
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, conformational studies, and biological properties of phosphonomethoxyethyl derivatives of nucleobases with a locked conformation via a pyrrolidine ring†
Radek
Pohl
a,
Lenka
Poštová Slavětínská
a,
Wai Soon
Eng
b,
Dianne T.
Keough
b,
Luke W.
Guddat
b and
Dominik
Rejman
*a
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovo nám. 2, 166 10 Prague 6, Czech Republic. E-mail: rejman@uochb.cas.cz
bThe School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, 4072 QLD, Australia. E-mail: luke.guddat@uq.edu.au
First published on 13th March 2015
Abstract
Systematic structure–activity studies on a diverse family of nucleoside phosphonic acids has led to the development of potent antiviral drugs such as HPMPC (CidofovirTM), PMEA (AdefovirTM), and PMPA (TenofovirTM), which are used in the treatment of CMV-induced retinitis, hepatitis B, and HIV, respectively. Here, we present the synthesis of a novel class of acyclic phosphonate nucleotides that have a locked conformation via a pyrrolidine ring. NMR analysis of these compounds revealed that the pyrrolidine ring has a constrained conformation when in the cis-form at pD < 10 via hydrogen bonding. Four of these compounds were tested as inhibitors of the human and Plasmodium falciparum 6-oxopurine phosphoribosyltransferases. The most potent has a Ki of 0.6 μM for Plasmodium falciparum HGXPRT.
Introduction
Over the past few decades, there has been an enormous effort devoted to the synthesis and biochemical and biological evaluation of analogs of natural nucleosides and nucleotides. This has largely been due to their usefulness as tools in investigations aimed at a thorough understanding of metabolic processes. Such analogues exhibit their biological properties either via inhibition of enzymes of the nucleoside/nucleotide metabolism pathway or in the triphosphorylated form, where they can become incorporated into DNA or RNA via the action of polymerases or transcriptases. Among the most successful classes of nucleotide analogs are those which possess the enzymatically and chemically stable phosphonate moiety1 as a replacement for the phosphate group. The advantage of this modification is that it improves the stability by preventing hydrolysis of the phosphate ester bond.Systematic investigation of the structurally diverse nucleoside phosphonic acids has led to the development of potent antiviral drugs. Their chemistry is based on both the acyclic phosphonate nucleotides 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2,3 and cyclic counterparts 2 and 3 (Fig. 1).4 Specifically, acyclic compounds 1a–c were shown to inhibit the replication of DNA viruses and retroviruses, whereas cyclic compounds 2a–d and 3a–d exhibited favorable antiviral profiles against HIV strains. Thus, the modification of the sugar-phosphate moiety of the nucleotides is a successful approach in contributing to the pool of potential antivirals.
2,3 and cyclic counterparts 2 and 3 (Fig. 1).4 Specifically, acyclic compounds 1a–c were shown to inhibit the replication of DNA viruses and retroviruses, whereas cyclic compounds 2a–d and 3a–d exhibited favorable antiviral profiles against HIV strains. Thus, the modification of the sugar-phosphate moiety of the nucleotides is a successful approach in contributing to the pool of potential antivirals.
 | ||
| Fig. 1 Structures of nucleotide analogs. | ||
Furthermore, several types of aza-sugar nucleoside phosphonates, such as the pyrrolidine 4,5,6 isoxazolidine 57 and 6,8 and aziridine 79 ring-containing compounds, have been reported. However, among these only analogues 6a–e exerted significant inhibition of HIV reverse transcriptase comparable to AZT as well as maintained a low level of cytotoxicity. Pyrrolidine nucleosides 8 and 910 served as the starting point for the synthesis of pyrrolidine phosphonate analogs of nucleotides 10a–12, recently described by our group.11 Phosphonate derivative 13 has been found to be a potent inhibitor of thymidine phosphorylase from spontaneous SD-rat lymphoma cells exhibiting an IC50 of 11 nM12 and guanine derivative 14 exhibited nanomolar activity against human purine nucleoside phosphorylase.13
Herein, we present the synthesis and biological evaluation of phosphonomethoxyethyl derivatives of nucleobases 15a–e and 16a–c (Fig. 2) that are conformationally locked via a pyrrolidine ring. The conformational lock may, in principle, increase the entropy contribution to the binding energy of the ligand to its biological target. Herein we attempted to compare inhibition properties of PMEG and PMEHx with their conformationally locked counterparts 15b–c and 16b–c towards oxopurineribosyltransferases.
 | ||
| Fig. 2 Structures of target compounds 15a–e and 16a–c. | ||
Results and discussion
Synthesis
The synthesis of the title compounds could be divided into two parts: (A) synthesis of the pyrrolidine phosphonate intermediate containing either a hydroxyl (19) or primary amino group (23), and (B) attaching the nucleobase to the intermediate (via the Mitsunobu reaction in the case of the hydroxyl derivative or nucleobase assembly procedure on the amino moiety). Two routes to the synthesis of the amino intermediate 23 were evaluated (Scheme 1).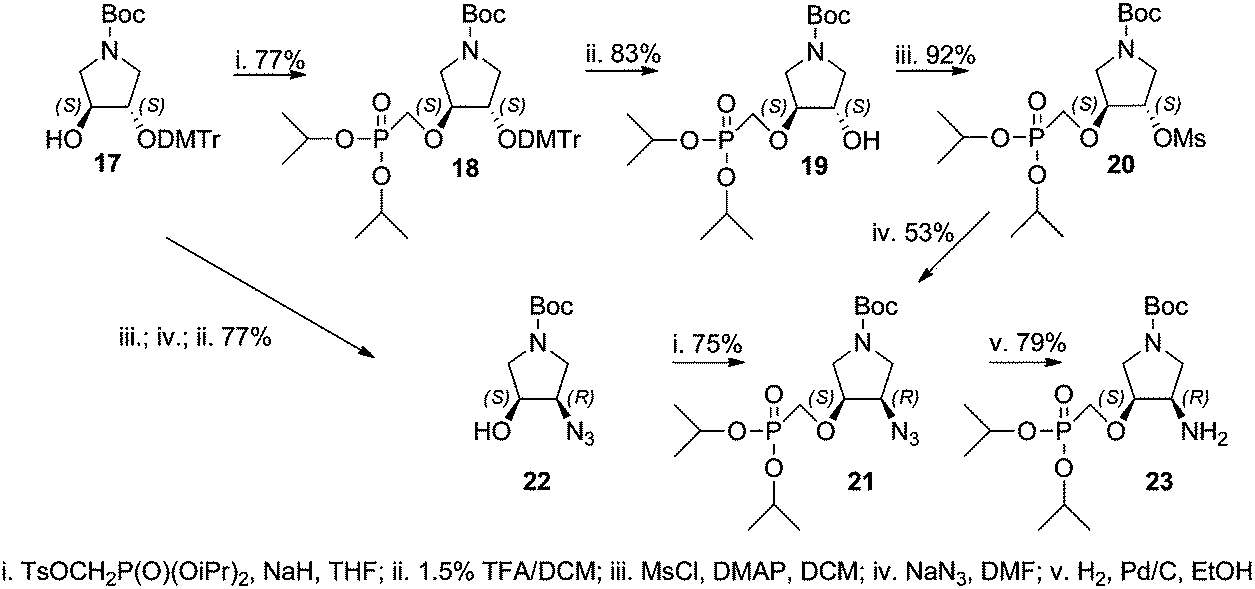 | ||
| Scheme 1 Synthesis of precursors 19 and 23. | ||
Monodimethoxytrityl derivative 17 was reacted with diisopropyl tosyloxymethanephosphonate to afford phosphonate 18 that was treated with 1.5% TFA in DCM to yield the first intermediate 19. Compound 19 was mesylated and treated with sodium azide giving azido derivative 21. This reaction was accompanied by removal of one isopropyl ester group decreasing the yield of 21. Thus, a different route to azido derivative 21 was explored. Monodimethoxytrityl derivative 17 was first converted to the azido derivative 22 that subsequently reacted with diisopropyl tosyloxymethanephosphonate. The obtained azido derivative 21 was finally converted to the amino derivative 23 by catalytic hydrogenation over a palladium catalyst. The chloropurine intermediate 25 was prepared by the Mitsunobu reaction of hydroxy derivative 19 with 6-chloropurine (24) (Scheme 2). Adenine derivative 15a was prepared from 25 by aminolysis with conc. aqueous ammonia and dioxane followed by stirring with 20% TFA in DCM (removal of the Boc protecting group) and finally by bromotrimethylsilane treatment (to remove isopropyl esters). The hypoxanthine derivative 15b was prepared from the same intermediate 25 by bromotrimethylsilane treatment followed by heating with aq. 3 M HCl.
 | ||
| Scheme 2 Synthesis of adenine and hypoxanthine derivatives 15a and 15b. | ||
The guanine nucleobase was formed on the amino moiety of 23 using a standard procedure employing 2,5-diamino-4,6-dichloropyrimidine (26) according to Scheme 3.14
 | ||
| Scheme 3 Synthesis of guanine derivative 15c. | ||
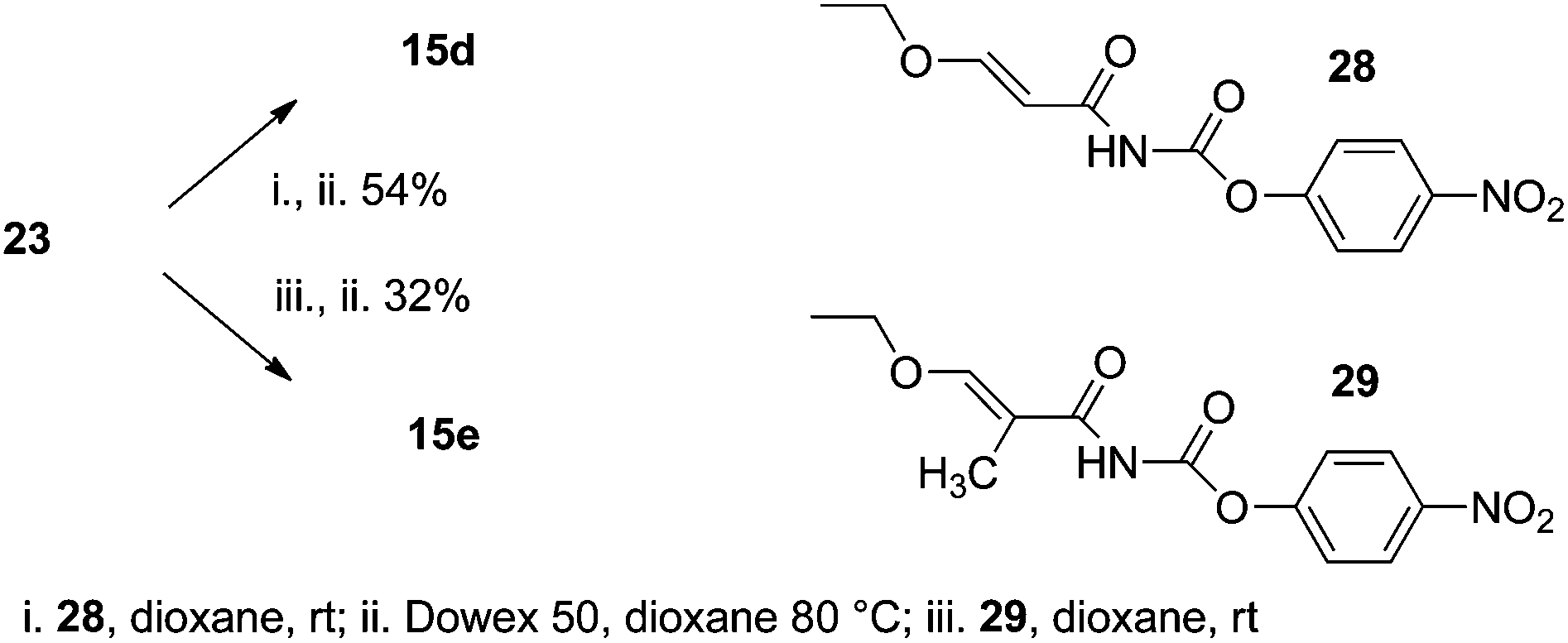
The reaction of amine 23 with reagent 2815 leads to the formation of a linear intermediate with a high yield (Scheme 4). This intermediate, after silica gel chromatography purification, was dissolved in dioxane and heated with Dowex 50 in H+ for 5 h. The treatment with Dowex accomplished the cyclisation of the uracil moiety, removal of the Boc protecting group and, surprisingly, removal of both isopropyl ester groups, thus leading to the final uracil derivative 15d. The thymine derivative 15e was prepared by the same procedure except that reagent 28 was replaced by reagent 2915 (Scheme 4).
 | ||
| Scheme 4 Synthesis of uracil and thymine derivatives 15d and 15e. | ||
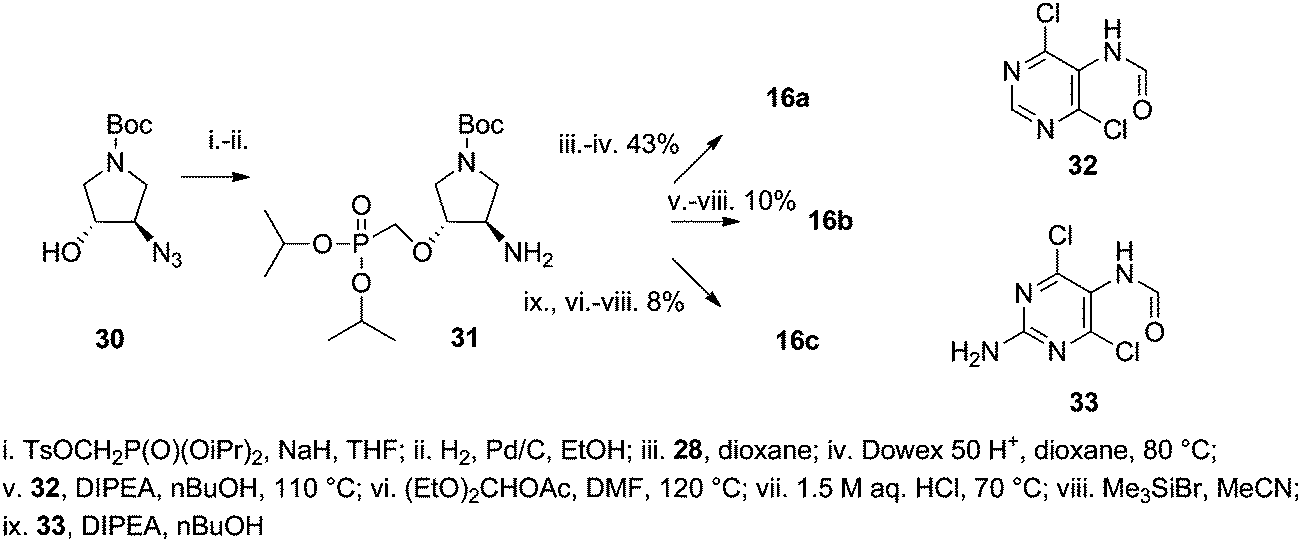
The uracil derivative 16a with a trans configuration was prepared using the same synthetic procedure as for derivative 15d (Scheme 5). The starting azido derivative 30 was prepared according to our previously published procedure.13 Hypoxanthine derivative 16b and guanine derivative 16c were prepared using the nucleobase assembly approach adopted from ref. 16 (employing 4,6-dichloro-5-formamidopyrimidine (32) and 2-amino-4,6-dichloro-5-formamidopyrimidine (33) respectively) followed by bromotrimethylsilane promoted isopropyl ester group removal. The reaction of amino derivative 31 with 32 and 33 did not lead to purine ring closure so additional treatment with diethoxymethyl acetate in DMF at elevated temperatures was required. It appears that the nucleobase assembly on a primary amino group is the preferred procedure for introduction of thymine or uracil but for purine bases the Mitsunobu alkylation is the method of choice.
 | ||
| Scheme 5 Synthesis of uracil, hypoxanthine, and guanine derivatives 16a–c. | ||
NMR conformational analysis
The final compounds 15a–e and 16a–c were fully characterized by 1H, 13C and 31P NMR in D2O solutions. The cis and trans relative configuration of uracil derivatives 15d and 16a was determined by inspection of the H,H-ROESY spectrum. Thus, a strong NOE cross-peak of H-6 from the uracil nucleobase and H-3′ from the pyrrolidine moiety can be found in the H,H-ROESY spectrum of trans-derivative 16a. This NOE interaction is missing in the case of cis-derivative 15d. In addition, cis and trans isomers differ significantly in the magnitudes of 3J(H,H) coupling constants of pyrrolidine protons. Characteristic values of 3J(3′,4′) that can be used for determination of the relative configuration directly from 1H NMR spectra are 4.1–5.3 Hz for cis-derivatives 15a–e and 1.2–2.0 Hz for trans-derivatives 16a–c.Since the original acyclic phosphonate moiety in 15a–e and 16a–c is conformationally restricted by the five-membered pyrrolidine ring we were interested in conformation preferences of such pyrrolidine derivatives. Taking into account that molecules contain both acidic (phosphonic acid) and basic (pyrrolidine component) moieties, we first examined at which pD deuteration/dedeuteration transitions take place (Fig. 3).
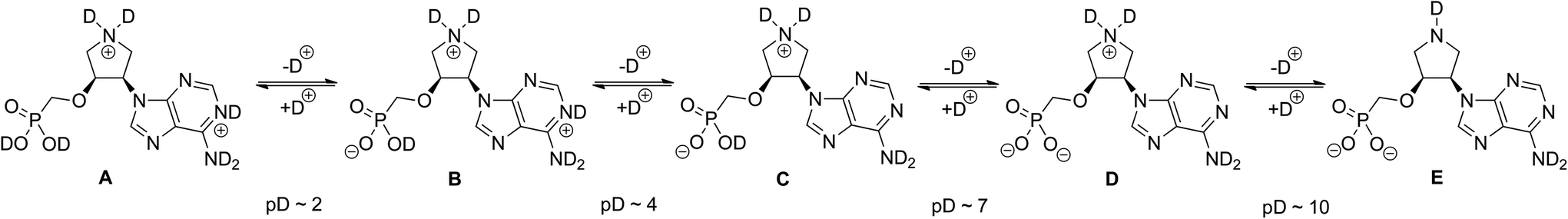 | ||
| Fig. 3 Deuteration/dedeuteration transitions of 15a in D2O at different pD values. | ||
Therefore, D2O solutions of 15a and 16c were titrated with diluted solutions of DCl in D2O or NaOD in D2O and 1H, 13C and 31P NMR spectra were acquired (see ESI†). Based on the titration curves five different deuterated/dedeuterated forms A–E of 15a can be observed at different pD values (Fig. 3). The pyrrolidine nitrogen remains in positively charged deuterated form C until pD ∼ 10. This is manifested by the H-2′ and H-5′ 1H chemical shift changes or C-2′ and C-5′ 13C chemical shift changes. At pD ∼ 4, dedeuteration of adenine nitrogen N-1 was observed by the changes in the 13C chemical shift of C-2 and C-6. We have also found that deuteration/dedeuteration of other derivatives 15b–e and 16a–b follows the same trends resulting in dedeuteration of positively charged pyrrolidine nitrogen at pD ∼ 10.
The protonation/deprotonation or deuteration/dedeuteration of the pyrrolidine nitrogen can influence the conformation of the five-membered pyrrolidine ring (Fig. 4).
 | ||
| Fig. 4 Pyrrolidine pseudorotation pathway (P = 0° to 360°) of PME derivatives 15a–e and 16a–c. The sign B stands for a nucleobase and R means a phosphonomethyl moiety. | ||
The particular conformation is described by two pseudorotation parameters: by the phase angle (P) and by the maximum puckering amplitude (ϕmax).17 The phase angle is a periodic variable indicating which ring atoms are situated outside the ring plane and can reach 0°–360°. The maximum puckering amplitude describes the degree of distortion of the five-membered ring out of the plane and its value is usually in the range of 35°–45°. Therefore, we examined conformation preferences of the pyrrolidine ring in cis-adenine derivative (15a) and trans-guanine derivative (16c) at low (<2.0) and high (>12.0) pD values. The conformation analysis based on the concept of pseudorotation17 was performed using 3J(H,H) spin–spin couplings of pyrrolidine ring protons within the Matlab Pseudorotation GUI program18 and the methodology developed for the conformational analysis of pyrrolidine nucleotide analogues we have published previously.19 In trans-derivative 16c, we observed only negligible changes in 3J(H,H) of pyrrolidine protons upon pD change indicating little or no change in the conformation of the pyrrolidine ring. This assumption was later confirmed by the conformation analysis of 16c (Fig. 5) that revealed the existence of very similar conformations at both high and low pD.
 | ||
| Fig. 5 Conformation of the pyrrolidine ring of derivative 16c at different pD values in a D2O solution examined by NMR. | ||
Changes in 3J(H,H) of pyrrolidine protons of cis-derivative 15a upon pD change (Fig. 6) on the other hand suggest that the dedeuteration of the pyrrolidine ring at pD ∼ 10 may result in changes of the pyrrolidine ring conformation.
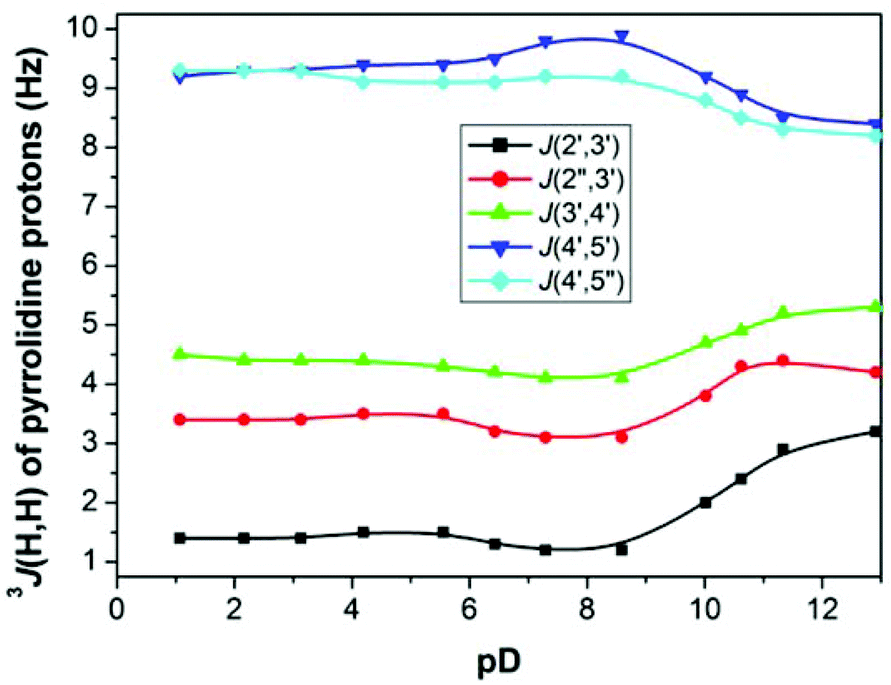 | ||
| Fig. 6 Changes in the values of 3J(H,H) coupling constants of derivative 15a upon pD change. | ||
The conformation analysis of cis-derivative 15a at pD < 10 revealed the exclusive existence of one conformer (P = 26°) constrained by strong hydrogen bonding between the phosphonate moiety and the deuterated positively charged pyrrolidine nitrogen (Fig. 7). This hydrogen bonding is weakened as a consequence of dedeuteration at pD > 10, which results in an equilibrium of two conformers (P1 = 26° (75%), P2 = 253° (25%)) in D2O solution. Similar behavior was also observed for hypoxanthine and guanine derivatives 15b and 15c, respectively.
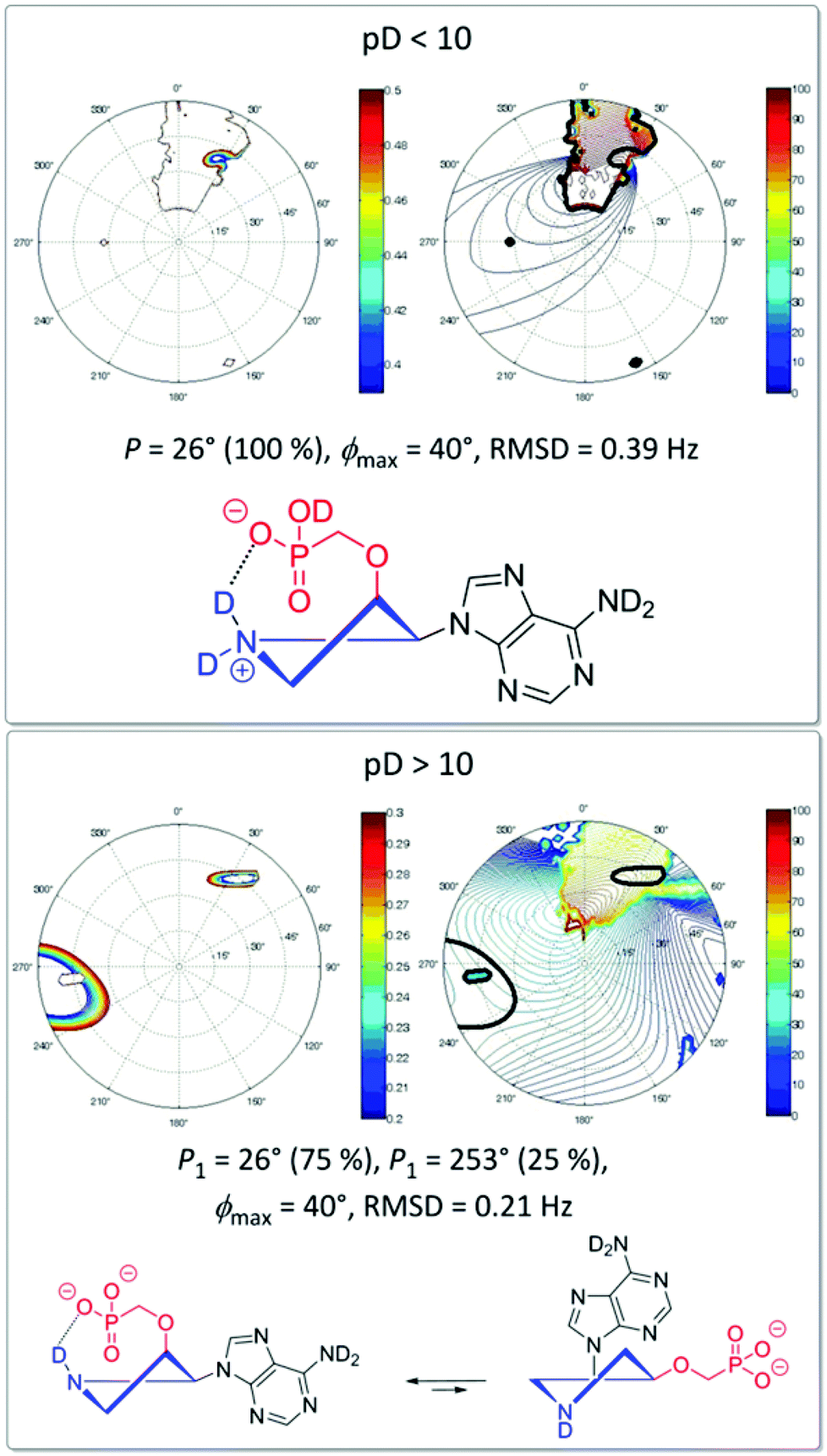 | ||
| Fig. 7 Conformation of the pyrrolidine ring of derivative 15a at different pD values in a D2O solution examined by NMR. | ||
Biological activity
Compounds 15a–e and 16a–c were tested for their cytostatic (HepG2, HL60, HeLa S3, CCRF-CEM), antimicrobial, and antifungal (Escherichia coli CCM 3954, Enterococcus faecalis CCM 4224, Pseudomonas aeruginosa CCM 3955, Staphylococcus aureus CCM 4223, Bacillus subtilis, Streptococcus agalactiae, Candida albicans, and Candida krusei) activities but no significant effects were observed.Compounds 15a–e and 16a–b did not exhibit any inhibitory activity against human mitochondrial (mdN) and cytosolic (cdN) 5′(3′)-deoxynucleotidases.20 Compounds 15a and 15e were tested in a HCV replicon assay and did not exhibit any activity at a concentration below 50 μM.
Inhibition of human HGPRT and PfHGXPRT by pyrrolidine derivatives of PME derivatives of the acyclic nucleoside phosphonates
The Ki values of four compounds were determined for human hypoxanthine-guanine phosphoribosyltransferase (HGPRT) and P. falciparum (Pf) hypoxanthine-guanine-xanthine phosphoribosyltransferase (HGXPRT) – a potential drug target for treatment of malaria (Table 1). There are two chemical differences between these compounds: (i) the purine base is either guanine or hypoxanthine; and (ii) there are two isomers. One has the S configuration at the carbon atom of the five membered ring and the second has the R configuration.The data show that compounds as the S-isomer have lower Ki values for the parasite enzyme while those that are the R-isomer have lower Ki values for the human enzyme. The inhibitors containing hypoxanthine (15bvs.15c and 16bvs.16c) as the base have lower Ki values for the parasite enzyme but the reverse is true for the human enzyme as it favours compounds with guanine as the base.
Structural analysis
Docking studies were undertaken to try to understand how the pyrrolidine derivatives bind in the active site.22 The crystal structures of human HGPRT in complex with 9-2-[-2(phosphonoethoxy)ethyl]guanine (PEEG) and 9-2-[-2(phosphonoethoxy)ethyl]hypoxanthine (PEEHx) (PDB: 3GGC and 3GGJ, respectively) were used as the model template.21 The PEE compounds contain an extra carbon atom in the linker connecting the N9 atom of the purine ring with the phosphorus atom of the phosphonate group compared with 15b, 15c, 16b and 16c. However, they are similar in that they both contain an oxygen atom two atoms distal to the N9. The acyclic nucleoside phosphonates (PEEG and PEEHx) bind to two key regions in the active site of human HGPRT: the purine binding site and the 5′-phosphate binding pocket (D137-T141).21 To validate this approach, we first docked PEEG and PEEHx into the protein devoid of the ligand. The results showed that all the highest scoring docking poses correlated with the position observed in the crystal structure. The rmsd for all atoms in the ligands was <0.2 Å (Fig. 8a and b). The docked structures of 15b and 16b are compared in Fig. 8c and those of 15c and 16c are compared in Fig. 8d. | ||
| Fig. 8 Molecular docking models and the crystal structure of the nucleoside phosphonates in the active site of human HGPRT. (a) Comparison of the location of PEEHx in the crystal structure (carbon atoms in cyan) with that of the docked compound (magenta). (b) Comparison of the location of PEEG in the crystal structure (carbon atoms in tan) with that of the docked compound (bright green). (c) 15b (yellow) and 16b (carbon atoms in cream). (d) 15c (pale green) and 16c (carbon atoms in white). | ||
These data show that the phosphonate group in all four compounds is found in the 5′-phosphate binding pocket, but it can have two different orientations depending on the isomer. Thus, the chemical structure of the two isomers appears to be responsible for the location of the phosphonate group. Therefore, the nature of the base itself does not affect the location of the phosphonate group (cf.15b with 15c and 16b with 16c). The differences in affinity for each isomer with either guanine or Hx as the base only relates to the binding of the base itself as human HGPRT has a higher affinity for guanine over hypoxanthine but PfHGXPRT binds hypoxanthine better than guanine.23 For the weaker inhibitors of human HGPRT (15b and 15c), the phosphonate group does not reach as far into the 5′-phosphate binding pocket as it does in 16b and 16c. This location of the phosphonate group in the active site could be one of the contributing factors for the differences in the Ki values between the human and Pf enzymes for the two isomers. In comparison, PfHGXPRT favours the S-isomers of the pyrrolidine nucleoside phosphonates over their R-isomer counterparts. The docking studies suggest that the “open” structure of these pyrrolidine derivatives is preferred to the “locked” structure when these compounds bind in the active site of the 6-oxopurine phosphoribosyltransferases.
Conclusions
Eight PME derivatives of nucleobases with a locked conformation via a pyrrolidine ring have been synthesized. Pyrimidine derivatives were prepared in good yields via nucleobase construction on a primary amine. In the case of purine bases, Mitsunobu coupling with a hydroxy derivative appeared to be a better approach than construction of the nucleobase on a primary amino group. NMR conformation analysis revealed that the conformation of the pyrrolidine ring in cis-series 15 is pH dependent. It was found that protonation of the pyrrolidine ring at pH < 10 is responsible for the constraining of the conformation and the exclusive existence of one conformer. Derivatives bearing hypoxanthine and guanine nucleobases were tested as inhibitors of the human and Plasmodium falciparum 6-oxopurine phosphoribosyltransferases. The most potent compound 15b has a Ki of 0.6 μM for Pf HGXPRT with the selectivity in favour of the Pf enzyme over its human counterpart of approx. 120-fold (S-isomer). However, when the purine base is the same but the isomer is different (R-isomer), this selectivity changes in favour of the human enzyme (14-fold in favour of the human enzyme). Docking studies suggest that the R-isomer is favoured for the human HGPRT because the phosphonate group reaches further into the 5′-phosphate binding pocket. This allows the phosphoryl oxygens to form more hydrogen bonds with the amino acid side chain or main chain atoms in the flexible loop surrounding this group. These findings will help us in designing better and more selective inhibitors of parasite HGXPRT.Experimental
Unless stated otherwise, all used solvents were anhydrous. The final products were lyophilized from water, and dried over phosphorus pentoxide at 50–70 °C and 13 Pa. TLC was performed on silica gel pre-coated aluminium plates silica gel/TLC-cards, UV 254 (Fluka), and compounds were detected with UV light (254 nm), by heating (detection of the dimethoxytrityl group; orange color), by spraying with a 1% solution of ninhydrin to visualize amines, and by spraying with a 1% solution of 4-(4-nitrobenzyl)pyridine in ethanol followed by heating and treating with gaseous ammonia (blue color of mono- and diesters of phosphonic acid). Preparative column chromatography was carried out on silica gel (40–60 μm; Fluka) neutralized with triethylamine (1 ml 100 g−1), and elution was performed at the flow rate of 40 ml min−1. The following solvent systems were used for TLC and preparative chromatography: toluene–ethyl acetate 1:1 (T); chloroform–ethanol 9:1 (C1); ethyl acetate–acetone–ethanol–water 6:1:1:0.5 (H3); ethyl acetate–acetone–ethanol–water 4:1:1:1 (H1). The concentrations of solvent systems are stated as volume percentages (%, v/v). Analytical RP HPLC was performed on an LC5000 Liquid Chromatograph (INGOS-PIKRON, CR) using a Luna C18 (2) column (4.6 × 150 mm) at a flow rate of 1 ml min−1 by gradient elution of methanol in 0.1 M TEAA pH 7.5 (A = 0.1 M TEAA; B = 0.1 M TEAA in 50% aqueous methanol; C = methanol). Mass spectra were recorded on a ZAB-EQ (VG Analytical) instrument, using FAB (ionization with Xe, accelerating voltage 8 kV). Glycerol and thioglycerol were used as matrices. NMR spectra were measured on Bruker AVANCE 600 (1H at 600.1 MHz, 13C at 150.9 MHz), Bruker AVANCE 500 and Varian UNITY 500 (1H at 500.0 and 499.8 MHz, 13C at 125.7 MHz, 31P at 202.3 MHz) spectrometers. Chemical shifts (in ppm, δ scale) were referenced to the solvent signal (CDCl3, 1H: 7.26 ppm, 13C: 77.0 ppm; DMSO-d6, 1H: 2.50 ppm, 13C 39.7 ppm), or to dioxane as the external standard when D2O solutions were used (1H: 3.75 ppm, 13C: 69.3 ppm). Coupling constants (J) are given in Hz. Complete assignment of protons and carbons was done by analysis of correlated homonuclear 2D-COSY and heteronuclear 1H-13C HSQC and 1H-13C HMBC spectra. The relative configuration was checked using DPFGSE-NOE and 2D-ROESY techniques.
[3S,4R]-(4-(Adenin-9-yl)pyrrolidin-3-yl)oxymethanephosphonic acid 15a
A mixture of 25 (0.72 g, 1.38 mmol), dioxane (14 ml) and conc. aq. ammonia (50 ml) was stirred at 50 °C in a sealed flask for five days. The mixture was concentrated in vacuo. The protected adenine intermediate (HRMS (FAB+) for C21H36N6O6P (M + H)+ calcd 499.2428, found 499.2429) was obtained by column chromatography on silica gel using a linear gradient of ethanol in chloroform.This intermediate (0.69 g, 1.384 mmol) was without further characterisation dissolved in 20% TFA in DCM (20 ml). The reaction mixture was stirred at rt overnight. The reaction mixture was diluted with chloroform (50 ml) and extracted with water (2 × 50 ml). The aqueous phase was applied to a column of Dowex 50 in H+ form. Dowex was washed with water (200 ml) and eluted with 3% aq. ammonia. The yellowish solution was evaporated. The residue was co-evaporated with ethanol (1 × 20 ml) and acetonitrile (2 × 20 ml) and dissolved in DMF (15 ml). Bromotrimethylsilane (1 ml, 7 mmol) was added under an argon atmosphere and the reaction mixture was stirred at rt for two days. The reaction mixture was concentrated in vacuo. 2 M aq. TEAB (5 ml) and ethanol (10 ml) were added. The solution was concentrated in vacuo. The title compound was obtained by preparative HPLC on the reversed phase using a linear gradient of methanol in 0.1 M aq. TEAB. After conversion to sodium salt by passing through a column of Dowex 50 in Na+ form (30 ml) the title compound was obtained in 58% (0.27 g, 0.803 mmol) yield as a fluffy solid (after lyophilisation from water).
1H NMR (499.8 MHz, D2O, 25 °C): 3.35 (dd, 1H, Jgem = 12.1, JH,P = 8.2, CHaHbP); 3.48 (dd, 1H, Jgem = 12.1, JH,P = 10.3, CHaHbP); 3.54 (dd, 1H, Jgem = 12.9, J2′b,3′ = 3.0, H-2′b); 3.80 (dd, 1H, Jgem = 12.0, J5′b,4′ = 9.7, H-5′b); 3.86 (dd, 1H, Jgem = 12.9, J2′a,3′ = 1.2, H-2′a); 3.91 (dd, 1H, Jgem = 12.0, J5′a,4′ = 9.1, H-5′a); 4.43 (ddd, 1H, J3′,4′ = 4.1, J3′,2′ = 3.0, 1.2, H-3′); 5.37 (ddd, 1H, J4′,5′ = 9.7, 9.1, J4′,3′ = 4.1, H-4′); 8.21 (s, 1H, H-2); 8.58 (s, 1H, H-8).
13C NMR (125.7 MHz, D2O, 25 °C): 48.39 (CH2-5′); 51.32 (CH2-2′); 57.10 (CH-4′); 69.95 (d, JC,P = 150.7, CH2P); 80.73 (d, JC,P = 12.1, CH-3′); 120.57 (C-5); 144.96 (CH-8); 152.08 (C-4); 155.29 (CH-2); 158.27 (C-6).
31P{1H} NMR (202.3 MHz, D2O, 25 °C): 12.86.
IR νmax(KBr) 2370 (w, vbr), 1644 (s), 1605 (s), 1576 (m), 1509 (w), 1477 (m), 1418 (w), 1374 (w), 1333 (w), 1301 (w), 1254 (w), 1224 (vw), 1115 (m, br, sh), 1075 (m, br), 970 (m), 798 (w), 648 (w).
HRMS (ESI+) for C10H16N6O4P (M + H)+: calcd 315.09652, found 315.09648.
[α]20 = +43.2 (c 0.389, H2O).
[3S,4R]-(4-(Hypoxanthin-9-yl)pyrrolidin-3-yl)oxymethanephosphonic acid 15b
Bromotrimethylsilane (0.66 ml, 5 mmol) was added to a solution of compound 25 (0.54 g, 1.04 mmol) in acetonitrile (10 ml) under an argon atmosphere. The reaction mixture was stirred at rt overnight. The reaction mixture was concentrated in vacuo, co-evaporated with toluene (2 × 10 ml) and dissolved in 3 M aq. HCl (30 ml). The mixture was stirred at 80 °C overnight, diluted with water (100 ml) and applied on a column of Dowex 50 in H+ form (100 ml). The resin was washed with water (150 ml) and the crude product was eluted with 3% aq. ammonia. The title compound was obtained in pure form by preparative HPLC, and converted to sodium salt by passing through a column of Dowex 50 in Na+ form. After lyophilisation from water 68% yield (0.24 g, 0.71 mmol) of the title compound was obtained in the form of a white amorphous solid.1H NMR (600.1 MHz, D2O, 25 °C): 3.05 (dd, 1H, Jgem = 12.5, JH,P = 8.3, CHaHbP); 3.25 (dd, 1H, Jgem = 12.5, JH,P = 9.6, CHaHbP); 3.26 (d, 2H, J2′,3′ = 3.8, H-2′); 3.30 (dd, 1H, Jgem = 11.8, J5′b,4′ = 8.7, H-5′b); 3.45 (dd, 1H, Jgem = 11.8, J5′a,4′ = 8.2, H-5′a); 4.27 (dt, 1H, J3′,4′ = 5.3, J3′,2′ = 3.8, H-3′); 5.06 (ddd, 1H, J4′,5′ = 8.7, 8.2, J4′,3′ = 5.3, H-4′); 8.15 (s, 1H, H-2); 8.30 (s, 1H, H-8).
13C NMR (150.9 MHz, D2O, 25 °C): 51.09 (CH2-5′); 52.68 (CH2-2′); 58.65 (CH-4′); 70.37 (d, JC,P = 151.1, CH2P); 82.60 (d, JC,P = 10.5, CH-3′); 125.17 (C-5); 143.35 (CH-8); 152.93 (C-4); 155.86 (CH-2); 170.01 (C-6).
31P{1H} NMR (202.3 MHz, D2O, 25 °C): 13.60.
IR νmax(KBr) 3415 (vs, br), 3260 9 (vs, vbr, sh), 3137 (vs, vbr), 1685 (s), 1559 (s), 1520 (m), 1470 (s), 1415 (m, sh), 1383 (m), 1335 (m), 1119 (s, br), 1051 (m, sh), 912 (w, sh), 896 (w, sh), 793 (vw), 652 (m).
HRMS (ESI-) for C10H13N5O5P (M − H)−: calcd 314.06598, found 314.06611.
[α]20 = +19.6 (c 0.73, H2O).
[3S,4R]-(4-(Guanin-9-yl)pyrrolidin-3-yl)oxymethanephosphonic acid 15c
A mixture of amino derivative 23 (1.95 g, 5.13 mmol), 2,5-diamino-4,6-dichloropyrimidine (1.8 g, 10.26 mmol) and TEA (3 ml, 22 mmol) in nBuOH (50 ml) was stirred in a sealed reactor at 150 °C overnight. The pyrimidine intermediate was obtained by chromatography on silica gel using a linear gradient of ethanol in chloroform (66% yield, 1.77 g, 3.37 mmol), dissolved in diethoxymethylacetate (20 ml) and stirred at rt for 20 h. The reaction mixture was stirred at 80 °C for an additional 2 h and at 110 °C overnight. Chloroaminopurine intermediate 27 was obtained by column chromatography on silica gel using a linear gradient of ethanol in chloroform in 39% yield (0.77 g, 1.33 mmol) in the form of a gray amorphous solid and used without further characterisation.TMSBr (0.88 ml, 6.65 mmol) was added to a solution of the intermediate 27 in DMF (15 ml) at rt under an argon atmosphere. The reaction mixture was stirred overnight. The mixture was concentrated in vacuo. The residue was dissolved in 1.5 M aq. HCl (50 ml) and stirred at 80 °C overnight. The reaction mixture was diluted with water (100 ml) and applied on a column of Dowex 50 in H+ form (80 ml). The Dowex was washed with water (150 ml) and the crude product was eluted with 3% aq. ammonia. The solvent was removed in vacuo and the title compound was obtained using preparative reversed phase HPLC, converted to its sodium salt by passing through a column of Dowex 50 in Na+ form and lyophilized from water in 17% overall yield (81 mg, 0.23 mmol) in the form of a white amorphous solid.
1H NMR (499.8 MHz, D2O, 25 °C): 3.20 (dd, 1H, Jgem = 12.6, JH,P = 8.2, CHaHbP); 3.29 (dd, 1H, Jgem = 12.9, J2′b,3′ = 4.4, H-2′b); 3.33 (dd, 1H, Jgem = 12.6, JH,P = 9.3, CHaHbP); 3.37 (m, 2H, H-2′a,5′b); 3.52 (dd, 1H, Jgem = 11.9, J5′a,4′ = 8.4, H-5′a); 4.32 (ddd, 1H, J3′,4′ = 5.0, J3′,2′ = 4.4, 3.0, H-3′); 4.97 (td, 1H, J4′,5′ = 8.4, J4′,3′ = 5.0, H-4′); 8.07 (s, 1H, H-8).
13C NMR (125.7 MHz, D2O, 25 °C): 50.47 (CH2-5′); 52.25 (CH2-2′); 57.95 (CH-4′); 70.25 (d, JC,P = 150.8, CH2P); 82.02 (d, JC,P = 10.5, CH-3′); 118.60 (C-5); 141.99 (CH-8); 154.55 (C-4); 159.62 (C-2); 165.61 (C-6).
31P{1H} NMR (202.3 MHz, D2O, 25 °C): 13.85.
IR νmax(KBr) 3431 (vs, br), 1682 (m, br), 1634 (s, br), 1571 (m), 1536 (w), 1480 (w), 1412 (w, br), 1111 (w, br, sh), 1080 (m, br), 973 (w), 802 (vw), 783 (w), 639 (w).
HRMS (ESI+) for C10H15N6O5PNa (M + Na)+: calcd 353.07338, found 353.07343.
[α]20 = +51.5 (c 0.307, H2O).
[3S,4R]-(4-(Uracil-1-yl)pyrrolidin-3-yl)oxymethanephosphonic acid 15d
Reagent 28 (0.36 g, 1.3 mmol) was added to a solution of amino derivative 23 (0.45 g, 1.18 mmol) in dioxane (12 ml). The reaction mixture was stirred at rt overnight. The mixture was concentrated in vacuo and a linear intermediate was obtained by column chromatography on silica gel using a linear gradient of ethanol in chloroform in the form of yellowish foam. Dowex 50 in H+ form (10 g) was added to a solution of the intermediate in dioxane (15 ml). The suspension was stirred at 85 °C for 5 h. The reaction mixture was filtered; the resin was washed with ethanol (50 ml) and eluted with 3% aq. ammonia (100 ml). The filtrate was concentrated and the desired product was obtained by preparative reverse phase HPLC with 54% overall yield (0.2 g, 0.64 mmol) after conversion to sodium salt by passing through a column of Dowex 50 in Na+ form and lyophilisation from water in the form of a white amorphous solid.1H NMR (500.0 MHz, D2O, 25 °C): 3.45 (dd, 1H, Jgem = 12.8, J2′b,3′ = 2.8, H-2′b); 3.46 (dd, 1H, Jgem = 12.1, JH,P = 8.4, CHaHbP); 3.50 (dd, 1H, Jgem = 12.1, JH,P = 10.3, CHaHbP); 3.59 (dd, 1H, Jgem = 12.2, J5′b,4′ = 9.9, H-5′b); 3.73 (dd, 1H, Jgem = 12.2, J5′a,4′ = 9.9, H-5′a); 3.79 (dd, 1H, Jgem = 12.8, J2′a,3′ = 0.7, H-2′a); 4.33 (ddd, 1H, J3′,4′ = 4.1, J3′,2′ = 2.8, 0.7, H-3′); 5.36 (td, 1H, J4′,5′ = 9.9, J4′,3′ = 4.1, H-4′); 5.83 (d, 1H, J5,6 = 8.1, H-5); 8.05 (d, 1H, J6,5 = 8.1, H-6).
13C NMR (125.7 MHz, D2O, 25 °C): 46.54 (CH2-5′); 51.46 (CH2-2′); 57.26 (CH-4′); 69.83 (d, JC,P = 150.8, CH2P); 80.57 (d, JC,P = 12.5, CH-3′); 104.30 (CH-5); 148.21 (CH-6); 155.48 (C-2); 169.22 (C-4).
31P{1H} NMR (202.3 MHz, D2O, 25 °C): 12.89.
IR νmax(KBr) 3189 (m, br), 2980 (s), 2936 (m), 1696 (vs, br), 1628 (m), 1480 (m, sh), 1457 (s), 1408 (s), 1387 (s), 1377 (s, sh), 1365 (s, sh), 1279 (s), 1244 (s, br), 1225 (s, sh), 1175 (s), 1142 (s), 1104 (s), 1011 (s, sh), 991 (vs), 888 (m), 768 (m).
HRMS (ESI+) for C9H14O6N3PNa (M + Na)+ calcd 314.05124, found 314.05123.
[α]20 = +88.9 (c 0.189, H2O).
[3S,4R]-(4-(Thymin-1-yl)pyrrolidin-3-yl)oxymethanephosphonic acid 15e
Reagent 29 (0.75 g, 2.56 mmol) was added to a solution of amino derivative 23 (0.65 g, 1.7 mmol) in dioxane (15 ml). The reaction mixture was stirred at rt overnight. The mixture was concentrated in vacuo and a linear intermediate was obtained by column chromatography on silica gel using a linear gradient of ethanol in chloroform in the form of yellowish foam. Dowex 50 in H+ form (15 g) was added to a solution of the intermediate in dioxane (25 ml). The suspension was stirred at 85 °C for 5 h. The reaction mixture was filtered; the resin was washed with ethanol (50 ml) and eluted with 3% aq. ammonia (100 ml). The filtrate was concentrated and the desired product was obtained by preparative reverse phase HPLC in 32% overall yield (175.6 mg, 0.54 mmol) after conversion to sodium salt by passing through a column of Dowex 50 in Na+ form and lyophilisation from water in the form of a white amorphous solid.1H NMR (500.0 MHz, D2O, 25 °C): 1.90 (d, 1H, 4J = 1.1, CH3); 3.45 (dd, 1H, Jgem = 12.9, J2′b,3′ = 3.1, H-2′b); 3.52 (dd, 1H, Jgem = 12.4, JH,P = 8.6, CHaHbP); 3.55 (dd, 1H, Jgem = 12.4, JH,P = 9.7, CHaHbP); 3.68 (dd, 1H, Jgem = 12.5, J5′b,4′ = 10.2, H-5′b); 3.74 (dd, 1H, Jgem = 12.5, J5′a,4′ = 9.6, H-5′a); 3.81 (d, 1H, Jgem = 12.9, H-2′a); 4.38 (bdd, 1H, J3′,4′ = 4.1, J3′,2′ = 3.1, H-3′); 5.31 (ddd, 1H, J4′,5′ = 10.2, 9.6, J4′,3′ = 4.1, H-4′); 7.83 (q, 1H, 4J = 1.1, H-6).
13C NMR (125.7 MHz, D2O, 25 °C): 14.23 (CH3); 46.50 (CH2-5′); 51.72 (CH2-2′); 57.70 (CH-4′); 69.67 (d, JC,P = 152.7, CH2P); 80.67 (d, JC,P = 11.9, CH-3′); 113.32 (C-5); 143.71 (CH-6); 155.31 (C-2); 169.14 (C-4).
31P{1H} NMR (202.3 MHz, D2O, 25 °C): 13.78.
IR νmax(KBr) 3260 (w, vbr, sh), 2831 (w, vvbr), 1695 (vs), 1663 (s, sh), 1521 (w, br), 1473 (w), 1442 (w), 1394 (w), 1375 (w, sh), 1283 (m), 1126 (m), 1072 (m, br), 970 (w), 789 (w), 769 (w).
HRMS (ESI+) for C10H15O6N3P (M + H)+: calcd 304.07039, found 304.06983.
[α]20 = +69.5 (c 0.364, H2O).
[3R,4R]-(4-(Uracil-1-yl)pyrrolidin-3-yl)oxymethanephosphonic acid 16a
The compound was prepared according to the experimental procedure for compound 15d starting from amino derivative 31 (0.37 g, 0.97 mmol) in 43% overall yield (0.132 g, 0.42 mmol) in the form of a white amorphous colorless solid.1H NMR (500.0 MHz, D2O, 25 °C): 3.47 (dd, 1H, Jgem = 11.8, JH,P = 10.0, CHaHbP); 3.54 (dd, 1H, Jgem = 11.8, JH,P = 9.7, CHaHbP); 3.57 (dd, 1H, Jgem = 12.7, J2′b,3′ = 2.4, H-2′b); 3.71 (dd, 1H, Jgem = 13.1, J5′b,4′ = 5.3, H-5′b); 3.80 (dd, 1H, Jgem = 12.7, J2′a,3′ = 5.4, H-2′a); 3.91 (dd, 1H, Jgem = 13.1, J5′a,4′ = 8.9, H-5′a); 4.53 (ddd, 1H, J3′,2′ = 5.4, 2.4, J3′,4′ = 2.0, H-3′); 4.76 (dd, 1H, J4′,5′ = 8.9, 5.3, J4′,3′ = 2.0, H-4′); 5.85 (d, 1H, J5,6 = 8.0, H-5); 7.72 (d, 1H, J6,5 = 8.0, H-6).
13C NMR (125.7 MHz, D2O, 25 °C): 49.39 (CH2-5′); 53.29 (CH2-2′); 68.82 (CH-4′); 69.51 (d, JC,P = 151.4, CH2P); 85.66 (d, JC,P = 13.6, CH-3′); 104.78 (CH-5); 149.73 (CH-6); 154.55 (C-2); 169.26 (C-4).
31P{1H} NMR (202.3 MHz, D2O, 25 °C): 13.25.
IR νmax(KBr) 3500–3000 (m, vbr), 2792 (m, vbr), 2630 (m, br, sh), 2630 (m, br, sh), 2454 (m, vbr), 1695 (vs, br), 1628 (m, br, sh), 1461 (m, sh), 1440 (m), 1389 (m), 1277 (m), 1160 (m, br, sh), 1105 (s, br, sh), 1063 (s, br), 971 (m), 914 (m, br), 767 (m).
HRMS (ESI+) for C9H14O6N3PNa (M + Na)+ calcd 314.05124, found 314.05124.
[α]20 = −66.0 (c 0.053, H2O).
[3R,4R]-(4-(Hypoxanthin-9-yl)pyrrolidin-3-yl)oxymethanephosphonic acid 16b
A mixture of amino derivative 31 (0.23 g, 0.61 mmol), 4,6-dichloro-5-formamidopyrimidine (32) (0.14 g, 0.73 mmol), and DIPEA (0.52 ml, 3.05 mmol) in nBuOH (10 ml) was stirred at 110 °C overnight. The reaction mixture was concentrated in vacuo, dissolved in DMF (5 ml), diethoxymethyl acetate (3 ml) was added, and the mixture was stirred at 120 °C overnight. The chloropurine intermediate was obtained by chromatography on silica gel using a linear gradient of ethanol in chloroform and was used in the next step without further characterisation (except for LCMS). The intermediate (0.31 g, 0.6 mmol) was stirred in 1.5 M aq. HCl (50 ml) at 75 °C overnight, applied on a column of Dowex 50 in H+ form (100 ml), washed with 50% aq. ethanol (150 ml) and eluted with 3% NH3 in 50% aq. ethanol (300 ml). The obtained yellow solution was evaporated and purified using preparative HPLC on the reversed phase. The obtained hypoxanthine diisopropyl ester (80 mg, 0.2 mmol) was co-evaporated with MeCN (3 × 10 ml), dissolved in the same solvent, and Me3SiBr (0.13 ml, 1 mmol) was added under an argon atmosphere. The reaction mixture was stirred under an argon atmosphere at rt overnight. The title compound was obtained by preparative reverse phase HPLC in 10% overall yield (21.8 mg, 65 μmol) – calculated from 31 – after conversion to sodium salt by passing through a column of Dowex 50 in Na+ form and lyophilisation from water in the form of a white amorphous solid.1H NMR (600.1 MHz, D2O, 25 °C): 3.60 (dd, 1H, Jgem = 12.1, JH,P = 10.0, CHaHbP); 3.69 (dd, 1H, Jgem = 12.1, JH,P = 9.5, CHaHbP); 3.76 (dt, 1H, Jgem = 13.3, J2′b,3′ = J2′b,4′ = 1.2, H-2′b); 3.84 (dd, 1H, Jgem = 13.3, J2′a,3′ = 4.2, H-2′a); 4.07 (dd, 1H, Jgem = 13.6, J5′b,4′ = 3.2, H-5′b); 4.15 (dd, 1H, Jgem = 13.6, J5′a,4′ = 7.7, H-5′a); 4.51 (dt, 1H, J3′,2′ = 4.2, 1.2, J3′,4′ = 1.2, H-3′); 5.47 (ddt, 1H, J4′,5′ = 7.7, 3.2, J4′,3′ = J4′,2′b = 1.2, H-4′); 8.18 (s, 1H, H-2); 8.25 (s, 1H, H-8).
13C NMR (150.9 MHz, D2O, 25 °C): 50.33 (CH2-5′); 52.42 (CH2-2′); 62.19 (CH-4′); 69.49 (d, JC,P = 152.9, CH2P); 86.50 (d, JC,P = 13.3, CH-3′); 126.74 (C-5); 144.33 (CH-8); 148.42 (CH-2); 151.25 (C-4); 161.39 (C-6).
31P{1H} NMR (202.3 MHz, D2O, 25 °C): 13.57.
IR νmax(KBr) 3434 (vs, br), 3264 (m, br, sh), 2923 (m), 2853 (m), 2790 (m, vbr, sh), 1695 (s), 1588 (m), 1550 (w), 1515 (w), 1470 (w, sh), 1418 (w), 1382 (vw), 1346 (vw), 1216 (w), 1190 (w, br), 1146 (w, vbr), 1112 (w, sh), 1051 (m, br), 912 (w, br), 896 (w, sh), 790 (w), 646 (w).
HRMS (ESI-) for C10H13N5O5P (M − H)−: calcd 314.06598, found 314.06585.
[α]20= −30.4 (c 0.184, H2O).
[3R,4R]-(4-(Guanin-9-yl)pyrrolidin-3-yl)oxymethanephosphonic acid 16c
The title compound was prepared from amino derivative 31 (0.23 g, 0.61 mmol) and 2-amino-4,6-dichloro-5-formamidopyrimidine (33) (0.15 g, 0.73 mmol) using the same procedure as for compound 16b in 8% overall yield (17.3 mg, 49 μmol) in the form of a white amorphous solid.1H NMR (500.0 MHz, D2O, 25 °C): 3.71 (dt, 1H, Jgem = 13.3, J2′b,3′ = J2′b,4′ = 1.4, H-2′b); 3.71 (dd, 1H, Jgem = 12.5, JH,P = 9.9, CHaHbP); 3.79 (dd, 1H, Jgem = 12.5, JH,P = 9.6, CHaHbP); 3.90 (dd, 1H, Jgem = 13.3, J2′a,3′ = 4.6, H-2′a); 4.04 (dd, 1H, Jgem = 13.5, J5′b,4′ = 3.4, H-5′b); 4.08 (dd, 1H, Jgem = 13.5, J5′a,4′ = 6.7, H-5′a); 4.52 (ddd, 1H, J3′,2′ = 4.6, 1.4, J3′,4′ = 1.9, H-3′); 5.29 (m, 1H, H-4′); 7.89 (s, 1H, H-8).
13C NMR (125.7 MHz, D2O, 25 °C): 50.19 (CH2-5′); 52.53 (CH2-2′); 61.61 (CH-4′); 68.89 (d, JC,P = 155.7, CH2P); 86.68 (d, JC,P = 13.1, CH-3′); 119.04 (C-5); 141.77 (CH-8); 153.73 (C-4); 156.3 (C-2); 161.56 (C-6).
31P{1H} NMR (202.3 MHz, D2O, 25 °C): 14.71.
IR νmax(KBr) 3311 (m, vbr), 3117 (m, br), 3022 (m, br, sh), 2749 (m, vbr), 2440 (w, vbr), 1690 (vs), 1658 (s), 1607 (m), 1580 (m, sh), 1536 (w), 1486 (w), 1415 (w), 1374 (w, br), 1117 (m), 1064 (m, br), 970 (w), 779 (w), 691 (vw), 640 (vw).
HRMS (ESI+) for C10H15N6O5PNa (M + Na)+: calcd 353.07338, found 353.07346.
[α]20= +48.8 (c 0.172, H2O).
[3S,4S] Diisopropyl 1-N-tert-butyloxycarbonyl-4-dimethoxytrityloxypyrrolidin-3-yloxymethylphosphonate 18
[3S,4S]-1-N-Boc-3-dimethoxytrityloxy-4-hydroxypyrrolidine (12 g, 34.42 mmol) diisopropyl tosyloxymethanephosphonate (17.19 g, 49 mmol) was dissolved in THF (300 ml). The solution was cooled to 0 °C and sodium hydride (2.8 g, 70 mmol) was added. The reaction mixture was stirred at rt for 3 days and then cooled to 0 °C and acetic acid (1.8 ml, 29 mmol) was added slowly. The temperature was allowed to rise to rt and the solvent was evaporated in vacuo. The title compound was obtained by chromatography on silica gel using a linear gradient of ethyl acetate in toluene as a viscous colorless oil in 77% yield (14.5 g, 22.11 mmol).NMR – (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 mixture of amide rotamers)
:1 mixture of amide rotamers)
1H NMR (500.0 MHz, CDCl3, 25 °C): 1.24, 1.27, 1.28, 1.29 (4 × d, 4 × 6H, Jvic = 6.2, (CH3)2CH); 1.41, 1.46 (2 × s, 2 × 9H, (CH3)3C); 2.92 (d, 1H, Jgem = 12.0, H-5b); 3.07 (dd, 1H, J3,4 = 4.7, J3,2a = 4.1, H-3); 3.08 (dd, 1H, Jgem = 12.0, J5a,4 = 4.7, H-5a); 3.25, 3.29 (2 × dd, 2 × 1H, Jgem = 13.4, JH,P = 9.7, CH2P); 3.30–3.33 (m, 3H, H-3,5); 3.34 (dd, 1H, Jgem = 13.2, JH,P = 9.7, CHaHbP); 3.37, 3.41 (2 × d, 2 × 1H, Jgem = 12.1, H-2b); 3.42 (dd, 1H, Jgem = 13.2, JH,P = 9.7, CHaHbP); 3.49, 3.56 (2 × dd, 2 × 1H, Jgem = 12.1, J2a,3 = 4.1, H-2a); 3.786, 3.789 (2 × s, 2 × 6H, CH3O-DMTr); 4.10 (bt, 2H, J4,3 = J4,5a = 4.7, H-4); 4.60–4.70 (m, 4H, CH(CH3)2); 6.83, 6.84 (2 × m, 2 × 4H, H-m-C6H4-DMTr); 7.13–7.36 (m, 14H, H-o-C6H4-DMTr, H-m,p-C6H5-DMTr); 7.43 (m, 4H, H-o-C6H5-DMTr).
13C NMR (125.7 MHz, CDCl3, 25 °C): 23.85, 23.97, 24.01 (d, JC,P = 4.0, (CH3)2CH); 28.39, 28.45 ((CH3)3C); 48.68, 49.48 (CH2-2); 50.72, 50.82 (CH2-5); 55.17 (CH3O-DMTr); 63.92, 64.03 (d, JC,P = 170.0, CH2P); 70.95, 71.03, 71.05, 71.13 (d, JC,P = 7.0, CH(CH3)2); 74.27, 75.06 (CH-4); 79.14, 79.24 (C(CH3)3); 83.65, 84.37 (d, JC,P = 13.0, CH-3); 87.10, 87.25 (C-DMTr); 113.22, 113.25 (CH-m-C6H4-DMTr); 126.99 (CH-p-C6H5-DMTr); 127.91 (CH-m-C6H5-DMTr); 128.18, 128.21 (CH-o-C6H5-DMTr); 130.10, 130.14 (CH-m-C6H4-DMTr); 136.11, 136.20, 136.30 (C-i-C6H4-DMTr); 144.97, 145.03 (C-i-C6H5-DMTr); 154.54, 154.66 (CO); 158.67, 158.70 (C-p-C6H4-DMTr).
31P{1H} NMR (202.3 MHz, CDCl3, 25 °C): 19.00, 19.05.
HRMS (FAB+) for C37H50NO9PNa (M + H + Na)+: calcd 706.3121, found 706.3146.
[3S,4S] Diisopropyl 1-N-tert-butyloxycarbonyl-4-hydroxypyrrolidin-3-yloxymethylphosphonate 19
2% TFA in DCM (200 ml) was added to compound 18 (14.5 g, 22.11 mmol) and the reaction mixture was stirred until the DMTr group was cleaved completely (followed by TLC, ∼20 min). NaHCO3 (20 g) and MeOH (50 ml) were added and the suspension was vigorously stirred until neutral pH. The suspension was filtered over celite and the filtrate was evaporated. The title compound was obtained by chromatography on silica gel using a linear gradient of ethanol in chloroform in 83% yield (6.97 g, 18.28 mmol) in the form of colorless oil that upon keeping in a refrigerator (at 4 °C) starts to crystallize after several days.NMR – (1:1 mixture of amide rotamers)
1H NMR (499.8 MHz, DMSO-d6, 25 °C): 1.22, 1.23, 1.24 (3 × d, 24H, Jvic = 6.2, (CH3)2CH); 1.38, 1.39 (2 × s, 2 × 9H, (CH3)3C); 3.14, 3.15 (2 × d, 2 × 1H, Jgem = 11.4, H-5b); 3.26, 3.29 (2 × dd, 2 × 1H, Jgem = 11.4, J5a,4 = 4.4, H-5a); 3.30–3.34 (m, 3H, 2 × H-2b, H-2a); 3.37 (dd, 1H, Jgem = 12.2, J2a,3 = 4.1, H-2a); 3.77 (dd, 1H, Jgem = 13.1, JH,P = 9.0, CHaHbP); 3.79 (d, 2H, JH,P = 9.0, CH2P); 3.81 (dd, 1H, Jgem = 13.1, JH,P = 9.0, CHaHbP); 3.83 (m, 2H, H-3); 4.08, 4.10 (2 × m, 2 × 1H, H-4); 4.58 (m, 4H, CH(CH3)2); 5.249, 5.253 (2 × d, 2 × 1H, JOH,3 = 3.6, OH).
13C NMR (125.7 MHz, DMSO-d6, 25 °C): 23.84, 23.86 (d, JC,P = 4.4, (CH3)2CH); 24.00, 24.01 (d, JC,P = 3.6, (CH3)2CH); 28.34 ((CH3)3C); 48.85, 49.23 (CH2-2); 51.97, 52.26 (CH2-5); 63.05, 63.19 (d, JC,P = 165.4, CH2P); 70.44, 70.46, 70.47 (d, JC,P = 6.3, CH(CH3)2); 70.99, 71.81 (CH-4); 78.52, 78.55 (C(CH3)3); 83.92 (d, JC,P = 12.3, CH-3); 84.83 (d, JC,P = 12.0, CH-3); 153.97, 153.99 (CO).
31P{1H} NMR (202.3 MHz, DMSO-d6,CDCl3, 25 °C): 20.12, 20.17.
IR νmax(CHCl3) 3610 (w), 3363 (m, vbr), 2983 (vs), 2936 (s), 1689 (vs), 1679 (vs, sh), 1478 (s), 1467 (m), 1455 (s), 1415 (vs, br), 1389 (s), 1377 (s), 1368 (s), 1245 (s, br), 1170 (vs), 1143 (s), 1105 (vs), 1002 (vs, vbr), 889 (m).
HRMS (ESI+) for C16H32NO7PNa (M + Na)+ calcd 404.18141, found 404.18153.
[3S,4S] Diisopropyl 1-N-tert-butyloxycarbonyl-4-mesyloxypyrrolidin-3-yloxymethylphosphonate 20
Mesyl chloride (4.26 ml, 55 mmol) was added to a solution of 19 (6.97 g, 18.28 mmol) and DMAP (6.7 g, 55 mmol) in DCM (130 ml) at 0 °C. The reaction mixture was stirred at rt for 2 h. The reaction mixture was washed with a saturated solution of sodium bicarbonate. The organic phase was evaporated and the title compound was obtained by chromatography on silica gel using a linear gradient of ethyl acetate in toluene as a colorless syrup in 92% yield (7.71 g, 16.78 mmol).NMR – (1:1 mixture of amide rotamers)
1H NMR (499.9 MHz, CDCl3, 25 °C): 1.33, 1.34, 1.35 (3 × d, 24H, Jvic = 6.2, (CH3)2CH); 1.47 (s, 18H, (CH3)3C); 3.09, 3.11 (2 × bs, 2 × 3H, CH3-Ms); 3.48–3.73 (m, 8H, H-2,5); 3.75–3.84 (m, 4H, CH2P); 4.27 (bm, 2H, H-3); 4.68–4.82 (m, 4H, CH(CH3)2); 5.13 (m, 2H, H-4).
13C NMR (125.7 MHz, CDCl3, 25 °C): 23.91, 23.94 (d, JC,P = 4.2, (CH3)2CH); 23.99, 24.01 (d, JC,P = 3.5, (CH3)2CH); 28.35 ((CH3)3C); 38.63 (CH3-Ms); 48.61 (CH2-2); 49.31 (CH2-2,5); 49.83 (CH2-5); 64.57, 64.63 (d, JC,P = 169.3, CH2P); 71.26–71.51 (CH(CH3)2); 79.45 (CH-4); 80.08 (C(CH3)3); 80.13 (CH-4); 82.10, 83.29 (d, JC,P = 10.4, CH-3); 154.02, 154.08 (CO).
31P{1H} NMR (202.3 MHz, CDCl3, 25 °C): 18.28, 18.30.
HRMS (ESI+) for C17H34NO9PSNa (M + Na)+ calcd 482.15896, found 482.15880.
[3S,4R] Diisopropyl 4-azido1-1-N-tert-butyloxycarbonylpyrrolidin-3-yloxymethylphosphonate 21
NMR – (1:1 mixture of amide rotamers)
1H NMR (500.0 MHz, CDCl3, 25 °C): 1.35, 1.35 (2 × d, 2 × 12H, Jvic = 6.1, (CH3)2CH); 1.45 (s, 18H, (CH3)3C); 3.40 (dd, 1H, Jgem = 11.0, J2b,3 = 4.6, H-2b); 3.41 (dd, 1H, Jgem = 10.6, J5b,4 = 4.6, H-5b); 3.48 (m, 2H, H-2b,5b); 3.54–3.60 (m, 4H, H-2a,5a); 3.79–3.89 (m, 4H, CH2P); 3.91, 3.95 (2 × m, 2 × 1H, H-4); 4.28, 4.29 (2 × m, 2 × 1H, H-3); 4.71–4.83 (m, 4H, CH(CH3)2).
13C NMR (125.7 MHz, CDCl3, 25 °C): 23.94 (d, JC,P = 4.5, (CH3)2CH); 24.03, 24.04 (d, JC,P = 3.9, (CH3)2CH); 28.37 ((CH3)3C); 47.59, 47.75, 48.16 (CH2-2,5); 59.73, 60.30 (CH-4); 64.64, 64.92 (d, JC,P = 167.9, CH2P); 71.36 (d, JC,P = 6.7, CH(CH3)2); 76.75, 77.00 (C(CH3)3); 80.08 (d, JC,P = 8.7, CH-3); 80.86 (d, JC,P = 9.2, CH-3); 154.10 (CO).
31P{1H} NMR (202.3 MHz, CDCl3, 25 °C): 18.41, 18.44.
HRMS (ESI+) for C16H31N4O6PNa (M + H + Na)+: calcd 429.1879, found 429.1876.
[3S,4R] 4-Azido-1-N-tert-butyloxycarbonyl-3-hydroxypyrrolidine 22
MsCl (20 ml, 260 mmol) was added dropwise to a solution of dimethoxytrityl derivative 17 (64.6 g, 127.77 mmol) and DMAP (32 g, 260 mmol) in DCM (1 L) at 0 °C. The reaction mixture was stirred at rt overnight. The mixture was washed with sat. aq. NaHCO3 (2 × 400 ml), 10% aq. citric acid (2 × 400 ml) and water (2 × 400 ml). The organic phase was dried over sodium sulfate, filtered and concentrated in vacuo. The residue was dissolved in DMF (1 L). Sodium azide (26 g, 400 mmol) was added and the reaction mixture was stirred at 110 °C overnight. The reaction mixture was concentrated in vacuo. Ethyl acetate (500 ml) was added to the residue. The slurry was filtered and concentrated in vacuo. 1.5% TFA in DCM (600 ml) was added and the mixture was stirred until complete removal of the DMTr group (followed by TLC in 50% EtOAc/toluene and 10% EtOH in chloroform) for ca. 2 h. Solid NaHCO3 (40 g) and MeOH (100 ml) were added in portions. The mixture was vigorously stirred until the pH was neutral (∼30 min). The suspension was filtered and the filtrate was concentrated in vacuo. The title compound was obtained by chromatography on silica gel using a linear gradient of ethyl acetate in toluene with 77% yield (over three steps) (22.6 g, 99 mmol) in the form of colorless oil that solidified on keeping in the refrigerator.1H NMR (499.8 MHz, DMSO-d6, 80 °C): 1.41 (s, 9H, (CH3)3C); 3.13 (dd, 1H, Jgem = 10.9, J2b,3 = 5.5, H-2b); 3.23 (dd, 1H, Jgem = 11.3, J5b,4 = 5.0, H-5b); 3.42 (dd, 1H, Jgem = 10.9, J2a,3 = 6.0, H-2a); 3.46 (dd, 1H, Jgem = 11.3, J5a,4 = 6.0, H-5a); 3.92 (ddd, 1H, J4,5 = 6.0, 5.0, J4,3 = 4.4, H-4); 4.33 (m, 1H, H-3); 5.36 (bs, 1H, OH).
13C NMR (125.7 MHz, DMSO-d6, 80 °C): 28.00 ((CH3)3C); 47.45 (CH2-5); 50.44 (CH2-2); 61.24 (CH-4); 70.52 (CH-3); 78.50 ((CH3)3C); 153.40 (CO).
IR νmax(KBr) 3340 (m), 2980 (m), 2942 (w), 2138 (m), 2124 (s, sh), 2095 (s), 1682 (s), 1479 (m), 1470 (m), 1455 (m, sh), 1420 (vs), 1391 (m, sh), 1369 (m), 1217 (w), 1257 (m), 1163 (s), 1093 (m, sh), 552 (w).
HRMS (ESI+) for C9H16N4O3Na (M + Na)+ calcd 251.1115, found 251.1114.
[3S,4R] Diisopropyl 4-amino-1-N-tert-butyloxycarbonylpyrrolidin-3-yloxymethylphosphonate 23
A solution of 21 (2.17 g, 5.34 mmol) in ethanol (50 ml) was hydrogenated in the presence of Pd/C (0.2 g) overnight. The suspension was filtered over celite and this was evaporated. The title compound was obtained without further purification in 79% yield (1.61 g, 4.232 mmol) in the form of colorless oil.NMR – (1:1 mixture of amide rotamers)
1H NMR (500.0 MHz, DMSO-d6, 25 °C): 1.21–1.26 (m, 24H, (CH3)2CH); 1.375, 1.378 (2 × s, 2 × 9H, (CH3)3C); 2.82, 2.84 (2 × dd, 2 × 1H, Jgem = 9.9, J5b,4 = 7.5, H-5b); 3.25 (dd, 2H, Jgem = 12.1, J2b,3 = 4.3, H-2b); 3.29–3.43 (m, 6H, H-2a,4,5a); 3.77–3.85 (m, 4H, H-3, CHaHbP); 3.88 (dd, 2H, Jgem = 13.9, JH,P = 8.9, CHaHbP); 4.54–4.66 (m, 4H, CH(CH3)2).
13C NMR (125.7 MHz, DMSO-d6, 25 °C): 23.87 (d, JC,P = 4.0, (CH3)2CH); 24.00, 24.05 (d, JC,P = 3.6, (CH3)2CH); 28.32 ((CH3)3C); 48.99, 49.40 (CH2-2); 50.47, 50.87 (CH2-5); 52.34, 53.18 (CH-4); 63.78, 64.01 (d, JC,P = 164.5, CH2P); 70.44–71.54 (CH(CH3)2); 78.51 (C(CH3)3); 80.94 (d, JC,P = 12.0, CH-3); 81.67 (d, JC,P = 11.6, CH-3); 153.69, 153.79 (CO).
31P{1H} NMR (202.3 MHz, DMSO-d6, 25 °C): 20.32, 20.36.
HRMS (ESI+) for C16H33N2O6PNa (M + H + Na)+: calcd 403.1968, found 403.1971.
[3S,4R] Diisopropyl 4-(6-chloropurin-9-yl)-1-N-tert-butyloxycarbonylpyrrolidin-3-yloxymethylphosphonate 25
DIAD (1.81 ml, 9.36 mmol) was added to a solution of triphenylphosphine (1.52 g, 9.36 mmol) in THF (30 ml). The mixture was stirred at rt for 3 h. A mixture of 19 (1.19 g, 3.12 mmol) and 6-chloropurine (0.58 g, 3.74 mmol) in THF (30 ml) was added. The suspension turned to a clear red-brown solution in 5 min. The reaction mixture was stirred at rt for an additional 2 h. The solvent was removed in vacuo and the title compound was obtained by column chromatography on silica gel using a linear gradient of ethyl acetate in toluene in 53% yield (0.85 g, 1.64 mmol).NMR – (1:1 mixture of amide rotamers)
1H NMR (600.1 MHz, DMSO-d6, 25 °C): 0.98, 1.00, 1.05, 1.07, 1.13 (5 × d, 24H, Jvic = 6.1, (CH3)2CH); 1.42, 1.44 (2 × s, 2 × 9H, (CH3)3C); 3.51–3.71 (m, 6H, H-2′, CHaHbP); 3.83–3.90 (m, 4H, H-5′b, CHaHbP); 3.956, 3.962 (2 × dd, 2 × 1H, Jgem = 9.9, J5′a,4′ = 8.9, H-5′a); 4.29–4.44 (m, 4H, H-3′, CHaHbP); 5.37, 5.38 (2 × td, 2 × 1H, J3′,4′ = 8.9, J3′,2′ = 8.9, 4.2, H-3′); 8.71, 8.72 (2 × s, 2 × 1H, H-8); 8.80 (s, 2H, H-2).
13C NMR (150.9 MHz, DMSO-d6, 25 °C): 23.62, 23.71, 23.73 (d, JC,P = 4.4, (CH3)2CH); 23.81, 23.87 (d, JC,P = 3.8, (CH3)2CH); 28.37 ((CH3)3C); 4.69, 46.90 (CH2-5′); 48.87, 49.34 (CH2-2′); 54.54, 55.02 (CH-3′); 63.08, 63.30 (d, JC,P = 164.0, CH2P); 70.36, 70.38 (d, JC,P = 6.0, CH(CH3)2); 70.49 (d, JC,P = 6.3, CH(CH3)2); 78.31, 79.10 (d, JC,P = 12.2, CH-3′); 79.34, 79.37 (C(CH3)3); 130.85, 130.90 (C-5); 147.06 (CH-8); 149.27 (C-6); 151.77 (CH-2); 152.52 (C-4); 153.76, 153.79 (CO).
31P{1H} NMR (202.3 MHz, DMSO-d6, 25 °C): 19.45, 19.54.
IR νmax (KBr) 2976 (s), 1705 (s, sh), 1679 (vs), 1596 (s), 1558 (m), 1492 (m), 1479 (m), 1465 (m), 1435 (s), 1415 (vs), 1399 (vs), 1388 (s, sh), 1372 (s), 1365 (s, sh), 1340 (s), 1259 (s), 1232 (s), 1228 (s), 1212 (m, sh), 1175 (s, sh), 1167 (s), 1140 (s), 1105 (s), 1008 (vs), 987 (vs), 933 (m), 884 (m), 790 (m), 644 (w, sh), 636 (m).
HRMS (ESI+) for C21H34N5O6ClP (M + H)+: calcd 518.1930, found 518.1928.
[3R,4R] Diisopropyl 4-amino-1-N-tert-butyloxycarbonylpyrrolidin-3-yloxymethylphosphonate 31
Starting from azido derivative 3013 (3.12 g, 13.67 mmol) the same synthetic procedures as for compound 23 were used. The title compound was obtained in 50% (2.76 g, 6.8 mmol) overall yield.
NMR – (1:1 mixture of amide rotamers)
1H NMR (500.0 MHz, DMSO-d6, 25 °C): 1.222, 1.23, 1.24 (3 × d, 24H, Jvic = 6.2, (CH3)2CH); 1.38, 1.39 (2 × s, 2 × 9H, (CH3)3C); 3.00 (bd, 2H, Jgem = 10.7, H-5b); 3.23–3.33 (m, 4H, H-2b,5a); 3.34 (bm, 2H, H-4); 3.44, 3.47 (2 × dd, 2 × 1H, Jgem = 12.0, J2a,3 = 4.4, H-2a); 3.68 (bm, 2H, H-3); 3.73–3.82 (m, 4H, CH2P); 4.53–4.63 (m, 4H, CH(CH3)2).
13C NMR (125.7 MHz, DMSO-d6, 25 °C): 23.85, 23.87 (d, JC,P = 4.4, (CH3)2CH); 24.01, 24.02 (d, JC,P = 3.6, (CH3)2CH); 28.37 ((CH3)3C); 48.94, 49.32 (CH2-2); 51.99, 52.32 (CH2-5); 53.82, 54.67 (CH-4); 63.10, 63.23 (d, JC,P = 165.5, CH2P); 70.36–70.47 (CH(CH3)2); 78.37, 78.39 (C(CH3)3); 85.25 (d, JC,P = 12.5, CH-3); 86.14 (d, JC,P = 12.0, CH-3); 153.95, 153.99 (CO).
31P{1H} NMR (202.3 MHz, DMSO-d6, 25 °C): 20.24, 20.39.
HRMS (ESI+) for C16H33N2O6PNa (M + H + Na)+: calcd 403.1968, found 403.1971.
Determination of kinetic constants
The Ki values for the ANPs were determined using a spectrophotometric method (J. Med. Chem. (2009) 52: 4390) with the concentration of the invariable substrate (guanine) being 60 μM and the concentration of the variable substrate (PRib-PP) ranging between 20 and 580 μM. The values were calculated from Hanes plots using the equations for either competitive or non-competitive inhibition.Docking studies
The three dimensional structures of 15c, 15d, 16c and 16d were obtained using the PRODRG2 server.24 The program GOLD was used for all the docking calculations.22 Coordinates for human HGPRT were obtained using chain A of the complex with 2-(phosphonoethoxy)ethyl guanine (PEEG, PDB: 3GGJ) and 2-(phosphonoethoxy)ethyl hypoxanthine (PEEHx, PDB: 3GGC).21 For setting up the protein template, the inhibitor, the water molecules and the magnesium ions were removed from the coordinates. Prior to docking simulations the enzyme was protonated using the GOLD default settings. The active site centre was defined by the coordinates of the phosphorus atom of PEEG or PEEHx when bound to human HGPRT. The search radius for all calculations was 15 Å. For each ligand twenty independent docking searches were performed. The GoldScore fitness function (default settings) was used to rank the poses. This algorithm takes into account the H-bonding energy, van der Waals energy and ligand torsion strain.Acknowledgements
Support by grants #13-24880S and #P207/11/0108 (Czech Science Foundation), National Health and Medical Research Council #1030353, is gratefully acknowledged.Notes and references
- A. Holý and I. Rosenberg, Collect. Czech. Chem. Commun., 1982, 47, 3447–3463 CrossRef.
- E. De Clercq, M. Baba, R. Pauwels, J. Balzarini, I. Rosenberg and A. Holý, Antiviral Res., 1987, 8, 261–272 CrossRef CAS.
- J. Balzarini, L. Naesens, P. Herdewijn, I. Rosenberg, A. Holý, R. Pauwels, M. Baba, D. G. Johns and E. De Clercq, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 332–336 CrossRef CAS.
- R. L. Mackman, C. G. Boojamra, V. Prasad, L. Zhang, K. Y. Lin, O. Petrakovsky, D. Babusis, J. Chen, J. Douglas, D. Grant, H. C. Hui, C. U. Kim, D. Y. Markevitch, J. Vela, A. Ray and T. Cihlar, Bioorg. Med. Chem. Lett., 2007, 17, 6785–6789 CrossRef CAS PubMed.
- M. R. Harnden and R. L. Jarvest, Tetrahedron Lett., 1991, 32, 3863–3866 CrossRef CAS.
- M. R. Harnden, M. J. Jarvest and R. L. Parratt, J. Chem. Soc., Perkin Trans. 1, 1992, 2259–2263 RSC.
- D. R. Adams, A. S. F. Boyd, R. Ferguson, D. S. Grierson and C. Monneret, Nucleosides, Nucleotides Nucleic Acids, 1998, 17, 1053–1075 CAS.
- U. Chiacchio, E. Balestrieri, B. Macchi, D. Iannazzo, A. Piperno, A. Rescifina, R. Romeo, M. Saglimbeni, M. T. Sciortino, V. Valveri, A. Mastino and G. Romeo, J. Med. Chem., 2005, 48, 1389–1394 CrossRef CAS PubMed.
- G. A. Sheikha, P. L. Colla and A. G. Loi, Nucleosides, Nucleotides Nucleic Acids, 2002, 21, 619–635 CAS.
- D. Rejman, P. Kočalka, M. Buděšínský, R. Pohl and I. Rosenberg, Tetrahedron, 2007, 63, 1243–1253 CrossRef CAS PubMed.
- D. Rejman, R. Pohl, P. Kočalka, M. Masojídková and I. Rosenberg, Tetrahedron, 2009, 65(18), 3673–3681 CrossRef CAS PubMed.
- P. Kocalka, D. Rejman, V. Vanek, M. Rinnová, I. Tomecková, S. Králíková, M. Petrová, O. Páv, R. Pohl, M. Budesínský, R. Liboska, Z. Tocík, N. Panova, I. Votruba and I. Rosenberg, Bioorg. Med. Chem. Lett., 2010, 20, 862–865 CrossRef CAS PubMed.
- D. Rejman, N. Panova, P. Klener, B. Maswabi, R. Pohl and I. Rosenberg, J. Med. Chem., 2012, 55, 1612–1621 CrossRef CAS PubMed.
- S. Kovačková, M. Dračínský and D. Rejman, Tetrahedron, 2011, 67, 1485–1500 CrossRef PubMed.
- D. Rejman, S. Kovačková, R. Pohl, M. Dračínský, P. Fiedler and I. Rosenberg, Tetrahedron, 2009, 65, 8513–8523 CrossRef CAS PubMed.
- M. Dejmek, H. Hřebabecký, M. Šála, M. Dračínský, E. Procházková, P. Leyssen, J. Neyts, J. Balzarini and R. Nencka, Bioorg. Med. Chem., 2014, 22(11), 2974–2983 CrossRef CAS PubMed.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1972, 94, 8205–8212 CrossRef CAS.
- P. Hendrickx and J. Martins, Chem. Central J., 2008, 2, 20 CrossRef PubMed.
- L. Poštová Slavětínská, D. Rejman and R. Pohl, Beilstein J. Org. Chem., 2014, 10(22), 1967–1980 CrossRef PubMed.
- C. Mazzon, C. Rampazzo, M. C. Scaini, L. Gallinaro, A. Karlsson, C. Meier, J. Balzarini, P. Reichard and V. Bianchi, Biochem. Pharmacol., 2003, 66, 471–479 CrossRef CAS.
- D. T. Keough, D. Hocková, A. Holý, L. M. J. Naesens, T. S. Skinner-Adams, J. de Jersey and L. W. Guddat, J. Med. Chem., 2009, 52, 4391–4399 CrossRef CAS PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 2(67), 727–748 CrossRef PubMed.
- D. T. Keough, T. Skinner-Adams, K. M. Jones, A. L. Ng, I. M. Brereton, L. W. Guddat and J. de Jersey, J. Med. Chem., 2006, 49(25), 7479–7486 CrossRef CAS PubMed.
- A. W. Schüttelkopf and D. M. F. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 1355–1363 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00097a |
| This journal is © The Royal Society of Chemistry 2015 |