substructure in 1a provided pyrimidothiazinone 10 with no anti-HIV activity. In contrast, benzo[4,5]isothiazolo[2,3-a]pyrimidine 2a was stable in acidic and basic solutions, and the assay medium containing fetal calf serum.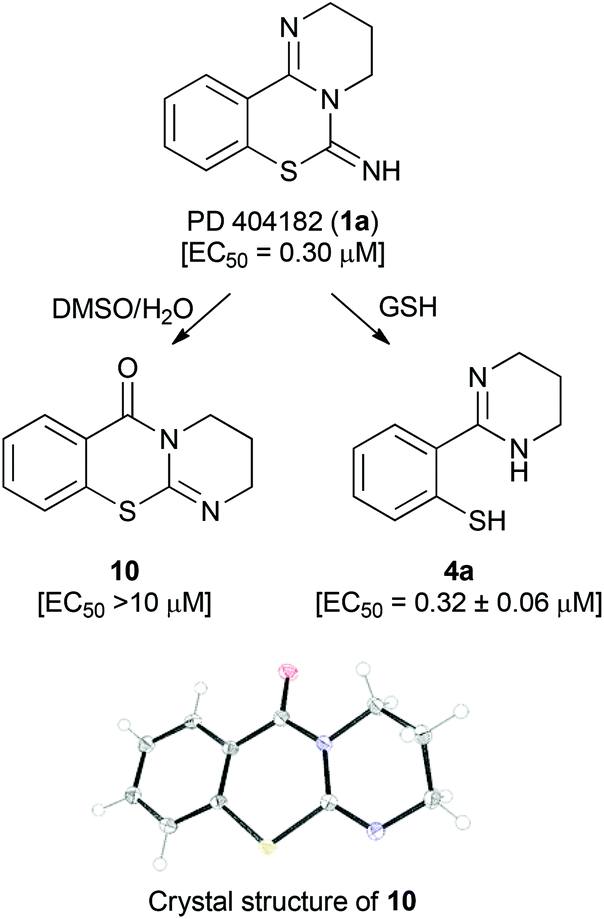 | ||
| Fig. 2 Transformations of PD 404182 under assay conditions and under reductive conditions in the presence of GSH at 37 °C. EC50 values represent the concentration of the compound required to inhibit the HIV-1 infection by 50%. The data were obtained from three independent experiments by the NCK assay. | ||
Transformation of PD 404182 and benzo[4,5]isothiazolo[2,3-a]pyrimidine derivatives under reductive conditions in the presence of GSH
We investigated the conversion of PD 404182 (1a) in the presence of high glutathione (GSH) concentrations that mimic the intracellular environment.17 Interestingly, the imino group of 1a decomposed in 10 mM GSH solution at 37 °C, and was converted into thiophenol 4a within 1 h (Fig. 2 and 3).18 This thiophenol derivative 4a exhibited equipotent anti-HIV activity as the parent compound 1a (EC50 = 0.32 ± 0.06 μM). The mechanism of conversion from 1a into thiophenol 4a could be via the release of cyanylated glutathione, which was detected by ESI-MS analysis.19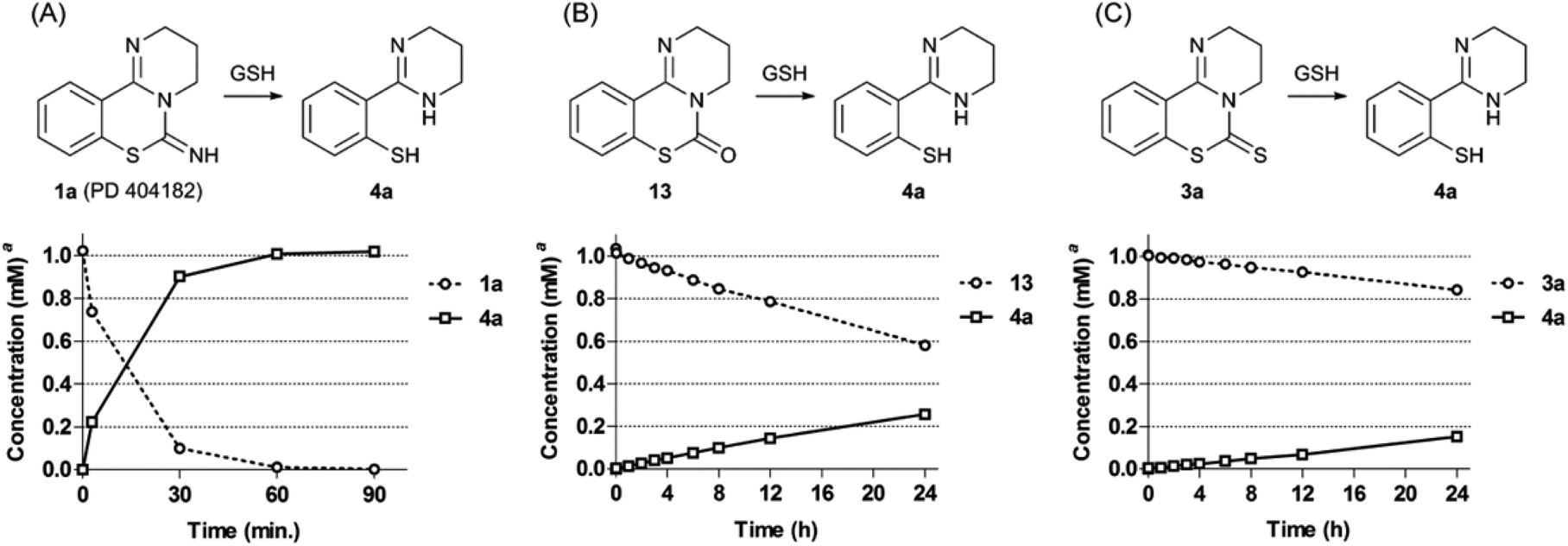 | ||
| Fig. 3 Transformation of compounds 1a (A), 13 (B) and 3a (C) under reductive conditions in the presence of GSH. aThe concentrations of compounds were calculated by HPLC analysis using previously determined calibration curves. | ||
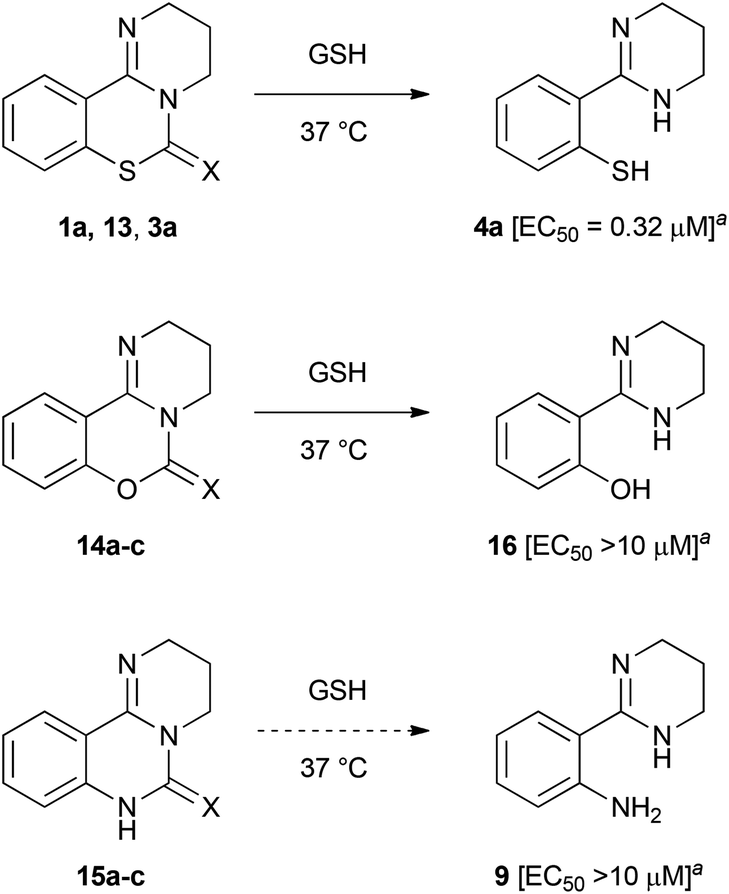
Our previous structure–activity relationship study of 1a demonstrated that the 6-imino group and the 7-sulfur atom in 1a were indispensable for the anti-HIV activity (Table 2).7 For example, substitution of the imino group in compound 1a with a carbonyl (13) or thiocarbonyl (3a) group resulted in a significant decrease or loss of activity. Neither pyrimido[1,2-c][1,3]benzoxazines (14a–c) nor pyrimido[1,2-c]quinazolines (15a–c) exhibited anti-HIV activity (Table 2). In the presence of GSH, the carbonyl (13) and thiocarbonyl (3a) derivatives were slowly converted to compound 4a in 26 and 15% yield, respectively, after 24 h (Table 2, Fig. 3). This is in contrast with the rapid and quantitative conversion from 1a within 1 h, indicating that a more reactive imino group in 1a was a prerequisite for the efficient generation of 4a with potent anti-HIV activity. In the absence of GSH, compound 13 was converted into 4a in only 5% yield for 24 h, and 3a did not produce 4a, indicating that degradation of 13 and 3a was mainly mediated by GSH.20 Compound 14a was unstable in the presence or absence of GSH to give a cyclic carbamate 14b and a phenol derivative 16,7,21 which showed no anti-HIV activity. The carbamate 14b and thiocarbamate 14c were also degraded into an inactive phenol 16 in 78 and 71% yield, respectively, in GSH solution over 24 h. The pyrimido[1,2-c]quinazolines (15a–c) were stable under identical conditions, and thus, did not produce the possible aniline 9. In light of these results, the absent or diminished anti-HIV activity of 3a and 13–15 is likely attributable to decreased conversion to the potent thiophenol 4a or the poor anti-HIV activity of the parent compounds and ring-opened products.
|
|
|||
|---|---|---|---|
| Cmpd | X | EC50b,7 (μM) |
Conversion (time, yieldc) |
| a EC50 values represent the concentration of the compound required to inhibit the HIV-1 infection by 50%. The data were obtained from three independent experiments by the NCK assay. b EC50 values were obtained by the MAGI assay. c The product yields were calculated by HPLC analysis using previously determined calibration curves. d The reactions were also carried out in the absence of GSH: 5% conversion from 13 after 24 h; no reaction from 3a. e The accurate product yields of 14b and 16 by GSH-mediated conversion were not calculated because of the chemical instability of the substrate 14a. | |||
| PD 404182 (1a) | NH | 0.44 ± 0.08 | 4a (1 h, quant.) |
| 13 | O | 8.94 ± 1.07 | 4a (24 h, 26%)d |
| 3a | S | >10 | 4a (24 h, 15%)d |
| 14a | NH | >10 | 16 + 14be |
| 14b | O | >10 | 16 (24 h, 78%) |
| 14c | S | >10 | 16 (24 h, 71%) |
| 15a | NH | >10 | No reaction |
| 15b | O | >10 | No reaction |
| 15c | S | >10 | No reaction |
We next focused on the isothiazolopyrimidine scaffold of 2a with a characteristic S–N covalent bond, which is contained in several anti-HIV agents against HIV-1 nucleocapsid protein 7 (NCp7).22–24 We assessed the stability of compound 2a under identical GSH-containing conditions (Fig. 4). In 10 mM GSH solution at 37 °C, compound 2a was also immediately converted to a thiophenol derivative 4a with potent anti-HIV activity (<3 min). The two-fold more potent pyrimidobenzothiazine 1d or isothiazolopyrimidine 2d was also converted under identical reductive conditions into the corresponding thiophenol derivative 4d, which has similar anti-HIV activity to those of 1d and 2d.25 The thiophenol derivative 4g, which was obtained by conversion from the inactive pyrimidobenzothiazine 1g or isothiazolopyrimidine 2g under the GSH-mediated conditions, exhibited no anti-HIV activity. Thus, the anti-HIV activities of the PD 404182 derivatives (1a, 1d and 1g) and isothiazolopyrimidine derivatives (2a, 2d and 2g) corresponded, at least in part, to those of the thiophenol derivatives (4a, 4d and 4g). These results suggest that the tricyclic structures of 1a and 2a are the prodrug forms, which could be transformed into 4a under intracellular conditions of high GSH concentrations in host cells. Interestingly, inactive analogues with scaffolds similar to PD 404182 (1a) or compound 2a were not converted into thiophenol 4a (Fig. 5). For example, compound 10,26 thioanisole derivative 7, benzo[4,5]thiazolo[3,2-a]pyrimidine 11 (the structural isomer of 2a),27 and saccharin-like derivative 1228 were completely stable under high concentrations of GSH for 24 hours. Therefore, the pyrimidobenzothiazine scaffold in 1a and the isothiazolopyrimidine scaffold in 2a structurally satisfied two criteria, which would make them good prodrugs of 4a.
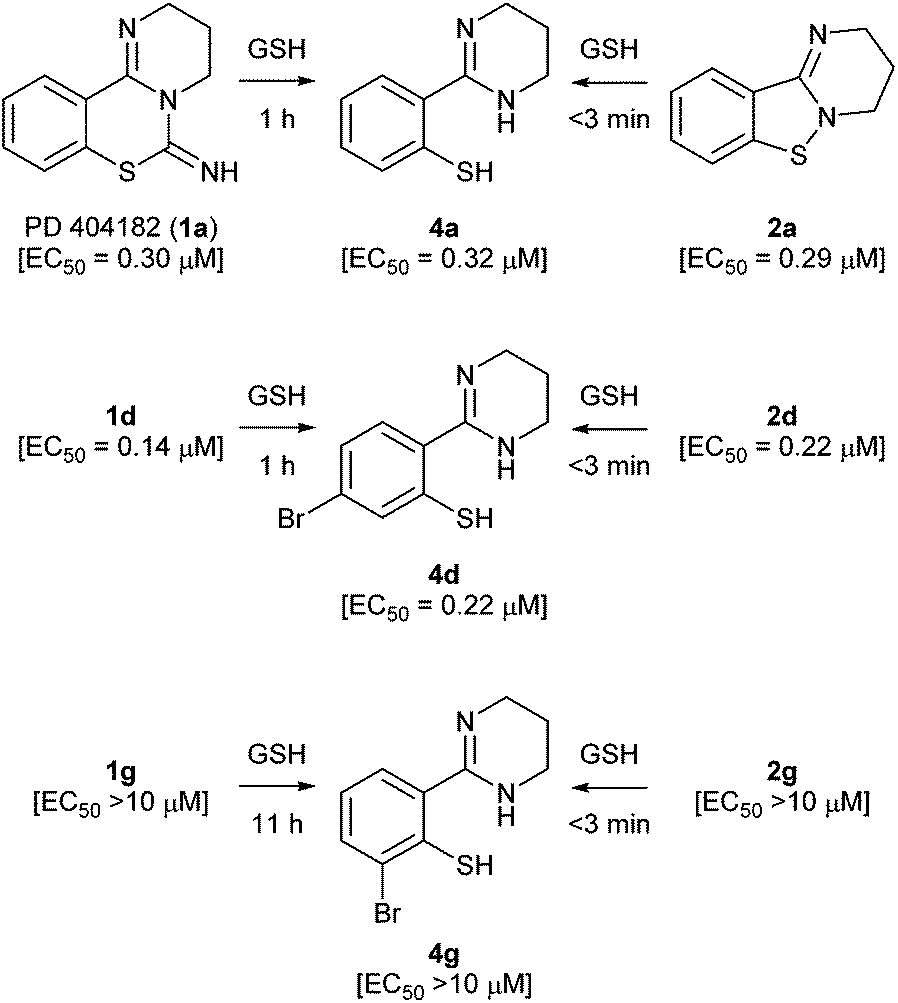 | ||
| Fig. 4 Reactions of potent and inactive pyrimido[1,2-c][1,3]benzothiazin-6-imine and benzo[4,5]isothiazolo[2,3-a]pyrimidine derivatives under reductive conditions in the presence of GSH at 37 °C. EC50 values represent the concentration of the compound required to inhibit the HIV-1 infection by 50%. The data were obtained from three independent experiments by the NCK assay. | ||
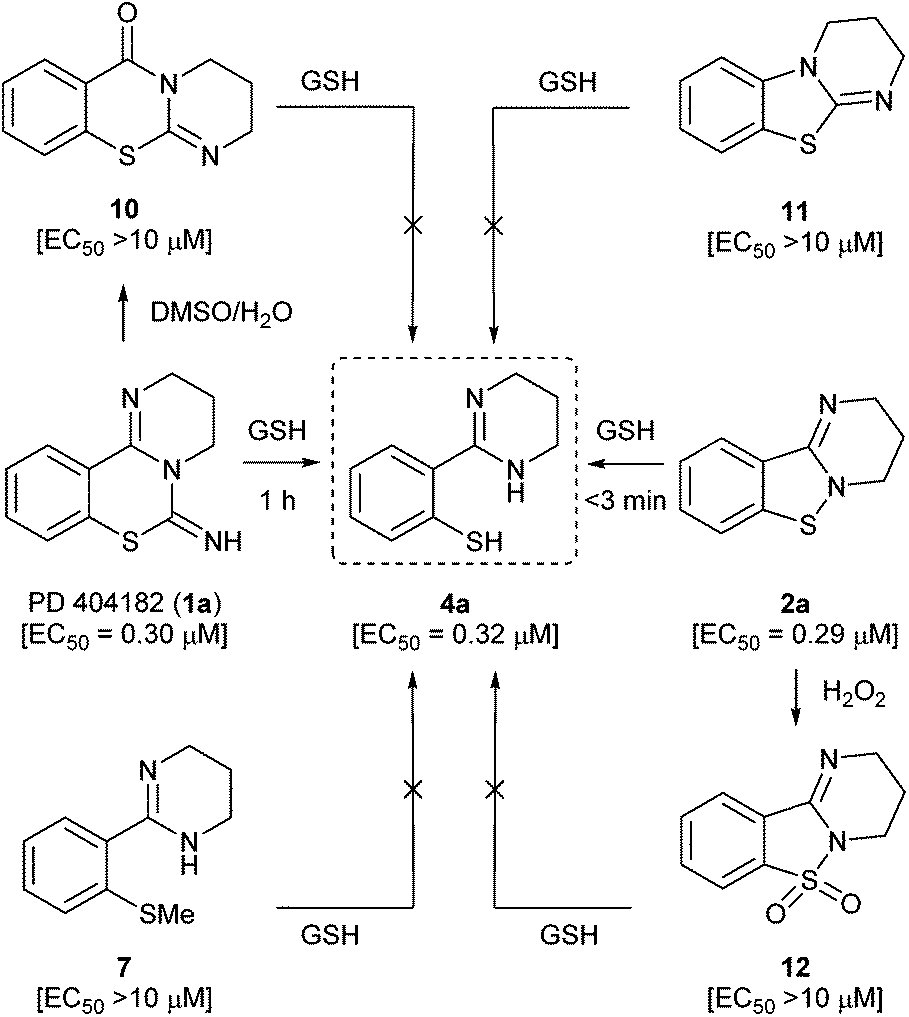 | ||
| Fig. 5 Reactions of PD 404182, benzo[4,5]isothiazolo[2,3-a]pyrimidines and the inactive analogues under reductive conditions in the presence of GSH at 37 °C. EC50 values represent the concentration of the compound required to inhibit the HIV-1 infection by 50%. The data were obtained from three independent experiments by the NCK assay. | ||
Taken together, the structure–activity relationships and antiviral profiles were similar between the PD 404182 derivatives 1 and benzo[4,5]isothiazolo[2,3-a]pyrimidine derivatives 2. Under the reductive conditions corresponding to the intracellular environment, these scaffolds were efficiently converted to the common ring-opened thiophenol derivatives with potent anti-HIV activity. In addition, these two scaffolds inhibited not only HIV-1 infection at an early stage, but also several RNA and DNA viruses as we and others recently reported.5–8,10,11 In light of these findings, it was suggested that PD 404182 and benzo[4,5]isothiazolo[2,3-a]pyrimidine derivatives may achieve their antiviral activities by acting through an identical intermediate or via an interaction with a similar target molecule(s) or mechanism of action in an intracellular compartment. A viral target molecule(s) or mechanism(s) has not yet been determined,14 but these heterocyclic compounds could conceivably regulate the defense mechanism(s) of the host cells.
Conclusions
We have unveiled the chemical transformations of pyrimido[1,2-c][1,3]benzothiazin-6-imine and benzo[4,5]isothiazolo[2,3-a]pyrimidine scaffolds, which yield compounds with potent antiviral activities. Under the reductive conditions meant to mimic intracellular GSH levels, the covalent S–N bond of the isothiazole substructure in 2a was cleaved to provide the ring-opened 2-(2-mercaptophenyl)tetrahydropyrimidine 4a with potent anti-HIV activity, which was also generated from PD 404182 (1a) under identical conditions. The similar structure–activity relationships and antiviral profiles between compounds 1a and 2a suggested that these could possibly be the prodrug forms for the common bioactive substance 4a that binds with the potential target molecule(s) in host cells.Experimental section
General
1H NMR spectra were recorded using a JEOL ECA-500 spectrometer. Chemical shifts are reported in δ (ppm) relative to Me4Si as an internal standard. 13C NMR spectra were referenced to the residual solvent signal. Exact mass (HRMS) spectra were recorded on a JMS-HX/HX 110A mass spectrometer or Shimadzu LC-ESI-IT-TOF-MS equipment. For flash chromatography, Wakogel C-300E (Wako) and CHROMATOREX NH DM1080 (Fuji Silysia) were employed. For analytical HPLC, a Cosmosil 5C18-ARII column (4.6 × 250 mm, Nacalai Tesque, Inc.) was employed with a linear gradient of CH3CN containing 0.1% (v/v) aq. TFA at a flow rate of 1 cm3 min−1, and eluting products were detected by UV at 254 nm. Preparative HPLC was performed using a Cosmosil 5C18-ARII preparative column (20 × 250 mm, Nacalai Tesque, Inc.) with a linear gradient of CH3CN containing 0.1% (v/v) aq. TFA at a flow rate of 8 cm3 min−1. The purity of the compounds was determined as no less than 95% by combustion analysis or HPLC analysis. Preparation of the compounds 1–3, 4d, 6, 8, 13–15 was already reported from our laboratory.7,8,11,15:0 to 9:1) to give the title compound 4a as yellow crystals (999 mg, quant.); mp 208–210 °C (from n-hexane–CHCl3–MeOH); IR (neat) νmax/cm−1: 3185–3092 (SH), 1590 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); δH (500 MHz, CD3OD): 2.05–2.09 (2H, m, CH2), 3.55 (4H, t, J 5.7, CH2), 6.87–6.91 (1H, m, Ar), 7.06 (1H, ddd, J1 = J2 8.0, J3 1.7, Ar), 7.28 (1H, dd, J1 7.7, J2 1.4, Ar), 7.56 (1H, dd, J1 8.0, J2 1.1, Ar); δC (125 MHz, DMSO-d6): 18.1, 38.1 (2C), 118.7, 122.8, 126.7, 128.8, 137.8, 158.9, 159.8; HRMS (FAB): m/z calcd for C10H13N2S [M + H]+ 193.0799; found: 193.0801.
N); δH (500 MHz, CD3OD): 2.05–2.09 (2H, m, CH2), 3.55 (4H, t, J 5.7, CH2), 6.87–6.91 (1H, m, Ar), 7.06 (1H, ddd, J1 = J2 8.0, J3 1.7, Ar), 7.28 (1H, dd, J1 7.7, J2 1.4, Ar), 7.56 (1H, dd, J1 8.0, J2 1.1, Ar); δC (125 MHz, DMSO-d6): 18.1, 38.1 (2C), 118.7, 122.8, 126.7, 128.8, 137.8, 158.9, 159.8; HRMS (FAB): m/z calcd for C10H13N2S [M + H]+ 193.0799; found: 193.0801.
Method B: 3,4-dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazine-6-thione 3a15 (1.15 g, 4.90 mmol) was suspended in 0.1 N NaOH in MeOH–H2O (9:1, 98.0 cm3). After being stirred under reflux for 12 h, the mixture was quenched with 1 N HCl until pH was adjusted to 7. The whole mixture was extracted with CHCl3–MeOH (95:5), and dried over MgSO4. After concentration, the resulting solid was recrystallized from Et2O–CHCl3–MeOH to give the title compound 4a as an yellow solid (655 mg, 70%): spectral data were in good agreement with those of the compound that was synthesized using method A.
N); δH (500 MHz, DMSO-d6): 1.88–1.93 (2H, m, CH2), 3.44 (4H, t, J 5.7, CH2), 6.58 (1H, dd, J1 = J2 7.7, Ar), 7.29 (1H, d, J 8.0, Ar), 7.53 (1H, d, J 8.0, Ar), 10.85 (2H, br s); δC (125 MHz, DMSO-d6): 17.8, 38.2 (2C), 117.5, 126.2, 128.1, 132.8, 133.4, 160.1, 160.7; anal. calcd for C10H11BrN2S: C, 44.29; H, 4.09; N, 10.33. Found: C, 44.12; H, 4.08; N, 10.31.
:0 to 10:1) followed by recrystallization from n-hexane–CHCl3 to give the title compound 7 as colorless crystals (294 mg, 48%): mp 110–112 °C; IR (neat) νmax/cm−1: 1621 (CN); δH (500 MHz, CDCl3): 1.82–1.87 (2H, m, CH2), 2.43 (3H, s, CH3), 3.45 (4H, t, J 5.7, CH2), 4.68 (1H, br s, NH), 7.12 (1H, ddd, J1 = J2 7.4, J3 1.1, Ar), 7.20 (1H, d, J 6.9, Ar), 7.29 (1H, ddd, J1 = J2 7.6, J3 1.5, Ar), 7.36 (dd, J1 7.4, J2 1.7, 1H, Ar); δC (125 MHz, CDCl3): 16.0, 20.6, 42.1 (2C), 124.6, 125.5, 128.3, 129.1, 136.7, 137.0, 154.8; HRMS (ESI): m/z calcd for C11H15N2S [M + H]+ 207.0956; found: 207.0954.
N); δH (500 MHz, CDCl3): 1.81–1.86 (2H, m, CH2), 3.48 (4H, t, J 5.7, CH2), 4.71 (1H, br s, NH), 5.72 (2H, br s, NH2), 6.61–6.66 (2H, m, Ar), 7.07–7.10 (1H, m, Ar), 7.23–7.26 (1H, m, Ar); δC (125 MHz, CDCl3): 20.7, 42.0 (2C), 116.56, 116.61, 119.3, 126.3, 129.9, 147.1, 155.1; anal. calcd for C10H14N3: C, 68.54; H, 7.48; N, 23.98. Found: C, 68.28; H, 7.57; N, 23.75.
O), 1608 (CN); δH (500 MHz, CDCl3): 1.98–2.03 (2H, m, CH2), 3.57 (2H, t, J 5.7, CH2), 4.02 (2H, t, J 6.0, CH2), 7.17 (1H, d, J 8.0, Ar), 7.26–7.29 (1H, m, Ar), 7.47 (1H, ddd, J1 = J2 7.7, J3 1.6, Ar), 8.24 (1H, dd, J1 8.0, J2 1.1, Ar); δC (125 MHz, CDCl3): 21.1, 42.3, 46.1, 122.7, 124.0, 126.0, 131.0, 133.2, 134.3, 147.0, 161.7; HRMS (FAB): m/z calcd for C11H11N2OS [M + H]+ 219.0592; found: 219.0584. Spectral data were in good agreement with those previously reported.26
Crystal structure: C11H10N2OS, Mw: 218.27, primitive monoclinic, a = 10.7456(6), b = 10.9466(6), c = 16.6473(7) Å, β = 97.422(2)°, V = 1941.78(17) Å3, space group P21/n (no. 14), Z = 8. The data were collected with a Rigaku R-AXIS RAPID diffractometer using graphite monochromated Mo-Kα radiation at −93 K. 18534 Reflections were measured, 4420 unique (Rint = 0.0220) which were used in all calculations. The final wR was 0.0796. The substance was crystallized from toluene. The CCDC deposition number: 1037500.
O), 1632 (CN); δH (500 MHz, CD3OD): 2.31–2.36 (2H, m, CH2), 3.70 (2H, t, J 5.7, CH2), 4.27 (2H, t, J 6.0, CH2), 7.42–7.45 (1H, m, Ar), 7.57–7.61 (2H, m, Ar), 7.84 (1H, d, J 8.0, Ar); δC (125 MHz, CD3OD): 19.2, 41.3, 44.0, 113.5, 118.0 (q, J 291.5), 123.1, 124.1, 126.7, 129.2, 140.0, 162.4 (q, J 36.0), 166.0; HRMS (FAB): m/z calcd for C10H11N2S [M + H]+ 191.0643; found: 191.0641.
:2 to 1:1) to give the title compound 12 as colorless crystals (25.2 mg, 71%): mp 146–148 °C (from Et2O–MeOH); IR (neat) νmax/cm−1: 1670 (CN); δH (500 MHz, CDCl3): 2.01–2.05 (2H, m, CH2), 3.70 (2H, t, J 5.7, CH2), 3.79 (2H, t, J 6.0, CH2), 7.70–7.76 (2H, m, Ar), 7.85–7.88 (1H, m, Ar), 7.98–8.01 (1H, m, Ar); δC (125 MHz, CDCl3): 19.5, 36.5, 44.2, 120.7, 122.8, 129.6, 132.4, 133.6, 134.8, 142.7; anal. calcd for C10H10N2O2S: C, 54.04; H, 4.54; N, 12.60. Found: C, 53.89; H, 4.74; N, 12.36.
:0 to 10:1) to give the title compound 16 as colorless crystals (497 mg, 28%): mp 259–261 °C (from Et2O–MeOH); IR (neat) νmax/cm−1: 3212–3050 (OH), 1620 (CN); δH (500 MHz, DMSO-d6): 1.83–1.88 (2H, m, CH2), 3.40 (4H, t, J 5.7, CH2), 6.27–6.30 (1H, m, Ar), 6.46 (1H, d, J 8.6, Ar), 7.04–7.08 (1H, m, Ar), 7.45 (1H, dd, J1 8.0, J2 1.7, Ar), 12.10 (1H, br s, OH); δC (125 MHz, CD3OD): 20.0, 39.3 (2C), 111.2, 114.5, 124.4, 126.3, 135.1, 160.9, 172.5; anal. calcd for C10H12N2O: C, 68.16; H, 6.86; N, 15.90. Found: C, 68.13; H, 7.03; N, 16.09.
Determination of anti-HIV activity
The anti-HIV activity of a series of compounds against HIV-1IIIB was determined by the NCK assay.29 The target cells (NCK45-β-Gal; 104 cells per well) were plated in 96-well flat microtiter culture plates. On the following day, the cells were inoculated with HIV-1 (60 NCK U per well, giving 60 blue cells after 48 h of incubation) and cultured in the presence of various concentrations of the test compounds in fresh medium. Forty-eight hours after viral exposure, all the blue cells stained with X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) were counted in each well. Cytotoxic effects were not observed at 10 μM except for compound 2g.The activity of test compounds was determined as the concentration that blocked HIV-1 infection by 50% (50% effective concentration [EC50]). EC50 was determined by using the following formula:
| EC50 = 10[log(A/B)×(50−C)/(D−C)+log(B)], |
Glutathione-mediated transformation of pyrimidobenzothiazine and isothiazolopyrimidine derivatives
The compound (1 mM) was incubated in 10 mM GSH (in a mixture of 50 mM phosphate buffer (pH 7.4) and MeCN [75:25 (v/v) or 1:1 (v/v)]) at 37 °C. The sample was analyzed by HPLC and the peak area was recorded by UV detection at 254 nm. The concentrations of the products were calculated using previously determined calibration curves. Of note, the GSH-mediated transformation from 1a was slightly accelerated by the addition of MeCN in phosphate buffer.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research from JSPS, Japan; Platform for Drug Discovery, Informatics, and Structural Life Science from MEXT, Japan; a Grant-in-Aid for Research on HIV/AIDS from the Ministry of Health and Welfare of Japan. T.M. is grateful for JSPS Research Fellowships for Young Scientists.References
- PD 404182 (1a) was previously reported to be an enzyme inhibitor against 3-deoxy-D-manno-octulosonic acid 8-phosphate synthase2 and phosphopantetheinyl transferase.3 In addition, it was recently reported that PD 404182 (1a) inhibits human dimethylarginine dimethylaminohydrolase isoform 1 (DDAH).4.
- M. R. Birck, T. P. Holler and R. W. Woodard, J. Am. Chem. Soc., 2000, 122, 9334–9335 CrossRef CAS.
- C. R. Vickery, N. M. Kosa, E. P. Casavant, S. Duan, J. P. Noel and M. D. Burkart, ACS Chem. Biol., 2014, 9, 1939–1944 CrossRef CAS PubMed.
- Y. T. Ghebremariam, D. A. Erlanson and J. P. Cooke, J. Pharmacol. Exp. Ther., 2014, 348, 69–76 CrossRef PubMed.
- K. Chockalingam, R. L. Simeon, C. M. Rice and Z. Chen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3764–3769 CrossRef CAS PubMed.
- A. M. Chamoun, K. Chockalingam, M. Bobardt, R. Simeon, J. Chang, P. Gallay and Z. Chen, Antimicrob. Agents Chemother., 2012, 56, 672–681 CrossRef CAS PubMed.
- T. Mizuhara, S. Oishi, H. Ohno, K. Shimura, M. Matsuoka and N. Fujii, Org. Biomol. Chem., 2012, 10, 6792–6802 CAS.
- T. Mizuhara, S. Oishi, H. Ohno, K. Shimura, M. Matsuoka and N. Fujii, Bioorg. Med. Chem., 2012, 20, 6434–6441 CrossRef CAS PubMed.
- T. Mizuhara, S. Oishi, H. Ohno, K. Shimura, M. Matsuoka and N. Fujii, Bioorg. Med. Chem., 2013, 21, 2079–2087 CrossRef CAS PubMed.
- A. M. Chamoun-Emanuelli, M. Bobardt, B. Moncla, M. K. Mankowski, R. G. Ptak, P. Gallay and Z. Chen, Antimicrob. Agents Chemother., 2014, 58, 687–697 CrossRef PubMed.
- S. Okazaki, T. Mizuhara, K. Shimura, H. Murayama, H. Ohno, S. Oishi, M. Matsuoka and N. Fujii, Bioorg. Med. Chem., 2015, 23, 1447–1452 CrossRef CAS PubMed.
- M. Baba, D. Schols, R. Pauwels, H. Nakashima and E. De Clercq, J. Acquired Immune Defic. Syndr., 1990, 3, 493–499 CrossRef CAS PubMed.
- T. Matthews, M. Salgo, M. Greenberg, J. Chung, R. DeMasi and D. Bolognesi, Nat. Rev. Drug Discovery, 2004, 3, 215–225 CrossRef CAS PubMed.
- Of note, it was reported that PD 404182 showed the antiviral activities by the virucidal effect via the physical disruption of virions, see ref. 6 and 10.
- T. Mizuhara, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2010, 75, 265–268 CrossRef CAS PubMed.
- M. Ishihara and H. Togo, Tetrahedron, 2007, 63, 1474–1480 CrossRef CAS PubMed.
- For a review on the function of glutathione, see: R. Franco, O. J. Schoneveld, A. Pappa and M. I. Panayiotidis, Arch. Physiol. Biochem., 2007, 113, 234–258 CrossRef CAS PubMed and references cited therein.
- Compound 4a was also identified from HTS as an antibacterial agent that inhibits localization of lipoproteins (Lol) during cell wall biosynthesis, see: H. Ito, A. Ura, Y. Oyamada, H. Yoshida, J. Yamagishi, S. Narita, S. Matsuyama and H. Tokuda, Microbiol. Immunol., 2007, 51, 263–270 CrossRef CAS PubMed.
- During the course of our investigations, an initial report describing GSH-mediated degradation of PD 404182 appeared, see ref. 4.
- In the absence of GSH, compound 1a was converted into pyrimidothiazinone 10 as discussed above.
- R. Mitsuhashi, T. Suzuki and Y. Sunatsuki, Inorg. Chem., 2013, 52, 10183–10190 CrossRef CAS PubMed.
- J. A. Loo, T. P. Holler, J. Sanchez, R. Gogliotti, L. Maloney and M. D. Reily, J. Med. Chem., 1996, 39, 4313–4320 CrossRef CAS PubMed.
- T. Vercruysse, B. Basta, W. Dehaen, N. Humbert, J. Balzarini, F. Debaene, S. Sanglier-Cianférani, C. Pannecouque, Y. Mély and D. Daelemans, Retrovirology, 2012, 9, 95–108 CrossRef CAS PubMed.
- C. Pannecouque, B. Szafarowicz, N. Volkova, V. Bakulev, W. Dehaen, Y. Mély and D. Daelemans, Antimicrob. Agents Chemother., 2010, 54, 1461–1468 CrossRef CAS PubMed.
- The transformations from 1f and 2f into the ring-opened thiophenol derivative were also observed in the presence of GSH.
- D. Chen, J. Wu, J. Yang, L. Huang, Y. Xiang and W. Bao, Tetrahedron Lett., 2012, 53, 7104–7107 CrossRef CAS PubMed.
- P. A. Woods, L. C. Morrill, T. Lebl, A. M. Z. Slawin, R. A. Bragg and A. D. Smith, Org. Lett., 2010, 12, 2660–2663 CrossRef CAS PubMed.
- S. Sivaramakrishnan, A. H. Cummings and K. S. Gates, Bioorg. Med. Chem. Lett., 2010, 20, 444–447 CrossRef CAS PubMed.
- K. Kajiwara, E. Kodama, Y. Sakagami, T. Naito and M. Matsuoka, J. Clin. Microbiol., 2008, 46, 792–795 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1037500. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00301f |
| This journal is © The Royal Society of Chemistry 2015 |