
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of the Ugi reaction with multiple amino acid-derived components: synthesis and conformational evaluation of piperazine-based minimalist peptidomimetics†
Mattia
Stucchi
a,
Silvia
Cairati
a,
Rengul
Cetin-Atalay
b,
Michael S.
Christodoulou
a,
Giovanni
Grazioso
c,
Gennaro
Pescitelli
d,
Alessandra
Silvani
*a,
Deniz Cansen
Yildirim
e and
Giordano
Lesma
a
aDipartimento di Chimica, Università degli Studi di Milano, via Golgi 19, 20133 Milano, Italy. E-mail: alessandra.silvani@unimi.it; Tel: +39 0250314080
bCancer Systems Biology Laboratory, Graduate School of Informatics, METU, 06800, Ankara, Turkey
cDipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, via L. Mangiagalli 25, 20133 Milano, Italy
dDipartimento di Chimica e Chimica Industriale, Università di Pisa, via Moruzzi 3, 56124 Pisa, Italy
eDepartment of Molecular Biology and Genetics, Faculty of Science, Bilkent University, Ankara, Turkey
First published on 19th March 2015
Abstract
The concurrent employment of α-amino acid-derived chiral components such as aldehydes and α-isocyanoacetates, in a sequential Ugi reaction/cyclization two-step strategy, opens the door to the synthesis of three structurally distinct piperazine-based scaffolds, characterized by the presence of L-Ala and/or L-Phe-derived side chains and bearing appropriate functionalities to be easily applied in peptide chemistry. By means of computational studies, these scaffolds have been demonstrated to act as minimalist peptidomimetics, able to mimic a well defined range of peptide secondary structures and therefore potentially useful for the synthesis of small-molecule PPI modulators. Preliminary biological evaluation of two different resistant hepatocellular carcinoma cellular lines, for which differentiation versus resistance ability seem to be strongly correlated with well defined types of PPIs, has revealed a promising antiproliferative activity for selected compounds.
Introduction
Many proteins exert their biological roles as components of complexes, and their functions are often determined by specific protein–protein interactions (PPIs). In the past decade, PPIs have begun to gain attention as viable targets for therapeutic intervention, despite the fact that they do not involve endogenous small molecule ligands that could provide leads for discovery programmes. In particular, the targeting of protein–protein interactions relevant to cancer is of fundamental importance, as the tumor-promoting function of several aberrantly expressed proteins in the cancerous state is directly correlated to their ability to interact with a protein-binding partner.1A minor fraction of the protein–protein interface residues can account for the majority of the free energy of binding between proteins. Statistical analyses of structurally characterized protein–protein interfaces have shown that side-chain substituents account for about 80% of the interactions, with the polyamide backbone accounting for much less.2 Since it is well known that the majority of PPIs are mediated by three main recognition motifs (α-helix, β-turn, or β-strand), an attractive approach for the discovery of PPI modulators is to mimic the key interaction residues using small molecule mimetics of these recognition motifs.3,4
In recent years, an important development in peptide mimicry has been the emergence of analogues of peptide secondary structures that mainly present selected side-chains, with the main-chain polyamide backbone abbreviated or totally absent. This approach is appealing because small molecules without polyamide backbones are more likely to be orally bioavailable and proteolytically stable. Compounds that present only selected side-chains to resemble peptide secondary structures were referred to, for the first time, as minimalist mimics by Burgess and coworkers in 2011.5
Minimalist peptidomimetics are likely to be most useful for targets where exact binding conformations are unknown. In these situations such compounds can have the intrinsic ability to adjust conformations via rotation around a few significant degrees of freedom, allowing such mimics to easily adapt. Minimalist mimics must be amenable to synthetic diversification and conveniently accessible with side-chains that correspond to the protein amino acids in order to be generally useful. In this context, reactions that transform protein amino acids into carbocyclic or heterocyclic backbones that have only a few significant degrees of freedom are particularly useful.
Following our interest in the multicomponent reaction (MCR)/cyclization approach to the synthesis of conformationally constrained peptidomimetics,6 in this work we demonstrate for the first time the application of this kind of strategy to the synthesis of minimalist peptidomimetics.
The atom economy of MCRs,7 their convergent character, operational simplicity, and the structural diversity and complexity of the resulting molecules make this chemistry exceptionally useful for drug discovery processes. In particular, Ugi-MCRs have undergone developments over the years, and various modifications in classic isocyanide-based multicomponent reactions (IMCRs) have been achieved by the introduction of unusual building blocks,8 by transformation of the IMCR products using post-condensation reactions9,10 or by performing intramolecular IMCRs with bifunctional inputs.11–13
With regard to the synthesis of peptidomimetics,14 replacement of the amine or acid component in the Ugi reaction with natural amino acids has had a number of successful outcomes.15,16 However, only a few reports make use of amino acid-derived chiral isocyanides in IMCR, probably because of the believed, but surmountable, configurationally instability of chiral α-substituted isocyanoacetates.17 Moreover, the use of amino acid-derived chiral aldehydes as the carbonyl component has almost not been documented at all,18 despite its potential for enabling secondary reactions in order to constrain or improve ‘drug-likeness’ of the initial flexible peptide-like products.
By employing amino acid-derived chiral α-isocyanoacetates and aldehydes, we pursued the synthesis of minimalist peptidomimetics relying on three different piperazine-based cores, spanning side-chains corresponding to two chemically diverse amino acids, namely L-Ala and L-Phe. Piperazines and their keto analogs are considered privileged scaffolds in medicinal chemistry, thanks to their versatile binding properties and frequent recurrence among positive hits encountered in biological screens.
We also accomplished a computational evaluation of their secondary structure mimicking properties and a biological screening within the COST Action CM 1106, entitled Chemical Approaches to Targeting Drug Resistance in Cancer Stem Cells.19
Results and discussion
As the carbonyl moiety for the Ugi reaction, we focused our attention on enantiomerically pure N-methyl-N-Boc amino aldehydes 1 and 2 (Scheme 1), easily prepared in good yields starting from the corresponding commercial N-Boc amino acids. Among the reported synthetic procedures for the preparation of isocyanides from α-amino acid ester hydrochlorides, we selected a two-step sequence, involving formylation of the precursor by reaction with trimethyl orthoformate without the use of a solvent, followed by dehydration of the obtained α-N-formylamino acid esters, by means of triphosgene as a mild dehydrating agent and N-methyl morpholine as a base.20 In this way, enantiomerically pure isocyanide components 3 and 4 have been prepared. The carboxylic acid and the amine Ugi components were properly chosen as bifunctional substrates in order to mediate highly selective outcomes in the post-Ugi cyclization steps.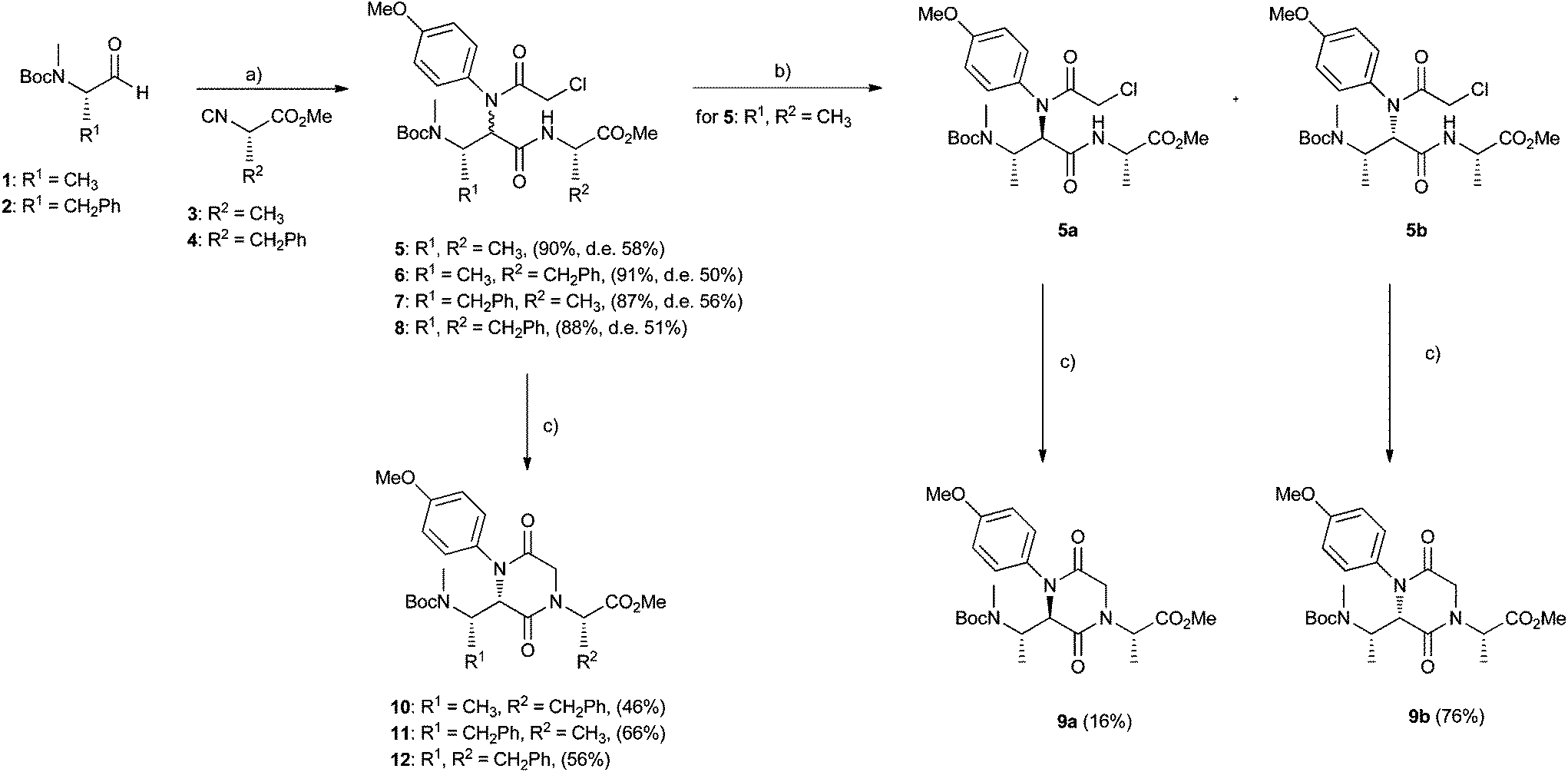 | ||
| Scheme 1 Reagents and conditions: (a) p-anisidine, MeOH, rt, 2 h; then chloroacetic acid, 60 h. (b) Flash chrom. (c) Cs2CO3, 10% LiI, CH3CN, rt (or 60 °C for 5a to 9a), 20 h. | ||
First of all, using chloroacetic acid and p-anisidine, we could access a small family of 2,5-diketopiperazine-based peptidomimetics, as reported in Scheme 1. The Ugi reaction was conducted after a precondensation time of two hours between p-anisidine and the amino aldehyde (1 or 2), as suggested by Carney and coworkers21 in order to avoid the risk of loss of optical purity of the isocyanoacetates. Ugi compounds 5–8 were all obtained in good yields (87–91%), but as unseparable diastereoisomeric mixtures, with the exception of compounds 5, which could be separated by flash chromatography (d.r. 5a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5b 21:79). The two Ugi diastereoisomers 5a and 5b were separately cyclized by means of cesium carbonate in dry acetonitrile,22 to afford 2,5-diketopiperazines 9a and 9b, whose overall stereochemistry has been assigned on the basis of computational, NMR and CD studies (vide infra). Cyclization performed better for compound 5b, affording 9b in 76% yield, whereas it proved to be quite difficult for 5a, so that compound 9a could be isolated only in 16% yield, under heating. Evidently, steric factors play a key role in the more or less smooth achievement of a crowded trisubstituted 2,5-diketopiperazine ring. The same cyclization reaction was then applied to Ugi adducts 6–8 to afford the corresponding compounds 10–12, which proved to be isolable as unique diastereoisomers in moderate yields (46–56%). This result can be rationalized in the light of the observed extremely different propensity of separated Ugi diastereoisomers 5 to cyclize to 9.
:5b 21:79). The two Ugi diastereoisomers 5a and 5b were separately cyclized by means of cesium carbonate in dry acetonitrile,22 to afford 2,5-diketopiperazines 9a and 9b, whose overall stereochemistry has been assigned on the basis of computational, NMR and CD studies (vide infra). Cyclization performed better for compound 5b, affording 9b in 76% yield, whereas it proved to be quite difficult for 5a, so that compound 9a could be isolated only in 16% yield, under heating. Evidently, steric factors play a key role in the more or less smooth achievement of a crowded trisubstituted 2,5-diketopiperazine ring. The same cyclization reaction was then applied to Ugi adducts 6–8 to afford the corresponding compounds 10–12, which proved to be isolable as unique diastereoisomers in moderate yields (46–56%). This result can be rationalized in the light of the observed extremely different propensity of separated Ugi diastereoisomers 5 to cyclize to 9.
Starting again from aldehydes 1 and 2 and isocyanoacetates 3 and 4 and varying the acid and amine components in the Ugi reaction, we could also access strictly related 2,6-diketopiperazine-based peptidomimetics 17–20, as reported in Scheme 2.
 | ||
| Scheme 2 Reagents and conditions: (a) glycine benzylester, MeOH, rt, 2 h; then AcOH, 60 h. (b) Flash chrom. (c) H2, 10% Pd/C, MeOH, rt, 2 h; then CDI, THF, 75 °C to rt, 4 h. | ||
In this case the Ugi reaction was performed employing acetic acid and glycine benzylester as the acid and amine component respectively, affording intermediates 13–16, with yields up to 89% and d.e. up to 84% (from 1H NMR). Catalytic removal of the benzylester group, followed by activation with carbonyl diimidazole in tetrahydrofuran,23 allowed us to obtain the desired products 17–20 in good yields. Only for the Ugi intermediate 13 it was possible to perform a chromatographic separation of diastereoisomers (d.r. 13a:13b 11.5:88.5) and achieve distinct compounds 17a and 17b, in 15% and 59% yield respectively. Clearly, as for the above case of 2,5-diketopiperazines, also the cyclizations of Ugi adducts 13a and 13b, to give the 2,6-diketopiperazines 17a and 17b, are subject to quite different steric restrictions. 2,6-diketopiperazines 18–20 were obtained in good yields (71–77%) and high d.e. (80–95%, from 1H NMR).
Finally, the two-step Ugi/cyclization strategy employing a bifunctional amine was also applied to the synthesis of 3,4-dihydropyrazin-2(1H)-one-based peptidomimetics 23–26 (Scheme 3). In this case, N-Cbz amino aldehydes 21 and 22 were employed in the Ugi reaction under usual conditions, together with isocyanoacetates 3 and 4, 2,2-diethoxyethanamine and benzoic acid. The obtained Ugi products proved to be highly unstable and therefore were not isolated, but directly cyclized to give final compounds 23–26 in good overall yields. In this case, all diastereoisomeric compounds 23a–26a and 23b–26b could be easily separated by chromatography, allowing us to estimate very high diastereoisomeric ratios, up to b:a > 93:7.
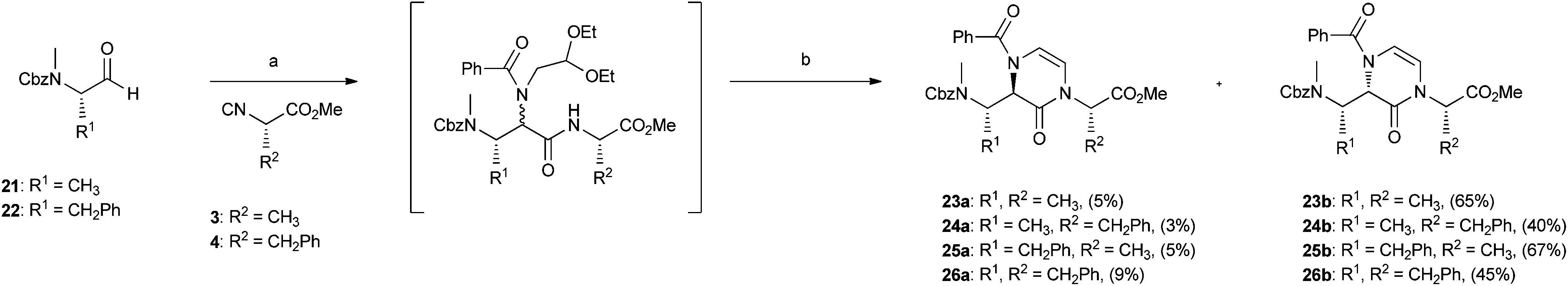 | ||
| Scheme 3 Reagents and conditions: (a) 2,2-diethoxyethanamine, MeOH, rt, 2 h; then benzoic acid, 60 h. (b) 50% TFA, DCM, rt, 24 h; then flash chrom. | ||
In order to rationalize the stereochemical outcome of the realized Ugi/cyclization processes, we relied on theoretical conformational analysis, NMR and CD spectra. Unfortunately, any attempt to obtain crystals suitable for X-ray diffraction analysis proved to be unsuccessful for all compounds in a wide range of solvents and crystallization conditions.
We focused our analysis on compounds 9 and performed a theoretical conformational search, which was run by the Monte-Carlo algorithm and molecular mechanics (MMFF force field), including all rotatable bonds and the puckering of 6-membered ring atoms. All structures thus obtained were geometry-optimized with the DFT method at the B3LYP/6-31G(d) level in vacuo. All structures obtained with population >1% at room temperature were considered in the following. The calculations were run independently for the C-2(R) and C-2(S) isomers of 9 (for atom numeration, see the structure in Table 1).
|
|
||||
|---|---|---|---|---|
| NOE | Predicted for C-2(S) | Predicted for C-2(R) | Exp. for 9b (major diast.) | Exp. for 9a (minor diast.) |
| a Legend: s, strong; m, medium; w, weak; —, no NOE. | ||||
| NCH3/H-2 | m | s | w | s |
| NCH3/H-27 | — | m | — | m |
| CH3-17/H-27 | m/s | — | m | — |
| tBu/H-27 | w | s | — | m |

The conformational analysis for the C-2(R) isomer revealed a strong preference for conformations with anti orientation between H-2 and H-15 and a pseudo-axial position of the C-2 appendage. Practically all populated structures at 300 K show a consistent conformation around the C-2/C-15 bond (Fig. 1). In this conformational family, aromatic ortho hydrogens are expected to give NOE with the tBu group of the Boc moiety and with NCH3, but not with CH3-17.
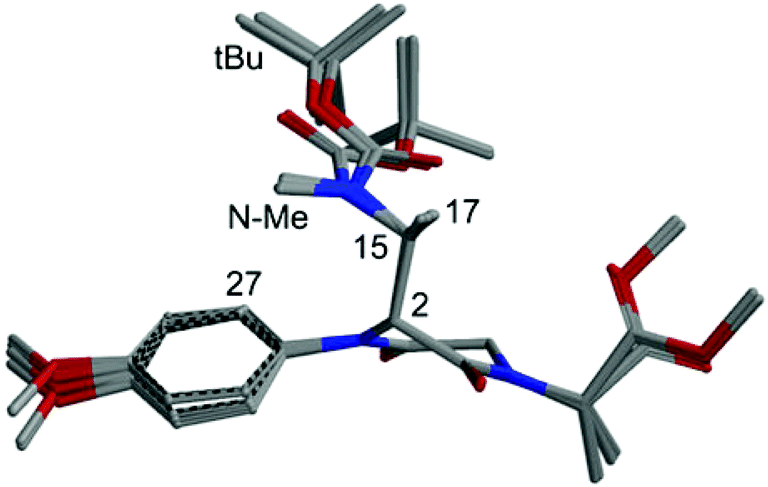 | ||
| Fig. 1 Superposition of the first 8 low-energy structures of isomer C-2(R) of compound 9, calculated at the B3LYP/6-31G(d) level. | ||
The conformational situation for the C-2(S) isomer is less clear-cut. While the C-2 appendage occupies again a pseudo-axial orientation, there are at least two main conformational families, one (more favored, Fig. 2a) with gauche orientation between H-2 and H-15 and a second (less favored, Fig. 2b) with anti orientation. Although the NOE analysis is less straightforward, we can infer that aromatic ortho hydrogens are expected to give a sizable NOE with CH3-17, and smaller or no NOE with the Boc moiety and NCH3, respectively.
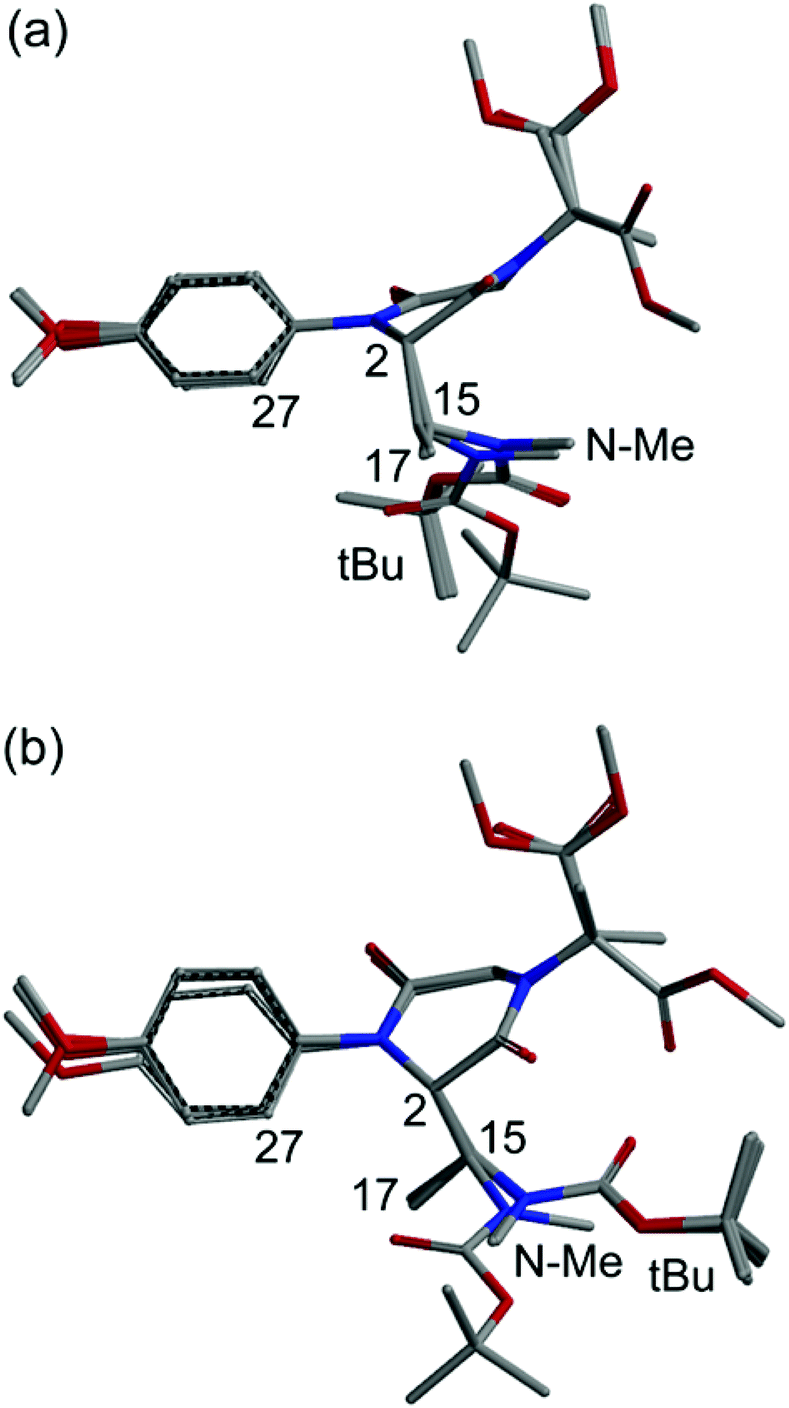 | ||
| Fig. 2 Superposition of the first 9 low-energy structures of isomer C-2(S) of compound 9, calculated at the B3LYP/6-31G(d) level (a, more favored; b, less favored). | ||
Comparison of the expected NOEs based on molecular modeling with the experimentally observed data obtained from ROESY correlation peaks (Table 1) offers a strong indication to assign a C-2(S) configuration to the major diastereoisomer 9b and, conversely, a C-2(R) one to the minor diastereoisomer 9a.
The configuration of the stereogenic carbon C-2 of the major diastereoisomer 9b as C-2(S) was also studied by electronic circular dichroism (CD).24 The CD spectrum of 9b, measured in acetonitrile solution, displays a series of relatively weak bands above 185 nm (ESI†). CD spectra were calculated with the TDDFT method on DFT-optimized input structures for the two C-2(R) and C-2(S) isomers. Unfortunately, the two isomers led to a similar sequence of bands which in both cases reproduces the experimental spectrum of 9b; therefore a safe discrimination based only on CD spectra is impossible (ESI†). However, since CD spectroscopy does not contradict the NMR results discussed above, we are confident of definitely attributing the C-2(S) stereochemistry to the major diastereoisomer 9b and the C-2(R) one to the minor diastereoisomer 9a.
In accordance with this attribution, the observed diastereoselectivity in the Ugi reaction (Scheme 1) can be rationalized by considering a Felkin–Ahn-like model, where the isocyanoacetate reagent approaches towards the preformed iminium intermediate from the less hindered side (Fig. 3), affording preferentially 5b over 5a.
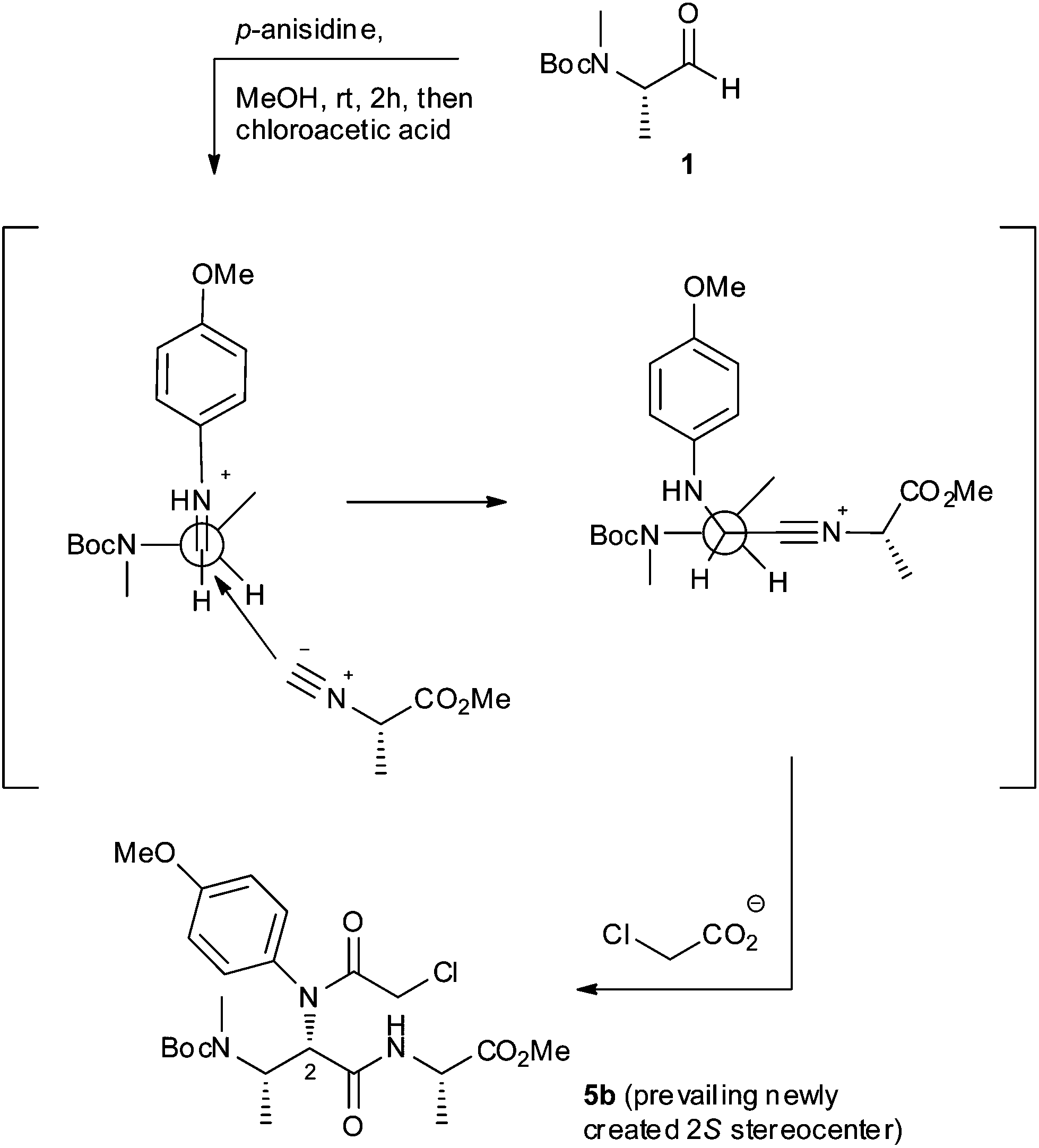 | ||
| Fig. 3 Proposed model for the diastereoselective formation of 5b in Scheme 1. | ||
The proposed model can also explain the trend of diastereoselectivity which was observed for strictly related 6–8 Ugi products. Reasoning that in all described Ugi/cyclization sequences (Schemes 2 and 3) the partial diastereocontrol is likely to come from the addition of the isocyanoacetate reagent to closely similar iminium intermediates, the reported diastereoselective outcomes could be reasonably explained by the application of Felkin–Ahn-like models similar to those depicted in Fig. 3.
In order to assess our compounds as minimalist peptidomimetics, a computational study was performed. Structures a, b and c were chosen as representative models of compounds 9b, 17b and 23b (Fig. 4), all characterized by the (S) stereochemical configuration on the piperazine-based ring and by L-Ala-derived side chains. Models a, b and c contain the minimalist core provided with N-Ac and CO-NHMe terminal groups, aimed to mimic the insertion of the peptidomimetic into a putative peptide.
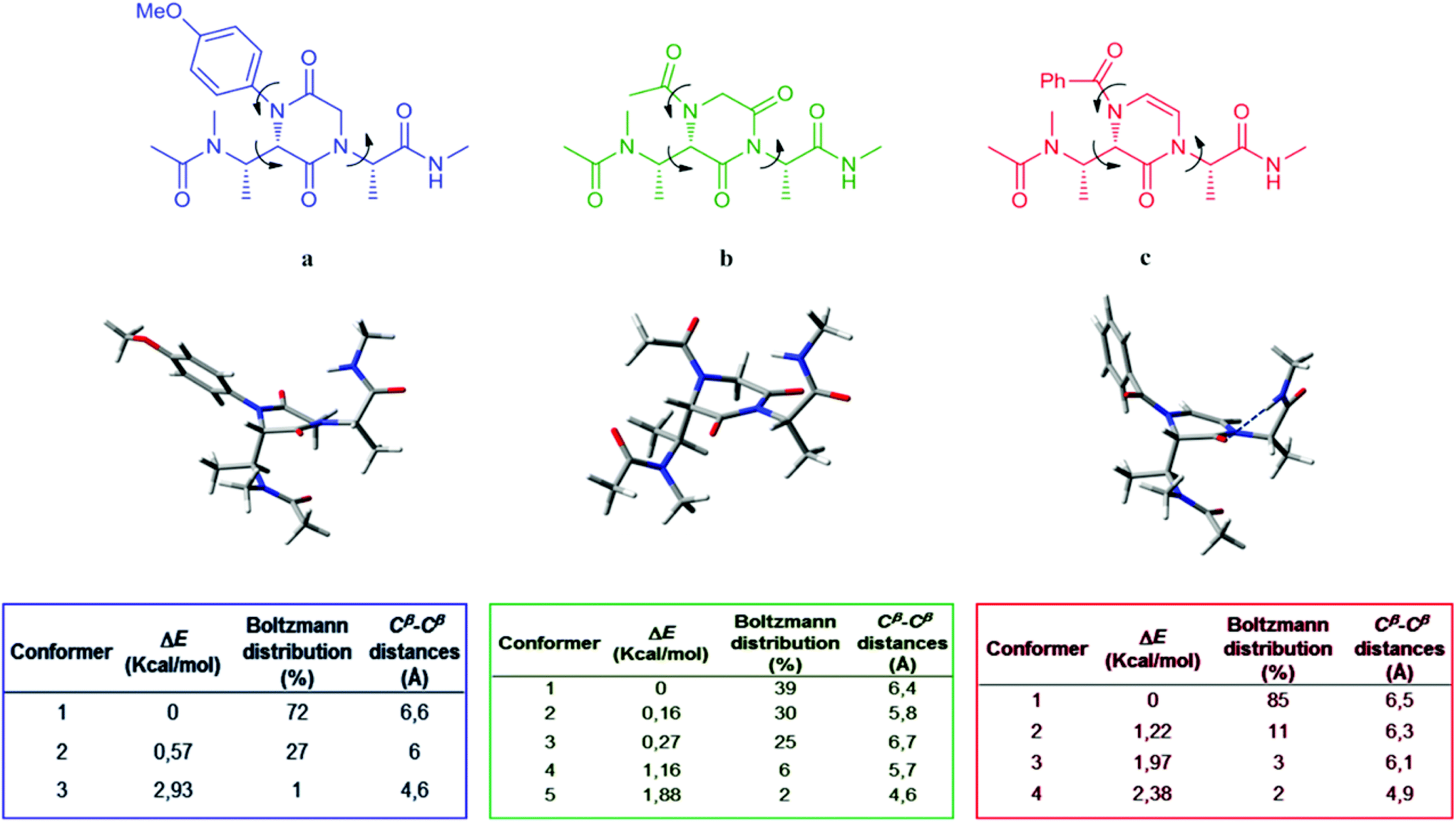 | ||
| Fig. 4 Above: 2D structures of models a, b and c with their low energy conformers 3D images. Blue dotted line on model c represents the internal hydrogen-bond. Below: tables reporting the Boltzmann distributions and the Cβ–Cβ distances. | ||
In this approach, the spatial mobility of the side chains connected to the central hetero-aromatic ring of the three models was analyzed by minimizing, equilibrating and heating up the starting models to 300 K, 700 K and 1000 K for a short period (2 ns). Finally, passing through an intermediate equilibration step at 700 K, 20 ns of MD simulations at room temperature (300 K) were performed.5b The algorithm of the AMBER12 package25 was executed for the entire calculation of each model, using the GB implicit water solvent model.26 A frame every 10 ps of MD simulations was recorded, acquiring 2800 conformational states of each mode (see the Experimental section). Analyzing the dihedral angle fluctuation (arrows in Fig. 4) over the obtained trajectories, the different families of conformations for models a, b and c were recognized.
Furthermore, the geometries representing those families of conformations were energy minimized by GAUSSIAN0927 at the quantum-mechanics DFT/B3LYP/6-31g(d)/CPCM-water level of calculation. The application of the Boltzmann equation provided the conformers distribution percentage (tables in Fig. 4). Finally, in order to establish the correspondence of Cβ–Cβ distances for peptidomimetics models a, b and c with common secondary structures, the distances between the Cβ atoms of the side chains were measured (tables in Fig. 4).
The results indicate that model b, in comparison with a and c, possesses the highest number of thermodynamically accessible conformers at room temperature. This can be due to the minor steric hindrance of the N-substituent on the piperazine core, with the acetyl group in model b allowing major mobility for the vicinal side chains in comparison with the 4-methoxy-phenyl and benzoyl groups of models a and c.
Comparing the Cβ–Cβ distances measured in the lowest energy conformers of a, b and c with those reported by Burgess and co-workers5 for typical secondary structures, we were able to predict the secondary structures potentially mimicked by our compounds (Table 2). Model compounds a and c can assume conformations compatible with secondary structures like α-helix, β-sheet (anti-parallel) and γ-turn (classic). In contrast, the populated conformers of model b may mimic all the secondary structures reported in Table 2. Moreover, it has to be underlined that model c mimics predominantly the α-helix and the β-sheet (anti-parallel) secondary structures. This is due to an internal hydrogen-bond stabilizing nearly only one structural geometry (Fig. 4).

|
Because of our particular interest in cancer drug resistance, all compounds have been preliminarily evaluated for their antiproliferative effects on two different hepatocellular carcinoma (HCC) cellular lines, namely Huh7 (well differentiated cells) and Mahlavu (PTEN deficient poorly differentiated cells), for which differentiation versus resistance ability seems to be strongly correlated with well defined types of PPIs.28 A significant antiproliferative effect, in the micromolar range (see ESI†), has been observed for 12 and 25b and deserves further studies, which are currently in progress.
Conclusion
We have demonstrated the feasibility of a two-step multicomponent synthesis of three complex, yet structurally distinct, piperazine-based scaffolds, in which bifunctional substrates mediate highly selective outcomes based on Ugi/cyclization sequences. All scaffolds are characterized by the presence of L-Ala and/or L-Phe-derived amino acid side chains and have been demonstrated to act as minimalist peptidomimetics, able to mimic a range of secondary structures and therefore potentially useful for the synthesis of small-molecule PPI modulators. Preliminary biological evaluation of cancer resistant cellular lines has revealed a promising antiproliferative activity for selected compounds, for which further investigation is currently underway.Experimental section
General information
All commercial materials (Aldrich, Fluka) were used without further purification. All solvents were of reagent grade or HPLC grade. All reactions were carried out under a nitrogen atmosphere unless otherwise noted. All reactions were monitored by thin layer chromatography (TLC) on precoated silica gel 60 F254; spots were visualized with UV light or by treatment with a 1% aqueous KMnO4 solution. Products were purified by flash chromatography on silica gel 60 (230–400 mesh). 1H NMR spectra and 13C NMR spectra were recorded on 300 and 400 MHz spectrometers. Chemical shifts are reported in parts per million relative to the residual solvent. 13C NMR spectra have been recorded using the APT pulse sequence. Multiplicities in 1H NMR are reported as follows: s = singlet, d = doublet, t = triplet, m = multiplet, br s = broad singlet. High-resolution MS spectra were recorded with an FT-ICR (Fourier Transform Ion Cyclotron Resonance) instrument, equipped with an ESI source. UV-Vis spectra were recorded with a Jasco V-650 spectrophotometer. CD spectra were recorded with a JASCO J-715 spectropolarimeter; the conditions are reported in the legend of Fig. 2.General procedure for the synthesis of compounds 5–8
Aldehyde (1 or 2) (1.0 mmol, 1 eq.) was dissolved in 1 mL of dry methanol under nitrogen, 4-methoxy aniline (1.0 mmol, 123 mg, 1 eq.) was added and the resulting mixture was kept under stirring for 2 h at room temperature. 2-Chloroacetic acid (1.0 mmol, 94.5 mg, 1 eq.) and isocyanide (3 or 4) (1.2 mmol, 1.2 eq.) were sequentially added and the reaction was stirred for an additional 60 h at room temperature. The resulting mixture was then concentrated under reduced pressure, to give a residue which was purified by flash chromatography (FC) as indicated below.:1.5; yield: 5a (97 mg, 19%), 5b (356 mg, 71%). 5a: colorless oil; Rf 0.21 (1.5:1 n-hexane–EtOAc); [α]30D −16.3 (c 0.5, CHCl3); 1H NMR (300 MHz, CD3CN, 1:1 mixture of rotamers) δ 7.38–7.08 (m, 3H), 6.95 (d, J = 8.8 Hz, 2H), 4.45–4.09 (br, s, 1H), 3.85 (br, s, 2H), 3.81 (s, 3H), 3.82–3.75 (m, 1H), 3.69 (s, 1.5H), 3.71–3.63 (m, 1H), 3.67 (br, s, 1.5H), 2.65 (s, 3H), 1.46–1.31 (m, 12H), 1.27 (br, d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CD3CN) δ 178.1, 173.0 (2C), 165.4, 160.2, 136.5, 136.2, 135.5, 120.0, 119.8, 84.8 and 84.6 (1C), 60.6, 60.5, 57.2, 53.5, 53.4, 48.5, 48.3, 34.3, 33.0 (3C), 22.2 and 21.9 (1C), 19.7 and 19.6 (1C); HRMS (ESI) calcd for C23H34ClN3NaO7+ [MNa]+ 522.1977, found 522.1983. 5b: colorless oil; Rf 0.18 (1.5:1 n-hexane–EtOAc); [α]30D +8.5 (c 0.5, CHCl3); 1H NMR (300 MHz, CD3CN, 1:1 mixture of rotamers) δ 7.40–7.08 (br, m, 1H), 7.20 (m, 2H), 6.95 (d, J = 9.1 Hz, 2H), 4.41–4.12 (br, m, 1H), 3.86 (br, s, 2H), 3.81 (s, 3H), 3.79 (m, 1H), 3.67 (br, s, 1.5H), 3.65 (m, 1H), 3.64 (s, 1.5H), 2.67 (s, 3H), 1.38 (s, 9H), 1.32 (m, 3H), 1.27 (br, d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.5 and 173.4 (1C), 169.4–168.7 (2C), 160.6, 157.0 and 156.8 (1C), 131.4, 130.8, 130.2, 115.7, 115.5, 81.0 and 80.6 (1C), 56.1 (2C), 53.0, 52.9, 48.9 (2C), 43.1 and 43.0 (1C), 29.1 (4C), 18.2, 16.1; HRMS (ESI) calcd for C23H34ClN3NaO7+ [MNa]+ 522.1977, found 522.1961.
:1.5; yield 524 mg, (91%) as an inseparable mixture of diastereoisomers (d.e. 50%, NMR analysis): pale yellow oil; Rf 0.24 (1.5:1 n-hexane–EtOAc); 1H NMR (300 MHz, CD3CN, rotameric mixture of a mixture of diastereoisomers) δ 7.47–6.99 (m, 8H), 6.89 (br, d, J = 8.8 Hz, 2H), 4.80–4.40 (br, m, 1H), 3.81 (br, s, 2H), 3.80–3.74 (m, 4H), 3.69 (s, 0.5H), 3.69–3.63 (m, 3.5H), 3.24–3.05 (br, m, 1H), 3.03–2.80 (br, m, 1H), 2.62 (s, 2.75H), 2.58 (s, 0.25H), 1.36 (s, 9H), 1.28–1.21 (m, 3H); HRMS (ESI) calcd for C29H38ClN3NaO7+ [MNa]+ 598.2290, found 598.2305.
:1.5; yield 501 mg, (87%) as an inseparable mixture of diastereoisomers (d.e. 56%, NMR analysis): pale yellow oil; Rf 0.22 (1.5:1 n-hexane–EtOAc); 1H NMR (300 MHz, CD3CN, rotameric mixture of a mixture of diastereoisomers) δ 7.40–7.04 (m, 8.4H), 6.96 (br, d, J = 8.4 Hz, 1.6H), 4.59–4.40 (br, m, 1H), 3.93–3.64 (m, 10H), 3.33–2.87 (m, 1H), 2.80 (s, 0.66H), 2.76 (s, 2.34H), 2.64 (m, 1H), 1.52–1.26 (m, 12H); HRMS (ESI) calcd for C29H38ClN3NaO7+ [MNa]+ 598.2290, found 598.2305.
:1.5; yield 573 mg, (88%) as an inseparable mixture of diastereoisomers (d.e. 51%, NMR analysis): oil; Rf 0.44 (1.5:1 n-hexane–EtOAc); 1H NMR (400 MHz, CDCl3, rotameric mixture of a mixture of diastereoisomers) δ 7.38–7.06 (m, 11.4H), 7.02–6.80 (br, m, 3.6H), 5.05–4.67 (br, m, 1H), 3.93–3.58 (m, 10H), 3.32–3.18 (m, 1H), 3.05–2.86 (m, 2H), 2.78 (s, 0.75H), 2.75 (s, 2.25H), 2.65 (m, 1H), 1.40 (m, 9H); HRMS (ESI) calcd for C35H42ClN3NaO7+ [MNa]+ 674.2603, found 674.2622.
General procedure for the synthesis of compounds 9–12
To a solution of the Ugi product (5a, 5b, 6, 7 or 8) (0.5 mmol, 1 eq.) in dry acetonitrile (4.5 mL) under nitrogen, cesium carbonate (1 mmol, 326 mg, 4 eq.) and LiI (0.05 mmol, 7 mg, 0.1 eq.) were added, and the mixture was stirred for 20 h at room temperature (or at 60 °C, for 5a). The mixture was quenched with satd aq. NH4Cl (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated under reduced pressure, to give a residue, which was purified by flash chromatography (FC) as indicated below.:1.5; yield: 37 mg (16%); colorless oil; Rf 0.18 (1.5:1 n-hexane–EtOAc); [α]30D −12.7 (c 0.5, CHCl3); 1H NMR (300 MHz, CD3CN) δ 7.43 (br, m, 2H), 6.98 (d, J = 8.8 Hz, 2H), 5.03 (q, J = 7.0 Hz, 1H), 4.63 (br, m, 1H), 4.34 (br, m, 1H), 4.30 (br, d, J = 16.6 Hz, 1H), 3.88 (d, J = 16.7 Hz, 1H), 3.83 (s, 3H), 3.73 (s, 3H), 2.67 (s, 3H), 1.45 (s, 9H), 1.44 (d, J = 7.0 Hz, 3H), 1.24 (br, d, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CD3CN) δ 172.2, 168.4, 165.0 (2C), 160.7, 133.9, 131.4 (2C), 130.0, 115.3 (2C), 80.9, 68.5, 55.8, 52.5 (2C), 49.1 and 48.9 (1C), 48.0, 28.3 (3C), 17.4, 14.2; HRMS (ESI) calcd for C23H33N3NaO7+ [MNa]+ 486.2211, found 486.2227.
:1.5; yield: 176 mg (76%); colorless oil; Rf 0.16 (1.5:1 n-hexane–EtOAc); [α]30D +24.2 (c 1.0, MeOH); 1H NMR (400 MHz, CD3CN) δ 7.36 (br, d, J = 8.9 Hz, 2H), 6.98 (d, J = 9.2 Hz, 2H), 5.12 (q, J = 7.3 Hz, 1H), 4.73 (br, m, 1H), 4.29 (d, J = 16.7 Hz, 1H), 4.27 (br, m, 1H), 3.88 (br, d, J = 16.7 Hz, 1H), 3.83 (s, 3H), 3.69 (s, 3H), 2.68 (s, 3H), 1.46 (d, J = 7.3 Hz, 3H), 1.45 (s, 9H), 1.16 (br, d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CD3CN) δ 172.2, 165.9, 164.9, 159.1, 133.9, 130.0, 129.2 (2C), 114.6 (2C), 80.2, 68.4, 55.9, 52.7, 52.1, 51.5, 48.0, 29.5, 28.3 (3C), 16.5, 13.8; HRMS (ESI) calcd for C23H33N3NaO7+ [MNa]+ 486.2211, found 486.2216.
:7; yield: 124 mg (46%); wax; Rf 0.15 (7:3 n-hexane–EtOAc); [α]22D −55.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.34–7.23 (m, 3H), 7.21–7.16 (m, 2H), 7.14–7.05 (m, 2H), 6.90 (d, J = 8.9 Hz, 2H), 5.68 (dd, J = 11.4, 5.1 Hz, 1H), 4.70 (br, m, 1H), 4.41 (d, J = 17.3 Hz, 1H), 4.03 (d, J = 17.3 Hz, 1H), 3.88 (br, m, 1H), 3.81 (s, 3H), 3.80 (s, 3H), 3.40 (dd, J = 14.4 and 5.1 Hz, 1H), 2.99 (dd, J = 14.4, 11.4 Hz, 1H), 2.68 (s, 3H), 1.46 (s, 9H), 1.17 (d, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.7, 166.8, 164.7, 159.3, 156.7, 136.1, 133.0, 129.8 (2C), 129.4 (2C), 129.2 (2C), 127.7, 115.0 (2C), 80.9, 69.0, 56.6, 56.1, 53.0, 51.9, 47.6, 35.5, 29.9, 29.1 (3C), 17.1; HRMS (ESI) calcd for C29H37N3NaO7+ [MNa]+ 562.2524, found 562.2509.
:1.5; yield: 178 mg (66%); pale yellow oil; Rf 0.15 (1.5:1 n-hexane–EtOAc); [α]30D +2.2 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.35–7.24 (m, 2H), 7.24–7.12 (m, 3H), 7.12–7.05 (m, 2H), 6.92 (d, J = 8.6 Hz, 2H), 5.29 (q, J = 7.1 Hz, 1H), 4.97 (br, m, 1H), 4.43–4.30 (m, 2H), 3.99 (d, J = 16.7 Hz, 1H), 3.83 (s, 3H), 3.74 (s, 3H), 3.08 (br, m, 1H), 2.77 (br, m, 2H), 2.67 (s, 3H), 1.47 (d, J = 7.1 Hz, 3H), 1.44 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 171.8, 166.0, 164.7, 159.3, 157.1, 137.7, 132.9, 129.6 (2C), 129.1 (2C), 128.8 (2C), 127.2, 115.1 (2C), 81.0, 67.3, 57.7, 56.1, 52.3, 51.7, 47.6, 36.9, 30.8, 28.9 (3C), 14.5; HRMS (ESI) calcd for C29H37N3NaO7+ [MNa]+ 562.2524, found 562.2536.
:7; yield: 151 mg (56%); yellow oil; Rf 0.21 (7:3 n-hexane–EtOAc); [α]31D = −74.7 (c 1.0, MeOH); 1H NMR (400 MHz, CDCl3) δ 7.35–7.23 (m, 4H), 7.23–7.18 (m, 2H), 7.18–7.11 (m, 2H), 7.10–6.99 (m, 4H), 6.88 (d, J = 8.8 Hz, 2H), 5.73 (dd, J = 11.3 and 5.3 Hz, 1H), 4.94 (br, m, 1H), 4.40 (d, J = 17.2 Hz, 1H), 4.05 (d, J = 17.2 Hz, 1H), 4.03 (br, m, 1H), 3.83 (s, 3H), 3.81 (s, 3H), 3.44 (dd, J = 14.4 and 5.3 Hz, 1H), 3.03 (dd, J = 14.4 and 11.3 Hz, 1H), 3.05–2.77 (m, 2H), 2.64 (s, 3H), 1.40 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 170.6, 166.8, 164.7, 159.5, 157.0, 137.5, 136.1, 132.2, 129.7 (2C), 129.4–129.0 (8C), 127.8, 127.1 (2C), 115.1, 80.9, 67.1, 56.7, 56.1 (2C), 53.1, 47.4, 36.7, 35.4, 30.3, 28.9 (3C); HRMS (ESI) calcd for C35H41N3NaO7+ [MNa]+ 638.2837, found 638.2847.
General procedure for the synthesis of compounds 13–16
Aldehyde (1 or 2) (1.0 mmol, 1 eq.) was dissolved in 1 mL of dry methanol under nitrogen, O-benzyl glycine (1.0 mmol, 165 mg, 1 eq.) was added and the resulting mixture was kept under stirring for 2 h at room temperature. Acetic acid (1.0 mmol, 60 mg, 1 eq.) and isocyanide (3 or 4) (1.2 mmol, 1.2 eq.) were sequentially added and the reaction was stirred for an additional 60 h at room temperature. The resulting mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 5 mL). The combined organic layers were dried over Na2SO4 and the solvent was removed under reduced pressure to afford the crude Ugi product, which was purified by flash chromatography (FC) as indicated below.:1.5; yield: 13a (35 mg, 7%), 13b (276 mg, 54%). 13a: amber oil; Rf 0.18 (1:1.5 n-hexane–EtOAc); [α]30D −3.4 (c 0.5, CHCl3); 1H NMR (300 MHz, CDCl3, 1:1 mixture of rotamers) δ 7.34 (br, m, 5H), 6.62–6.27 (br, m, 1H), 5.16–5.08 (m, 2H), 5.06 (br, m, 1H), 4.79–4.61 (br, m, 1H), 4.47 (d, J = 19.5 Hz, 0.5H), 4.45 (d, J = 19.5 Hz, 0.5H), 4.23 (br, m, 1H), 4.01 (br, d, J = 19.5 Hz, 1H), 3.72 (s, 1.5), 3.71 (s, 1.5), 2.62 (br, s, 3H), 1.98 (br, s, 1.5), 1.95 (s, 1.5), 1.42 (s, 9H), 1.36 (d, J = 6.8 Hz, 3H), 1.14 (br, m, 3H); 13C NMR (75 MHz, CDCl3, mixture of rotamers) δ 173.0 and 172.9 (1C), 172.6 and 172.4 (1C), 168.2 and 168.1 (1C), 167.8, 155.8, 135.4, 128.6–128.2 (5C), 79.4, 66.9, 57.4–56.0 (br, 1C), 52.3, 48.4, 48.1, 47.1 and 46.7 (br, 1C), 28.7, 28.4 (3C), 21.6, 18.0, 15.2 and 14.8 (1C); HRMS (ESI) calcd for C25H37N3NaO8+ [MNa]+ 530.2473, found 530.2455. 13b: Rf 0.15 (1:1.5 n-hexane–EtOAc); [α]30D +8.8 (c 0.5, CHCl3); 1H NMR (300 MHz, CDCl3, 1:1 mixture of conformers) δ 7.35 (br, m, 5H), 6.65–6.28 (br, m, 1H), 5.17 (br, m, 3H), 4.51–4.19 (br, m, 4H), 3.70 (s, 1.5), 3.68 (s, 1.5), 2.81 (s, 3H), 2.03 (br, s, 3H), 1.42 (s, 9H), 1.32 (d, J = 6.8 Hz, 3H), 1.15 (d, J = 6.8 Hz, 1.5H), 1.12 (d, J = 6.8 Hz, 1.5H); 13C NMR (75 MHz, CDCl3, mixture of rotamers) δ 172.4–172.0 (2C), 169.6, 168.9 and 168.8 (1C), 155.4 and 155.2 (1C), 135.4 and 135.2 (1C), 129.1–128.0 (5C), 80.3, 67.4 and 67.2 (1C), 59.6, 52.6, 48.5, 48.1, 47.8, 29.7, 28.4 (3C), 21.6, 17.6, 15.7 and 15.3 (1C); HRMS (ESI) calcd for C25H37N3NaO8+ [MNa]+ 530.2473, found 530.2482.
:1; yield: 344 mg (59%), as an inseparable mixture of diastereoisomers (d.e. 80%, NMR analysis): pale yellow oil; Rf 0.27 (1:1 n-hexane–EtOAc); 1H NMR (300 MHz, CDCl3, rotameric mixture of two diastereoisomers) δ 7.41–7.03 (m, 11H), 5.28–5.10 (m, 3H), 4.78 (br, m, 0.9H), 4.78–3.93 (m, 3.1H), 3.68 (s, 0.3H), 3.65 (s, 2.7H), 3.09 (br, m, 1H), 3.01 (m, 1H), 2.75 (br, s, 2.7H), 2.58 (s, 0.3H), 1.97 (br, m, 3H), 1.42 (s, 0.9H), 1.39 (s, 8.1H), 1.09 (br, d, J = 6.8 Hz, 3H); HRMS (ESI) calcd for C31H41N3NaO8+ [MNa]+ 606.2786, found 606.2800.
:1; yield: 256 mg (44%), as an inseparable mixture of diastereoisomers (d.e. 84%, NMR analysis): pale yellow oil; Rf 0.20 (1:5 n-hexane–EtOAc); 1H NMR (300 MHz, CDCl3, rotameric mixture of two diastereoisomers) δ 7.46–7.04 (m, 11H), 5.34–5.10 (m, 3H), 4.88 (br, m, 0.9H), 4.62–3.91 (m, 3.1H), 3.73 (s, 0.24H), 3.65 (s, 2.76H), 3.32 (br, m, 1H), 3.07 (br, m, 1H), 2.81 (br, s, 2.76H), 2.65 (s, 0.24H), 2.00 (br, s, 2.76H), 1.97 (s, 0.24H), 1.43 (br, s, 9H), 1.36 (br, m, 3H); HRMS (ESI) calcd for C31H41N3NaO8+ [MNa]+ 606.2786, found 606.2761.
:1.5; yield: 587 mg (89%), as an inseparable mixture of diastereoisomers (d.e. 72%, NMR analysis): colorless oil; Rf 0.18 (1.5:1 n-hexane–EtOAc); 1H NMR (300 MHz, CDCl3, rotameric mixture of two diastereoisomers) δ 7.43–7.01 (m, 16H), 5.29–5.09 (m, 3H), 4.88 (m, 0.86H), 4.74 (m, 0.14H), 4.64–4.43 (m, 1.86H), 4.29 (m, 0.14H), 4.01 (br, d, J = 16.6 Hz, 1H), 3.74 (s, 0.24H), 3.67 (s, 1.26H), 3.65 (s, 1.5H), 3.54–2.84 (m, 4H), 2.70 (s, 2.52H), 2.63 (s, 0.48H), 2.07 (s, 0.24H), 1.98 (s, 1.5H), 1.95 (s, 1.26H), 1.41 (s, 9H); HRMS (ESI) calcd for C37H45N3NaO8+ [MNa]+ 682.3099, found 682.3114.
General procedure for the synthesis of compounds 17–20
Palladium (10 wt% on carbon, 70 mg) was added to a solution of the Ugi product (13a, 13b, 14, 15 or 16) (0.30 mmol, 1 eq.) in methanol (3 mL). The reaction mixture was degassed in vacuo, placed under an atmosphere of H2 (g), and stirred in the dark at rt for 2 h. The mixture was filtered through a pad of Celite eluting with methanol (10 mL), and the combined organic layers were concentrated in vacuo to give the crude carboxylic acid intermediate, which was directly used in the next step, as follows. 1,1′-Carbonyl diimidazole (0.30 mmol, 127 mg, 1 eq.) was added to a solution of the crude carboxylic acid in dry tetrahydrofuran (3 mL), under nitrogen, and the resulting mixture was refluxed for 60 min and then stirred for an additional 3 h at room temperature. The solvent was removed under reduced pressure to give a residue which was partitioned between CHCl3 (10 mL) and 1 M HCl (10 mL). The aqueous phase was extracted with CHCl3 (3 × 10 mL) and the combined organic layers were washed with H2O, dried over Na2SO4 and concentrated in vacuo to afford the crude product, which was purified by flash chromatography (FC), as indicated below.:3; yield: 18 mg (15%); colorless oil; Rf 0.33 (3:7 n-hexane–EtOAc); [α]30D −15.6 (c 0.5, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.63–3.90 (m, 5H), 3.72 (br, s, 3H), 2.82 (br, s, 3H), 2.08 (br, s, 3H), 1.45 (s, 9H), 1.37 (br, d, J = 6.8 Hz, 3H), 1.12 (br, d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.1, 169.1, 167.0, 166.5 and 166.4 (1C), 155.9, 80.5, 55.0, 52.5, 49.5, 48.8, 46.5, 29.0, 28.3 (3C), 21.1, 15.5 and 15.4 (1C), 14.2 and 13.9 (1C); HRMS (ESI) calcd for C18H29N3NaO7+ [MNa]+ 422.1898, found 422.1924.
:3; yield: 71 mg (59%); colorless oil; Rf 0.29 (3:7 n-hexane–EtOAc); [α]30D +39.9 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3, mixture of conformers 1.5:1) δ 5.36 (br, d, J = 9.9 Hz, 1H), 5.19 (m, 1H), 4.72 (m, 1H), 4.57 (d, J = 18.7 Hz, 0.6H), 4.52 (d, J = 18.7 Hz, 0.4H), 4.29 (d, J = 18.7 Hz, 0.4H), 4.24 (d, J = 18.7 Hz, 0.6H), 3.68 (s, 1.8H), 3.67 (s, 1.2H), 2.82 (br, s, 1.8H), 2.80 (s, 1.2H), 2.23–2.15 (m, 3H), 1.52 (d, J = 7.0 Hz, 1.8H), 1.45 (d, J = 7.0 Hz, 1.2H), 1.44–1.38 (m, 9H), 1.19 (br, d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 169.9, 169.0, 167.0, 166.4, 156.1, 80.2, 54.7, 52.4, 49.4, 48.7, 46.8, 28.9, 28.3 (3C), 21.4, 15.5, 14.1; HRMS (ESI) calcd for C18H29N3NaO7+ [MNa]+ 422.1898, found 422.1917.
:1; yield: 105 mg (74%), as an inseparable mixture of diastereoisomers (d.e. 80%, NMR analysis); foam; Rf 0.36 (1:1.5 n-hexane–EtOAc); 1H NMR (400 MHz, CD3CN, a mixture of two diastereoisomers) δ 7.34–7.03 (m, 5H), 5.56–5.37 (br, m, 1.9H), 4.59 (br, m, 1H), 4.57 (d, J = 18.7 Hz, 1H), 4.83–4.11 (m, 2H), 3.66 (br, s, 3H), 3.43 (m, 1H), 3.17–2.99 (m, 1H), 2.79 (s, 0.3H), 2.77 (s, 2.7H), 2.23 (s, 2.7H), 2.14 (s, 0.3H), 1.42 (s, 9H), 1.14 (m, 3H); 13C NMR (101 MHz, CD3CN), δ 170.2–167.8 (4C), 156.3 and 156.0 (1C), 138.2, 129.8 (2C), 129.0 (2C), 127.3 (1C), 80.4, 54.2–53.8 (2C), 52.8, 51.4 and 50.9 (1C), 47.1 and 46.9 (1C), 34.7, 30.4, 28.4 (3C), 21.3, 15.2 and 14.8 (1C); HRMS (ESI) calcd for C24H33N3NaO7+ [MNa]+ 498.5239, found 498.5223.
:1.5; yield: 110 mg (77%), as an inseparable mixture of diastereoisomers (d.e. 95%, NMR analysis); foam; Rf 0.13 (1.5:1 n-hexane–EtOAc); 1H NMR (400 MHz, CD3CN, 1:1 rotameric mixture) δ 7.37–7.19 (m, 5H), 5.50(m, 1H), 5.25 (m, 1H), 5.02–4.58 (m, 2H), 4.41 (d, J = 18.6 Hz, 1H), 3.66 (s, 1.5H), 3.65 (s, 1.5H), 3.04–2.77 (br, m, 2H), 2.79 (s, 3H), 2.18 (s, 3H), 1.45 (d, J = 6.9 Hz, 1.5H), 1.44 (d, J = 6.9 Hz, 1.5H), 1.35 (br, m, 9H); 13C NMR (100 MHz, CD3CN), δ 170.7 and 170.1 (1C), 170.0–169.7 (1C), 169.1, 167.5, 156.2 and 156.1 (1C), 137.8, 129.1, 128.9, 128.3 (2C), 126.5 and 126.4 (1C), 79.7 and 79.4 (1C), 56.4, 54.9 and 54.5 (1C), 52.0, 48.3, 46.7, 34.4 and 34.3 (1C), 27.9, 27.5 (3C), 20.9, 13.6 and 13.4 (1C); HRMS (ESI) calcd for C24H33N3NaO7+ [MNa]+ 498.2211, found 498.2225.
:1.5; yield: 117 mg (71%), as an inseparable mixture of diastereoisomers (d.e. 80%, NMR analysis); thick oil; Rf 0.39 (1.5:1 n-hexane–EtOAc); 1H NMR (400 MHz, CD3CN, rotameric mixture of two diastereoisomers) δ 7.36–7.07 (m, 10H), 5.56–5.40 (m, 1H), 5.14 (d, J = 9.3 Hz, 0.9H), 5.03 (d, J = 10.5 Hz, 0.1H), 4.95–4.80 (m, 0.9H), 4.65 (d, J = 18.5 Hz, 0.1H), 4.62 (d, J = 18.6 Hz, 0.9H), 4.42 (m, 0.1H), 4.33 (d, J = 18.5 Hz, 1H), 3.71 (s, 0.3H), 3.66 (s, 2.7H), 3.59–3.38 (m, 1H), 3.19–3.03 (m, 1H), 2.92–2.80 (m, 1H), 2.80–2.67 (m, 1H), 2.67 (s, 2.7H), 2.58 (s, 0.3H), 2.10 (s, 2.7H), 2.08 (s, 0.3H), 1.45 (br, s, 9H); 13C NMR (101 MHz, CD3CN), δ 169.7–168.5 (3C), 167.8 and 167.6 (1C), 154.7 and 154.6 (1C), 138.3–137.8 (1C), 137.2–137.0 (1C), 129.1 (4C), 128.4 and 128.3 (4C), 126.7–126.4 (2C), 79.7–79.4 (1C), 56.2, 54.9–54.6 (1C), 53.8, 52.1 and 52.0 (1C), 46.8 and 46.5 (1C), 34.4–34.2 (2C), 27.9, 27.7 (3C), 21.0 and 20.8 (1C); HRMS (ESI) calcd for C30H37N3NaO7+ [MNa]+ 574.2524, found 574.2506.
General procedure for the synthesis of compounds 23–26
Aldehyde (21 or 22) (1.0 mmol, 1 eq.) was dissolved in 1 mL of dry methanol under nitrogen, 2,2-diethoxyethanamine (1.0 mmol, 133 mg, 1 eq.) was added and the resulting mixture was kept under stirring for 2 h at room temperature. Benzoic acid (1.0 mmol, 122 mg, 1 eq.) and isocyanide (3 or 4) (1.2 mmol, 1.2 eq.) were sequentially added and the reaction was stirred for an additional 24 h at room temperature. The solvent was removed under reduced pressure to afford the unstable crude Ugi product, which was directly used in the next step. The crude was dissolved in 4.5 mL of 50% trifluoroacetic acid in dichloromethane, and the resulting solution was kept under stirring for 24 h. The solvent was removed under reduced pressure to give a residue which was dissolved in EtOAc (10 mL) and washed with satd aq. NaHCO3 (2 × 10 mL). The organic layer was dried over Na2SO4 and concentrated in vacuo to afford the crude product, which was purified by flash chromatography (FC) as indicated below.:3; yield: 23a (24 mg, 5%), 23b (311 mg, 65%). 23a: thick oil; Rf 0.30 (7:3 n-hexane–EtOAc); [α]30D −22.4 (c 0.5, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.60–7.20 (m, 10H), 6.04 (br, d, J = 5.9 Hz, 1H), 5.68 (d, J = 5.9 Hz, 1H), 5.28 (br, d, J = 9.2 Hz, 1H), 5.20–4.61 (m, 4H), 3.74 (s, 3H), 2.92 (s, 3H), 1.52 (d, J = 7.0 Hz, 3H), 1.24 (br, m, 3H); 13C NMR (75 MHz, CDCl3) δ 170.9, 168.1, 162.9, 155.0, 136.7, 132.3, 130.8, 128.5–127.7 (9C), 112.3, 110.6, 67.2, 56.8, 52.6, 52.0, 48.0, 29.7, 15.2, 14.9; HRMS (ESI) calcd for C26H29N3NaO6+ [MNa]+ 502.1949, found 502.1953. 23b: foam; Rf 0.37 (7:3 n-hexane–EtOAc); [α]20D +27.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, 1:1 mixture of rotamers) δ 7.61–7.40 (m, 5H), 7.39–7.27 (m, 5H), 5.96 (br, m, 0.5H), 5.88 (br, d, J = 5.4 Hz, 0.5H), 5.73 (m, 1H), 5.42–5.29 (m, 1H), 5.20–4.95 (m, 3H), 4.93–4.74 (m, 1H), 3.77 (s, 1.5H), 3.74 (s, 1.5H), 3.00 (s, 1.5H), 2.94 (s, 1.5H), 1.50 (d, J = 7.1 Hz, 1.5H), 1.38 (d, J = 7.3 Hz, 1.5H), 1.35–1.24 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 171.6 and 170.9 (1C), 168.7 and 168.6 (1C), 163.1 and 162.9 (1C), 156.6, 137.0, 134.0 and 133.9 (1C), 131.1 and 131.0 (1C), 128.6–127.4 (9C), 111.9 and 111.0 (1C), 111.4 and 110.3 (1C), 67.3 and 67.0 (1C), 57.9 and 57.1 (1C), 52.5, 52.0 and 50.3 (1C), 50.1 and 49.0 (1C), 29.1 and 28.8 (1C), 15.9–14.4 (2C); HRMS (ESI) calcd for C26H29N3NaO6+ [MNa]+ 502.1949, found 502.1961.
:7; yield: 24a (16 mg, 3%), 24b (223 mg, 40%). 24a: colorless oil; Rf 0.23 (7:3 n-hexane–EtOAc); [α]20D −13.6 (c 0.9, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.61–7.08 (m, 15H), 5.93 (br, d, J = 5.9 Hz, 0.7H), 5.75 (br, d, J = 5.9 Hz, 0.3H), 5.66 (d, J = 5.9 Hz, 0.7H), 5.60 (d, J = 5.9 Hz, 0.3H), 5.36–4.97 (m, 4H), 4.72 (m, 0.3H), 4.33 (dq, J = 10.7 and 6.8 Hz, 0.7H), 3.82 (s, 2.1H), 3.80 (s, 0.9H), 3.48 (dd, J = 14.7 and 4.9 Hz, 0.7H), 3.43 (dd, J = 14.7 and 4.9 Hz, 0.3H), 3.17 (dd, J = 14.7 and 12.7 Hz, 0.7H), 3.04 (dd, J = 13.8 and 11.7 Hz, 0.3H), 2.92 (s, 0.9H), 2.85 (s, 2.1H), 0.73 (d, J = 6.8 Hz, 2.1H), 0.64 (d, J = 6.8 Hz, 0.9H); 13C NMR (75 MHz, CDCl3) δ 170.0, 168.7, 163.7 and 163.2 (1C), 155.9, 136.0, 135.7, 133.8, 130.8, 129.1–127.2 (14C), 111.8 and 111.3 (1C), 111.0 and 110.0 (1C), 67.4 and 67.0 (1C), 57.7, 56.7, 55.0, 52.7, 34.8, 28.5, 14.6 and 14.3 (1C); HRMS (ESI) calcd for C32H33N3NaO6+ [MNa]+ 578.2262, found 578.2250. 24b: oil; Rf 0.27 (7:3 n-hexane–EtOAc); [α]20D +34.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.56–7.18 (m, 15H), 5.90 (d, J = 5.9 Hz, 0.7H), 5.78 (br, d, J = 5.7 Hz, 1H), 5.72 (dd, J = 11.3 and 5.4 Hz, 0.3H), 5.59 (m, 1H), 5.38 (br, d, J = 9.3 Hz, 0.7H), 5.23–5.01 (m, 2.3H), 4.83 (br, m, 1H), 3.73 (s, 2.1H), 3.70 (s, 0.9H), 3.45 (dd, J = 14.4 and 5.4 Hz, 0.3H), 3.40 (dd, J = 14.4 and 5.7 Hz, 0.7H), 3.02 (dd, J = 14.4 and 10.8 Hz, 1H), 2.90 (s, 2.1H), 2.86 (s, 0.9H), 1.25 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2, 168.6, 163.7 and 163.3 (1C), 156.6 and 156.0 (1C), 137.0 and 136.6 (1C), 135.7, 133.9, 131.0, 129.3–126.9 (14C), 112.7 and 111.6 (1C), 111.4 and 110.4 (1C), 67.0 and 66.9 (1C), 57.8 and 57.4 (1C), 54.6 and 54.4 (1C), 52.5, 49.9 and 49.7 (1C), 36.7, 28.9, 15.2 and 15.1 (1C); HRMS (ESI) calcd for C32H33N3NaO6+ [MNa]+ 578.2262, found 578.2248.
:1.5; yield: 25a (27 mg, 5%), 25b (372 mg, 67%). 25a: colorless oil; Rf 0.19 (1.5:1 n-hexane–EtOAc); [α]20D −5.9 (c 0.5, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.74–6.96 (m, 15 H), 6.47 (d, J = 6.8 Hz, 0.5H), 6.32 (d, J = 6.8 Hz, 0.5H), 6.09–6.01 (m, 0.5H), 5.93–5.84 (m, 0.5H), 5.36–4.83 (m, 4.5H), 4.49 (m, 0.5H), 3.75 (s, 1.5H), 3.70 (s, 1.5H), 3.18–2.69 (m, 5H), 1.32 (d, J = 7.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 173.0, 167.4, 162.8, 160.2 and 159.6 (1C), 137.2, 136.2, 133.0, 131.8, 129.4–127.1 (14C), 112.1–112.3 (2C), 68.1 and 66.8 (1C), 53.4, 52.8, 52.5, 48.3, 37.9 and 37.6 (1C), 29.1, 18.2; HRMS (ESI) calcd for C32H33N3NaO6+ [MNa]+ 578.2262, found 578.2270. 25b: pale yellow oil; Rf 0.35 (1.5:1 n-hexane–EtOAc); [α]20D −72.1 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.61–7.11 (m, 15H), 5.91 (br, d, J = 5.6 Hz, 0.6H), 5.82–5.74 (m, 1H), 5.70 (d, J = 5.8 Hz, 0.4H), 5.51 (d, J = 9.8 Hz, 0.6H), 5.42 (d, J = 7.6 Hz, 0.4H), 5.13 (m, 1H), 5.06–4.90 (m, 2.6H), 4.81 (d, J = 13.0 Hz, 0.4H), 3.77 (s, 1.8H), 3.74 (s, 1.2H), 3.17–2.92 (m, 1.6H), 2.97 (s, 1.8H), 2.87 (s, 1.2H), 2.78 (dd, J = 13.9 and 8.1 Hz, 0.4H), 1.49 (d, J = 7.2 Hz, 1.2H), J = 7.5 Hz, 1.8H); 13C NMR (100 MHz, CDCl3) δ 170.7, 169.3, 163.3, 157.5 and 157.0 (1C), 138.0 and 137.8 (1C), 137.3 and 137.1 (1C), 134.5 and 134.3 (1C), 132.0 and 131.7 (1C), 129.4–127.1 (14C), 113.4–111.1 (2C), 67.9 and 67.5 (1C), 59.3 and 58.4 (1C), 57.4 and 54.1 (1C), 53.2 and 52.9 (1C), 51.3 and 51.0 (1C), 36.2 and 35.6 (1C), 29.9 and 29.8 (1C), 15.5 and 15.0 (1C); HRMS (ESI) calcd for C32H33N3NaO6+ [MNa]+ 578.2262, found 578.2268.
:7; yield: 26a (56 mg, 9%), 26b (284 mg, 45%). 26a: colorless oil; Rf 0.22 (7:3 n-hexane–EtOAc); [α]30D −11.1 (c 0.5, CHCl3); 1H NMR (300 MHz, CDCl3, complex mixture of conformers) δ 7.68–6.81 (m, 20H), 6.38 (d, J = 5.8 Hz, 0.1H), 5.94–5.80 (m, 0.7H), 5.79–5.72 (m. 0.2H), 5.72–5.54 (m, 1H), 5.51–5.37 (m, 0.5H), 5.31–4.65 (m, 4.5H), 3.74 (s, 0.3H), 3.70 (s, 2H), 3.67 (s, 0.7H), 3.49–3.18 (m, 1H), 3.16–2.87 (m, 3H), 2.83 (s, 0.7H), 2.80 (s, 2H), 2.72 (s, 0.3H); 13C NMR (75 MHz, CDCl3) δ 170.2–169.6 (1C), 168.9–168.4 (1C), 163.6 and 163.1 (1C), 157.0 and 156.8 (1C), 137.3–135.7 (4C), 131.8–130.9 (1C), 129.3–126.5 (19C), 112.5–106.7 (2C), 68.1–66.8 (1C), 58.0 and 57.7 (1C), 57.3–57.0 (1C), 55.8–55.3 (1C), 54.1–52.2 (2C), 38.0–37.6 (1C), 35.3–34.7 (1C), 29.4 and 29.0 (1C); HRMS (ESI) calcd for C38H37N3NaO6+ [MNa]+ 654.2575, found 654.2561. 26b: foam; Rf 0.30 (7:3 n-hexane–EtOAc); [α]20D −139.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CD3CN, a mixture of two conformers 1.5:1) δ 7.64–7.01 (m, 20H), 6.01 (m, 0.8H), 5.96 (d, J = 5.9 Hz, 0.6H), 5.79 (d, J = 5.4 Hz, 0.6H), 5.63 (dd, J = 11.1 and 5.4 Hz, 0.4H), 5.53 (dd, J = 10.8 and 5.4 Hz, 0.6H), 5.15–4.87 (m, 3.6H), 4.69 (d, J = 12.9 Hz, 0.4H), 3.71 (s, 3H), 3.43 (dd, J = 14.7 and 5.4 Hz, 0.4H), 3.37 (dd, J = 14.7 and 5.4 Hz, 0.6H), 3.08 (dd, J = 14.7 and 11.1 Hz, 0.4H), 3.07 (dd, J = 14.7 and 0.81 Hz, 0.6H), 3.04–2.87 (m, 2H), 2.83 (s, 3H); 13C NMR (100 MHz, CD3CN) δ 170.1 and 169.9 (1C), 168.5 and 168.4 (1C), 163.6 and 163.3 (1C), 156.6 and 155.8 (1C), 138.0 and 137.4 (1C), 136.5 and 136.4 (2C), 134.2 and 133.8 (1C), 131.1 and 130.9 (1C), 129.1–126.4 (19C), 112.0–111.5 (2C), 66.5 and 66.3 (1C), 57.2 and 57.1 (1C), 56.0, 55.1–54.8 (2C), 52.2 and 52.1 (1C), 36.1 and 35.9 (1C), 34.9 and 34.6 (1C), 29.0 and 28.8 (1C); HRMS (ESI) calcd for C38H37N3NaO6+ [MNa]+ 654.2575, found 654.2558.
Computational studies
Biological evaluation
Notes and references
- L. Garner and K. D. Janda, Curr. Top. Med. Chem., 2011, 11, 258–280 CrossRef.
- (a) I. S. Moreira, P. A. Fernandes and M. J. Ramos, Proteins: Struct., Funct., Bioinf., 2007, 68, 803–812 CrossRef CAS PubMed; (b) S. Jones and J. M. Thornton, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13–20 CrossRef CAS.
- S. Butini, E. Gabellieri, P. Brady Huleatt, G. Campiani, S. Franceschini, M. Brindisi, S. Ros, S. Sanna Coccone, I. Fiorini, E. Novellino, G. Giorgi and S. Gemma, J. Org. Chem., 2008, 73, 8458–8468 CrossRef CAS PubMed.
- (a) A. Sacchetti, A. Silvani, G. Lesma and T. Pilati, J. Org. Chem., 2011, 76, 833–839 CrossRef CAS PubMed; (b) G. Lesma, N. Landoni, A. Sacchetti and A. Silvani, Tetrahedron, 2010, 66, 4474–4478 CrossRef CAS PubMed; (c) G. Lesma, N. Landoni, T. Pilati, A. Sacchetti and A. Silvani, J. Org. Chem., 2009, 74, 8098–8105 CrossRef CAS PubMed; (d) N. Landoni, G. Lesma, A. Sacchetti and A. Silvani, J. Org. Chem., 2007, 72, 9765–9768 CrossRef CAS PubMed; (e) G. Lesma, E. Meschini, T. Recca, A. Sacchetti and A. Silvani, Tetrahedron, 2007, 63, 5567–5578 CrossRef CAS PubMed.
- (a) E. Ko, J. Liu and K. Burgess, Chem. Soc. Rev., 2011, 40, 4411–4421 RSC; (b) E. Ko, J. Liu, L. M. Perez, G. Lu, A. Schaefer and K. Burgess, J. Am. Chem. Soc., 2011, 133, 462–477 CrossRef CAS PubMed.
- G. Lesma, R. Cecchi, S. Crippa, P. Giovanelli, F. Meneghetti, M. Musolino, A. Sacchetti and A. Silvani, Org. Biomol. Chem., 2012, 10, 9004–9012 CAS.
- (a) H. Eckert, Molecules, 2012, 17, 1074–1102 CrossRef CAS PubMed; (b) B. Ganem, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS PubMed.
- G. Lesma, F. Meneghetti, A. Sacchetti, M. Stucchi and A. Silvani, Beilstein J. Org. Chem., 2014, 10, 1383–1389 CrossRef PubMed.
- L. Banfi, A. Basso, V. Cerulli, G. Guanti, P. Lecinska, I. Monfardini and R. Riva, Mol. Diversity, 2010, 14, 425–442 CrossRef CAS PubMed.
- A. Kumar, Z. Li, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Org. Lett., 2013, 15, 1874–1877 CrossRef CAS PubMed.
- S. S. Van Berkel, B. G. M. Bögels, M. A. Wijdeven, B. Westermann and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2012, 3543–3559 CrossRef CAS.
- A. Shaabani, A. Maleki, A. Hossein Rezayan and A. Sarvary, Mol. Diversity, 2011, 15, 41–68 CrossRef CAS PubMed.
- E. Soleimani and M. Zainali, J. Org. Chem., 2011, 76, 10206–10311 CrossRef PubMed.
- G. Koopmanschap, E. Ruijter and R. V. A. Orru, Beilstein J. Org. Chem., 2014, 10, 544–598 CrossRef PubMed.
- (a) N. Narendra, T. M. Vishwanatha, G. Nagendra and V. V. Sureshbabu, Tetrahedron, 2012, 68, 1992–2000 CrossRef CAS PubMed; (b) W. Wang and A. Dömling, J. Comb. Chem., 2009, 11, 403–409 CrossRef CAS PubMed; (c) C. Faggi, S. Marcaggini, R. Pepino and M. C. Pozo, Synthesis, 2002, 2756–2760 CAS.
- M. Nikulnikov, A. Shumsky and M. Krasavin, Synthesis, 2010, 2527–2532 CAS.
- D. W. Carney, J. V. Troung and J. K. Sello, J. Org. Chem., 2011, 76, 10279–10285 CrossRef CAS PubMed.
- (a) A. Domling, B. Beck, U. Eichelberger, S. Sakamuri, S. Menon, Q.-Z. Chen, Y. Lu and L. A. Wessjohann, Angew. Chem., Int. Ed., 2006, 45, 7235–7239 CrossRef PubMed; (b) P. Tempest, V. Gore and C. Hulme, Tetrahedron Lett., 2003, 44, 1947–1950 CrossRef CAS.
- http://www.stemchem.org .
- J. Zhu, X. Wu and S. J. Danishefsky, Tetrahedron Lett., 2009, 50, 577–579 CrossRef CAS PubMed.
- D. W. Carney, J. V. Truong and J. K. Sello, J. Org. Chem., 2011, 76, 10279–10285 CrossRef CAS PubMed.
- G. Gerona-Navarro, M. A. Bonache, R. Herranz, M. T. García-López and R. González-Muñiz, J. Org. Chem., 2001, 66, 3538–3547 CrossRef CAS PubMed.
- M. Harfenist, D. C. Hoerr and R. Crouch, J. Org. Chem., 1985, 50, 1356–1359 CrossRef CAS.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2011, 40, 4603–4625 RSC.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- (a) G. D. Hawkins, C. J. Cramer and D. G. Truhlar, J. Phys. Chem., 1996, 100, 19824–19839 CrossRef CAS; (b) G. D. Hawkins, C. J. Cramer and D. G. Truhlar, Chem. Phys. Lett., 1995, 246, 122–129 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C.-H. Liu, T.-C. Chen, G.-Y. Chau, Y.-H. Jan, C. H. Chen, C.-N. Hsu, K.-T. Lin, Y.-L. Juang, P.-J. Lu, H.-C. Cheng, M.-H. Chen, C.-F. Chang, Y.-S. Ting, C.-Y. Kao, M. Hsiao and C.-Y. F. Huang, Mol. Cell. Proteomics, 2013, 12, 1335–1349 CAS.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- (a) F. Buontempo, T. Ersahin, S. Missiroli, S. Senturk, D. Etro, M. Ozturk and M. L. Neri, Invest. New Drugs, 2011, 29, 1303–1313 CrossRef CAS PubMed; (b) H. Yuzugullu, K. Benhaj, N. Ozturk, S. Senturk, E. Celik, A. Toylu and M. Ozturk, Mol. Cancer, 2009, 8, 90 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00218d |
| This journal is © The Royal Society of Chemistry 2015 |