
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Developing principles for predicting ionic liquid effects on reaction outcome. The importance of the anion in controlling microscopic interactions†
Sinead T.
Keaveney
,
Ronald S.
Haines
and
Jason B.
Harper
*
School of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: j.harper@unsw.edu.au; Fax: +61 2 9385 6141; Tel: +61 2 9385 4692
First published on 12th February 2015
Abstract
A series of ionic liquids containing anions of differing coordination strength were investigated as solvents for the condensation reaction of an alkyl amine and an aromatic aldehyde. As predicted, the rate constant of the process was found to increase with the proportion of the ionic liquid in the reaction mixture. Temperature-dependent kinetic analyses demonstrated that by varying the ability of the anion to interact with the cation the magnitude of both the enthalpy and entropy of activation could be controlled in a predictable manner, with the activation parameters being linearly dependent on the ionic liquid basicity. Interestingly, the unexpected trend in the rate constants observed when altering the anion of the ionic liquid highlighted the presence of more subtle secondary microscopic interactions involving the anion, further emphasizing the fragility of the enthalpy – entropy balance.
Introduction
The prospect of using ionic liquids, salts with melting points below 100 °C,1 as more environmentally-friendly alternatives to molecular solvents has received considerable attention in recent years due to their low vapour pressure,2–4 lack of flammability,4 and the possibility of recycling.5 Further, many reactions proceed more readily and/or more selectively in ionic liquids;6–15 although the lack of understanding of why these changes occur has hindered the application of these solvents; developing such an understanding is of interest to all seeking to use ionic liquids.By systematically investigating the effect that ionic liquids have on the outcome of a number of simple organic reactions,6,7,16–24 our group has been developing a set of principles for qualitatively predicting the effect of an ionic liquid on reaction outcome. These principles can be applied by considering the extent of charge development in the transition state and the nature of the starting materials;7,20–23,25,26 the proportion of the ionic liquid in the reaction mixture;7,20,27 and the components of the ionic liquid itself.19,24,25,27 A key outcome from this body of work, particularly relevant to the work described herein, is that for systems having: (i) a bimolecular rate determining step; (ii) involving some (but limited) charge development in the transition state;28 and (iii) a nitrogen nucleophile, there is a predictable rate enhancement due to an entropically favourable cation–nucleophile interaction.7,23,25
The effect that the nature of the cation had on the outcome of the reaction of 4-methoxybenzaldehyde 1 and hexan-1-amine 2 (Scheme 1) was investigated in a recent study by our group;27 the ionic liquids used were selected to include cations of varying steric and electronic nature, and all contained bis(trifluoromethanesulfonyl)imide as the counterion. Analogous to that seen for a bimolecular substitution reaction,25 the more charge dense and accessible the charged centre of the cation, the greater its interaction with the nitrogen lone pair on the nucleophile 2; importantly, this was reflected in a gradual increase in the enthalpy of activation across the series, which was offset by an increased entropy of activation. The rate constants observed were a result of the subtle interplay of entropic and enthalpic effects, where the importance of the former due to increased ordering in the ionic liquids containing methyl substituted imidazolium cations was shown.
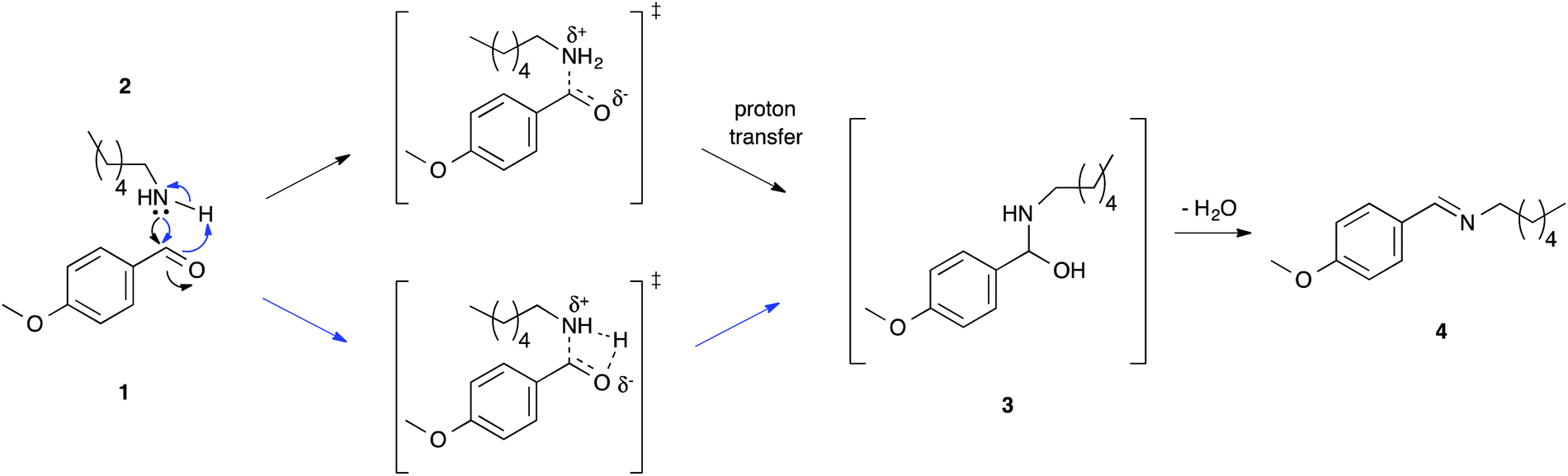 | ||
| Scheme 1 The condensation of the benzaldehyde 1 with an amine 2 to first give the intermediate 3, which proceeds to the imine 4, noting two possible mechanisms for the formation of the aminol 3; stepwise (top) and concerted (bottom). | ||
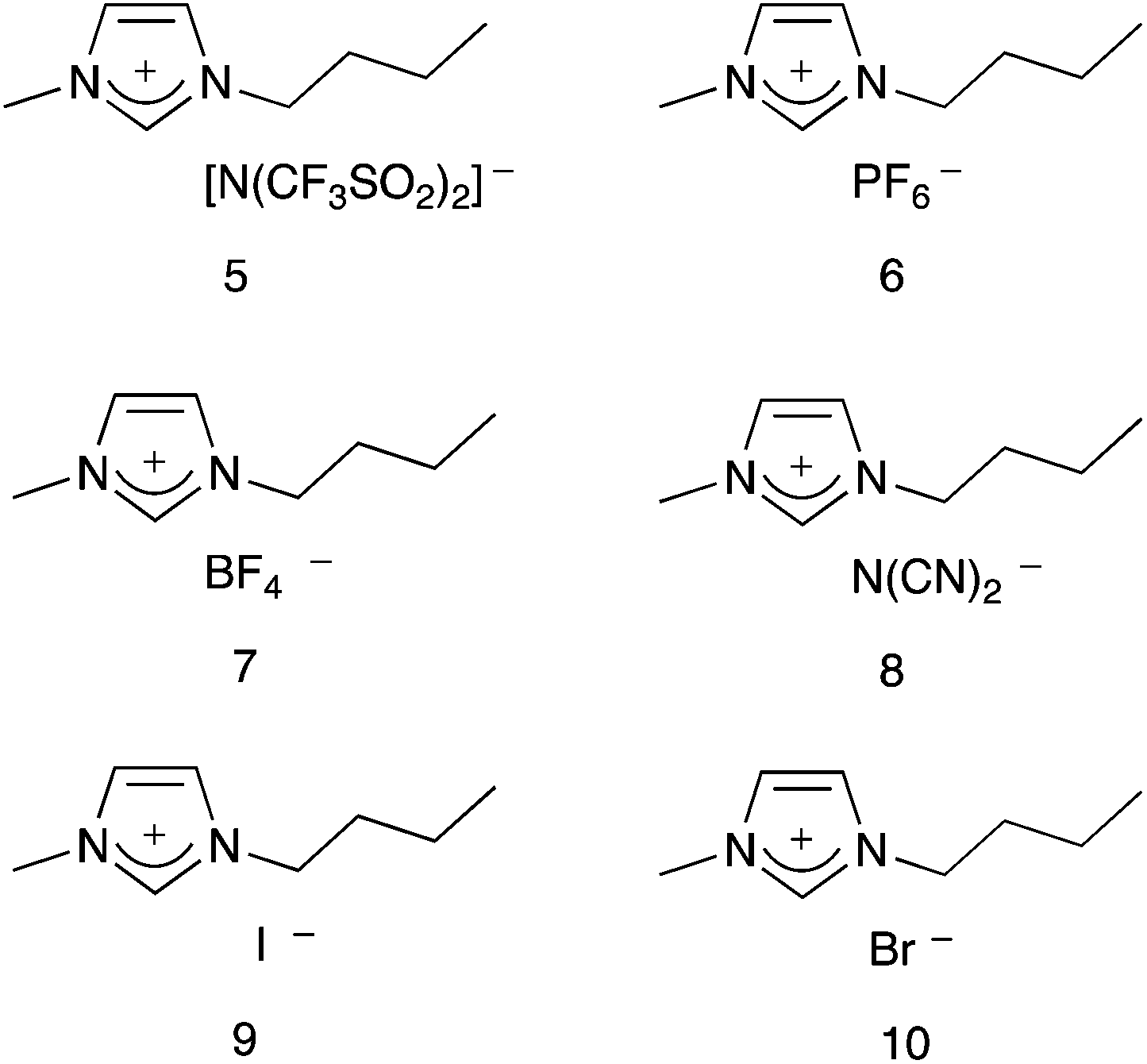
This manuscript builds on this previous work, and describes the effect that a series of imidazolium based ionic liquids 5–10, containing anions of different coordinating abilities, and across a range of proportions in a molecular solvent, has on the outcome of this reaction. It was envisaged that variation of the anion of the ionic liquid would alter the extent of anion–cation interactions, and hence modify the availability of the imidazolium cation for interaction with the nucleophile 2; this difference in cation–nucleophile interaction was predicted to cause changes in reaction outcome, which are discussed below.
Experimental
4-Methoxybenzaldehyde 1 and hexan-1-amine 2 were both commercially available, and each was distilled under reduced pressure then stored with molecular sieves at 253 K prior to use. Analytical grade deuterated acetonitrile was dried over molecular sieves for at least 48 h before use. The ionic liquids 5–10 were prepared according to literature methods;29–31 1-methylimidazole was treated with either butyl iodide or butyl bromide to afford 9 and 10, respectively. Ionic liquids 5–8 were prepared by treating the bromide 10 with the corresponding salt; either lithium bis(trifluoromethanesulfonyl)imide, potassium hexafluorophosphate, sodium tetrafluoroborate or sodium dicyanimide, to give the required ionic liquid. All ionic liquids were dried to constant weight at 70 °C under reduced pressure immediately before use, and were found to have <0.1% water using Karl Fischer titration methodology and salts 5–8 contained <0.2 mol% residual halide by ion chromatography.
Kinetic analyses were carried out in solutions containing the benzaldehyde 1 (ca. 0.02 mol L−1) and the amine 2 (ca. 0.25 mol L−1) at a given temperature and specific mole fraction of the ionic liquid, with the remaining solvent being made up by deuterated acetonitrile. For the ionic liquids 5–8 and 10, NMR samples containing both reagents 1 and 2, and the solvent mixture, were prepared and stored in liquid nitrogen prior to analysis. For the highly hygroscopic ionic liquid 9, samples were prepared under an inert atmosphere and the reagents 1 and 2 allowed to mix just prior to 1H NMR analysis. In each case the reaction was monitored in situ with the spectrometer set to the desired temperature for the duration of the reaction.
All reactions were followed until more than 95% of the starting material 1 was consumed, and all kinetic analyses were performed in triplicate. NMR spectra were processed using either the Bruker TOPSPIN 1.3 software or the MestReNova 7.1.1 software. The pseudo-first order rate constants for the reactions were calculated using integrations of the aldehyde signal in the starting material 1 at δ ca. 10.0, obtained from the processed 1H NMR spectra, by fitting the natural logarithm of the integrations to a linear function using the Microsoft Excel 14.4.3 LINEST function. Bimolecular rate constants were obtained from the pseudo-first order rate constants by dividing by the initial amine concentration in the reaction mixture. The activation parameters were then determined through fitting the obtained dated using the Microsoft Excel 14.4.3 LINEST function to the bimolecular Eyring equation shown below (eqn (1)).6,32
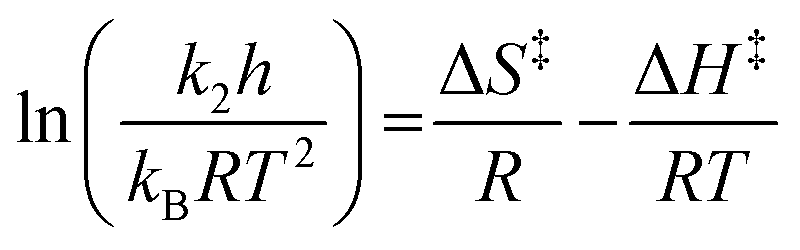 | (1) |
Tables containing the exact mole fraction, amine 2 concentration, temperature and rate constants for all the systems described below can be found in the ESI.†
Results and discussion
NMR spectroscopy was used to follow reaction progress, as has been done in previous studies.6,7,16–24 The use of this method allows convenient analysis of this reaction27 and circumvents any issues with work-up of the reaction mixture (such as selective extraction of components of the reaction mixture16).As this manuscript focuses on the solvent effects on this process, it is important to discuss the mechanism through which this reaction proceeds (Scheme 1). The reaction proceeds through nucleophilic attack of the amine 2 onto the carbonyl of the benzaldehyde 1 and proton transfer to give the intermediate aminol 3. This process can be considered to occur through either a stepwise process (demonstrated for aqueous solutions33) or a concerted mechanism (shown in the gas phase34). Experimental data from organic solvents demonstrates significant charge separation in the transition state,35 which is consistent with either mechanism. This addition step is then followed by the elimination of water to give the imine product 4.
In all cases (different ionic liquids and different temperatures) described here, no formation of the intermediate 3 was observed, indicating that the elimination step was significantly faster than the addition step, and the reaction was found to be first order with respect to the nucleophile 2 (see ESI†). This observation is consistent with previous studies on this reaction where, typically, the intermediate is not observed.33 It is important to note that the electron-donating methoxy substituent was chosen as it deactivates the carbonyl group in benzaldehyde 1 to nucleophilic attack, slowing down the addition step and making it rate limiting; for electron-withdrawing substituents the carbonyl group can be sufficiently activated (for example, with a nitro group) so that the second, elimination step becomes rate-determining.36
As the initial bimolecular addition step is rate-limiting it is necessary to consider any interaction between the ionic liquid and both reagents 1 and 2, and the transition state leading to the intermediate 3 when predicting any effects on the rate constant and activation parameter data. Previous work has shown that interaction between an ionic liquid and the amine 2 is the main interaction occurring,27 and while there may be interaction between the ionic liquid and transition state, this is not the major interaction; this idea will be elaborated further in the discussions below.
As mentioned above, this previous work27 has shown that rate enhancements for this reaction type, when using ionic liquids, are entropically driven. Ordering of the cation about, and stabilisation of, the nucleophile incurs an enthalpic cost, which is overcome by a more substantial entropic benefit that results from the reduction in the decrease in entropy on moving to the transition state. Variations in the electronic and steric nature of the cation were shown to affect the degree of interaction between the cation and the nucleophile in a predictable manner; with the extent of interaction having a marked effect on reaction outcome.27 For the series of ionic liquids considered herein, it was expected that when using a more coordinating anion, the extent of anion–cation interaction would increase. This increased interaction would, in turn, reduce the extent of cation–nucleophile interaction; this can be thought of in terms of the competing equilibria shown in Fig. 1. That is, if the anion is interacting to a greater extent with the cation, it will be more difficult for the amine 2 to access the cation, resulting in a reduction in the enthalpic cost and entropic benefit that is known to be associated with this interaction.27 The importance of considering preferential interaction of the ionic liquid components over ion–reagent interaction has been highlighted elsewhere in the literature.37–39
 | ||
| Fig. 1 Competing interactions in solution can be considered using a simple series of equilibria (left), noting that multiple interactions are possible with each cation. There are many possible sites of interaction for electron rich species with the imidazolium cation used, as shown in the organisational profile of bis(trifluoromethanesulfonyl)imide (right) reproduced from Yau et al.25 | ||
Given the above, it was predicted that the use of more coordinating anions would result in reduced enthalpic and entropic effects, when compared to ionic liquids containing less coordinating anions. Considering the ionic liquids 5–10, the extent to which the anion will interact with the surrounding [Bmim]+ cations can be ranked by considering both the coordinating ability and basicity of the anion. A previous study38 measured the coordination strength of a number of cation–anion combinations using electrospray ionisation mass spectrometry experiments. Of particular relevance here are the results from their experiments on [Bmim]+-based ionic liquids using acetonitrile as the eluent; it was found that the coordination strength increases in the order [N(CF3SO2)2]− < [PF6]− < [BF4]− < [N(CN)2]− < [Br]−.38 The [I]− anion was not included in their study, but it is reasonable to assume that this halide with a more diffuse charge will be slightly less coordinating than [Br]−; hence the order will be [N(CF3SO2)2]− < [PF6]− < [BF4]− < [N(CN)2]− < [I]− < [Br]−.
Also worthwhile considering are the Kamlet-Taft parameters for the different ionic liquids, which will allow the experimental results to be assessed quantitatively. Of particular interest is the β value, a measure of hydrogen bond acceptor ability, as this is very dependent on the anion type, with a higher β value indicating a better hydrogen bond accepting ability.40 The change in the β value follows a similar trend as seen for the coordinating abilities discussed above, with the only discrepancy being [N(CF3SO2)2]− and [PF6]− (Table 1). It seems that while [N(CF3SO2)2]− is less coordinating than [PF6]− it is a slightly better hydrogen bond acceptor. The similar trends seen for the coordinating ability and hydrogen bond accepting ability of these anions is unsurprising, given that when increasing the anions basicity, the anion will be better able to form hydrogen bonding interactions with the [Bmim]+ cations surrounding it.
To allow direct comparison with previous work on both a comparable bimolecular substitution reaction,7 and the condensation reaction of the unsubstituted benzaldehyde and the amine 2,27 the effect of the ionic liquid 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([Bmim][N(CF3SO2)2], 5) on the condensation reaction between compounds 1 and 2 was considered in reaction mixtures across a range of different mole fractions of the ionic liquid 5 in acetonitrile (Fig. 2). Based on previous work,7,27 it was anticipated that the rate constant would gradually increase as the proportion of an ionic liquid in the reaction mixture increased, with the major rate enhancements occurring at lower mole fractions and an asymptote approached at high concentrations; the same trend as was seen for both the substitution7 and condensation27 reactions previously studied. It was expected that the methoxy substituent would have little effect on reaction outcome across the range of reaction mixtures, other than a systematic reduction in the rate constant due to deactivation of the carbonyl group to nucleophilic attack.
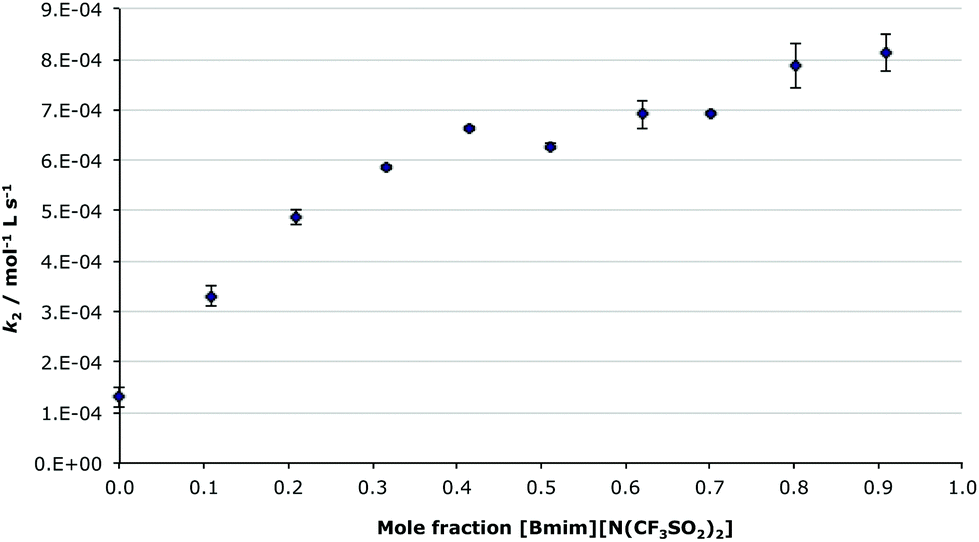 | ||
| Fig. 2 The bimolecular rate constants for the reaction between the benzaldehyde 1 and the amine 2 in different mole fractions of [Bmim][N(CF3SO2)2] 5 in acetonitrile, at 282 K. Uncertainties are reported as the standard deviation of three replicates. | ||
As shown in Fig. 2, the dependence of the bimolecular rate constant of the reaction of the methoxy substituted benzaldehyde 1 and the amine 2, with changing proportions of [Bmim][N(CF3SO2)2] 5 in acetonitrile, follows the expected trend, with rate changes comparable to that seen for benzaldehyde. In this case, there is a ca. 6-fold enhancement in the rate constant in the ionic liquid 5 when compared to acetonitrile, which is similar to the 8-fold increase seen for the unsubstituted benzaldehyde.27 The methoxy substituent uniformly reduces the rate constant by an order of magnitude across the range of ionic liquid/acetonitrile mixtures. Interestingly, there is a slight decrease in the rate constant at ca. χIL 0.5; this has appeared many times in previous work,7,27 and the microscopic origin of this phenomena is currently under investigation.
Another interesting outcome from the analysis of the effects of the different cations on the condensation of benzaldehyde with the amine 2 was that the imidazolium ionic liquids followed the same trend (regardless of methyl substitution on the imidazolium ring), when changing the solvent composition, as that described above.27 It was predicted that for the ionic liquids 5–10, which are all based on the same imidazolium cation, that the trend with respect to solvent composition would be the same as above due to the comparable nature of the cation–nucleophile interaction across the series; although the magnitude of the rate constant will be dependent on the balance of the enthalpic and entropic effects. As such, the effect of the ionic liquids 6–9 on the condensation of compounds 1 and 2 was examined in reaction mixtures across a range of different mole fractions of each of the ionic liquids in acetonitrile (Fig. 3, which also includes the ionic liquid 5 case for comparison, and S2†).
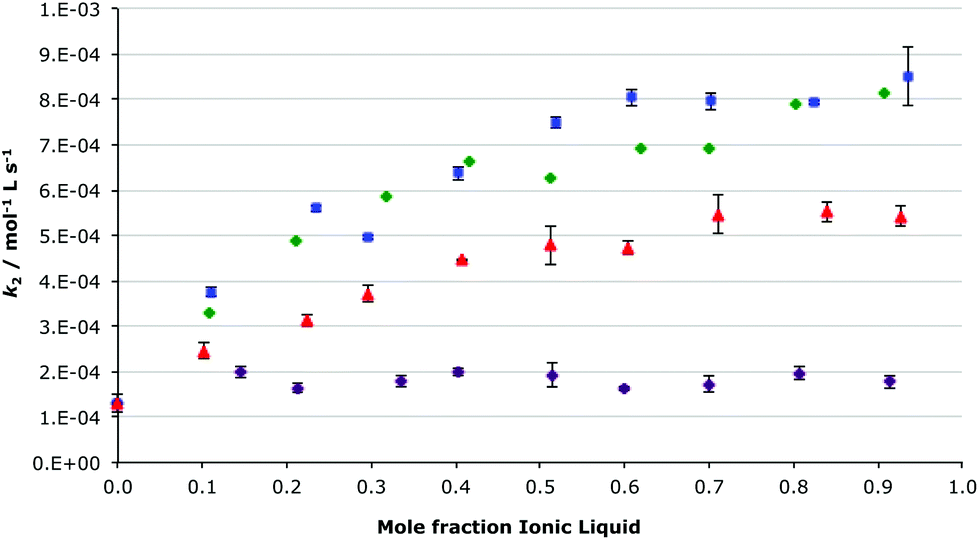 | ||
Fig. 3 The bimolecular rate constants for the reaction between benzaldehyde 1 and the amine 2 in different mole fractions of [Bmim][N(CF3SO2)2] 5 ( ), [Bmim][PF6] 6 ( ), [Bmim][PF6] 6 ( ), [Bmim][BF4] 7 ( ), [Bmim][BF4] 7 ( ) and [Bmim][N(CN)2] 8 ( ) and [Bmim][N(CN)2] 8 ( ) in acetonitrile, at 282 K. Uncertainties are reported as the standard deviation of three replicates. ) in acetonitrile, at 282 K. Uncertainties are reported as the standard deviation of three replicates. | ||
A mixture of [Bmim][Br] 10 in [Bmim][N(CF3SO2)2] 5 was of interest, as this mixture has been used to investigate the effects of a mixture of ionic liquids containing different anions on the bimolecular substitution reaction described above.25 At the temperature considered, it was not possible to investigate the reaction across different mole fractions of the ionic liquids 5 and 10 in acetonitrile as the bromide salt 10 is not sufficiently soluble in either the ionic liquid 5 or acetonitrile. The activation parameters of the condensation reaction were able to be determined in this mixture at slightly higher temperatures; this will be discussed in further detail below.
As can be seen in Fig. 3, the rate constant was affected in a similar manner by the ionic liquids [Bmim][N(CF3SO2)2] 5, 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4]) 7 and 1-butyl-3-methylimidazolium dicyanimide ([Bmim][N(CN)2]) 8; when increasing the proportion of the ionic liquid in the reaction mixture the rate constant of the reaction gradually increased. The most significant changes occurred between χIL 0 and χILca. 0.3–0.4; with further increases in ionic liquid concentration having little affect on the rate constant. Surprisingly the rate constant, at this temperature, remained essentially unchanged when using 1-butyl-3-methylimidazolium hexafluorophosphate ([Bmim][PF6]) 6, irrespective of the amount of the ionic liquid in the reaction mixture. This anomalous behaviour of the hexafluorophosphate salt 6, compared to the other ionic liquids, is consistent with results seen previously in a related bimolecular substitution process.24
The ionic liquid 1-butyl-3-methylimidazolium iodide ([Bmim][I]) 9 gave the greatest effect of those seen (Fig. S2†); the rate constant for the reaction shown in Scheme 1 in this ionic liquid was much larger (ca. 10–40 times larger) than in any of the other ionic liquids 5–8. Due to difficulties in monitoring such a fast reaction, rate constants for the complete range of ionic liquid mixtures were unable to be determined, however the same trend of increasing rate constant with higher proportions of ionic liquid in the reaction mixture, as seen for salts 5–8, is also seen for [Bmim][I] 9, with once again a characteristic ‘dip’ in the plot at χILca. 0.3.
It is worth noting that all of these rate enhancements may be the result of a change in mechanism for the process. Given the charged nature of an ionic liquid, it might be anticipated that such a medium would favour the formation of a zwitterionic intermediate and hence the stepwise mechanism over the concerted one. This, and other discussions, require activation parameters for the process.
Activation parameters were determined for the reaction between species 1 and 2 in the ionic liquids 5–9 at χILca. 0.2 and, where possible, χILca. 0.9 through temperature dependent kinetic studies (Fig. 4, 5 and Tables 2–5). A mixture of the bromide 10 in the ionic liquid 5 was also investigated. In all cases there was an increase in both the enthalpy and entropy of activation, as expected based on previous studies; note that this is exactly as was predicted and suggests that interaction between the cation and the nucleophile 2 is once again dominating reaction outcome.27 These changes in activation parameters are consistent with an increase in ordering of the solvent around the starting materials when using an ionic liquid, with the increased entropy (and hence rate constant) being the result of disruption of this ordering; the subtleties of the differences between the ionic liquids will be discussed below.
 | ||
Fig. 4 The bimolecular Eyring plot, from which the activation parameters in Table 2 were determined, for the reaction between methoxybenzaldehyde 1 and the amine 2 in either acetonitrile ( ) or one of the ionic liquids [Bmim][N(CF3SO2)2] 5 ( ) or one of the ionic liquids [Bmim][N(CF3SO2)2] 5 ( ), [Bmim][PF6] 6 ( ), [Bmim][PF6] 6 ( ), [Bmim][BF4] 7 ( ), [Bmim][BF4] 7 ( ), [Bmim][N(CN)2] 8 ( ), [Bmim][N(CN)2] 8 ( ), [Bmim][I] 9 ( ), [Bmim][I] 9 ( ) at ca. χIL 0.2 in acetonitrile. Exact mole fractions are outlined in Table 2. ) at ca. χIL 0.2 in acetonitrile. Exact mole fractions are outlined in Table 2. | ||
 | ||
Fig. 5 The bimolecular Eyring plot, from which the activation parameters in Table 3 were determined, for the reaction between methoxybenzaldehyde 1 and the amine 2 in either acetonitrile ( ) or one of the ionic liquids [Bmim][N(CF3SO2)2] 5 ( ) or one of the ionic liquids [Bmim][N(CF3SO2)2] 5 ( ), [Bmim][PF6] 6 ( ), [Bmim][PF6] 6 ( ), [Bmim][BF4] 7 ( ), [Bmim][BF4] 7 ( ), [Bmim][N(CN)2] 8 ( ), [Bmim][N(CN)2] 8 ( ) at ca. χIL 0.9, diluted only by the reagents 1 and 2, or in [Bmim][Br] 10/[Bmim][N(CF3SO2)2] 5 ( ) at ca. χIL 0.9, diluted only by the reagents 1 and 2, or in [Bmim][Br] 10/[Bmim][N(CF3SO2)2] 5 ( ). Exact mole fractions are outlined in Table 3. ). Exact mole fractions are outlined in Table 3. | ||
| Solvent | χ IL | ΔH‡/kJ mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
ΔS‡/J K−1 mol−1a |
|---|---|---|---|
| a Uncertainties quoted are from the fit of the linear regression. b From previous work.27 | |||
| Acetonitrileb | 0 | 14.2 ± 1.0 | −333.1 ± 3.3 |
| [Bmim][N(CF3SO2)2] 5 | 0.22 | 37.0 ± 0.9 | −242.1 ± 3.1 |
| [Bmim][PF6] 6 | 0.21 | 37.1 ± 1.5 | −249.4 ± 5.1 |
| [Bmim][BF4] 7 | 0.24 | 34.0 ± 0.8 | −250.3 ± 2.6 |
| [Bmim][N(CN)2] 8 | 0.22 | 27.3 ± 0.9 | −278.6 ± 2.9 |
| [Bmim][I] 9 | 0.23 | 21.6 ± 1.3 | −266.3 ± 4.9 |
| Solvent | χ IL | ΔH‡/kJ mol−1a |
ΔS‡/J K−1 mol−1a |
|---|---|---|---|
| a Uncertainties quoted are from the fit of the linear regression. b The ionic liquid was only diluted by the reagents 1 and 2. c From previous work.27 | |||
| Acetonitrilec | 0 | 14.2 ± 1.0 | −333.1 ± 3.3 |
| [Bmim][N(CF3SO2)2] 5 | 0.93 | 36.8 ± 1.5 | −238.5 ± 5.0 |
| [Bmim][PF6] 6 | 0.91 | 37.7 ± 1.2 | −246.5 ± 4.1 |
| [Bmim][BF4] 7 | 0.93 | 29.7 ± 1.7 | −262.1 ± 5.8 |
| [Bmim][N(CN)2] 8 | 0.93 | 22.2 ± 1.1 | −292.6 ± 3.7 |
| [Bmim][Br]/[N(CF3SO2)2] 10/5 | 0.32/0.60 | 14.1 ± 2.4 | −316.4 ± 7.8 |
| Solvent | Δ(ΔH‡)/kJ mol−1a |
|
|---|---|---|
| χ IL 0 → 0.2 | χ IL 0.2 → 0.9 | |
| a Uncertainties quoted are compounded using those from the fit of the linear regression. | ||
| [Bmim][N(CF3SO2)2] 5 | 22.8 ± 1.4 | −0.2 ± 1.8 |
| [Bmim][PF6] 6 | 22.9 ± 1.8 | 0.6 ± 1.9 |
| [Bmim][BF4] 7 | 19.8 ± 1.3 | −4.3 ± 1.8 |
| [Bmim][N(CN)2] 8 | 13.2 ± 1.3 | −5.2 ± 1.4 |
| [Bmim][I] 9 | 7.4 ± 1.7 | — |
| Solvent | Δ(ΔS‡)/J K−1 mol−1a |
|
|---|---|---|
| χ IL 0 → 0.2 | χ IL 0.2 → 0.9 | |
| a Uncertainties quoted are compounded using those from the fit of the linear regression. | ||
| [Bmim][N(CF3SO2)2] 5 | 91.0 ± 4.5 | 3.5 ± 5.9 |
| [Bmim][PF6] 6 | 83.7 ± 6.0 | 2.9 ± 6.5 |
| [Bmim][BF4] 7 | 82.8 ± 4.2 | −11.8 ± 6.3 |
| [Bmim][N(CN)2] 8 | 54.4 ± 4.4 | −14.0 ± 4.7 |
| [Bmim][I] 9 | 16.6 ± 8.5 | — |
Note that any change in mechanism from the concerted to the stepwise process (due to the nature of the transition state) might be anticipated to have the opposite effect on the activation parameters due to increased interaction with the more charge separated transition state. Since interactions with the nucleophile clearly dominate, no change in mechanism can be suggested, noting the ambiguity for the mechanism in acetonitrile as a starting point.
Immediately relevant to the variation of rate constant with the mole fraction of ionic liquid, Tables 4 and 5 show the difference in activation parameters between acetonitrile and χILca. 0.2, along with the difference in activation parameters between χILca. 0.2 and χILca. 0.9. For the ionic liquids 5–8 there is a much larger change in both the enthalpy and entropy of activation when moving from acetonitrile to χILca. 0.2, than the change incurred when moving from χILca. 0.2 to χILca. 0.9. These data confirm that as the proportion of ionic liquid in the reaction mixture increases, the most significant microscopic changes occur by χILca. 0.2, supporting the trends seen in Fig. 2 and 3.
The importance of the subtle balance of the enthalpic and entropic contributions is reinforced by the [Bmim][PF6] 6 case; the change in the activation parameters is in accordance with what is expected for an ionic liquid (and are comparable to [Bmim][N(CF3SO2)2] 5); yet the balance of the activation parameters results in a negligible change in the rate constant when compared to acetonitrile at 282 K, despite the very different activation parameters between the ionic liquid and acetonitrile cases!
The above results are consistent with the observed trends on varying the solvent composition being largely due to the nature of the cation, although the magnitude of this effect is dependent on the specific anion–cation combination. Further consideration of the activation parameters for the reaction of species 1 and 2 in the ionic liquids 5–10 allows the origin of the differing rate constants to be better understood and the validity of the predictions made above to be further considered.
Firstly, the activation parameters at ca. χIL 0.2 for ionic liquids 5–9 will be discussed. As described briefly above, for each of the ionic liquids 5–9 the enthalpies and entropies of activation are larger than those for acetonitrile (Table 2). Importantly, as the coordinating ability of the anion increases there is a gradual decrease in the enthalpy of activation, as was predicted! This is consistent with the fact that as the coordinating ability of the anion is changed, the extent of coordination between the cation and the anion, and hence between the cation and the nucleophile 2, can be controlled in a predictable fashion. The effect of the ionic liquids 5–9 at ca. χIL 0.2 on the entropy of activation was not as straightforward to understand; with a decrease in enthalpy of activation it was expected that there would be an associated reduction in the entropy of activation; while the activation entropy generally decreases on moving down Table 2, the trend is not as systematic as expected. Consideration of the ‘neat’ ionic liquid cases (χILca. 0.9, diluted only by reagents) – a better representation of the true ionic liquid environment – is thus important.
As shown in Table 3, the activation parameters for ionic liquids 5–8 at χILca. 0.9 further reinforces the results presented above and validates the predictive model; there is a clear correlation between the coordinating ability of the anion and the enthalpy of activation. The effect on the entropy of activation was also as anticipated; when increasing the coordinating ability of the anion there is a gradual reduction in the entropy of activation. This demonstrates the predictable nature of these interactions; the coordinating ability of the anion can be exploited to control the extent of cation–anion, and hence cation–nucleophile interactions, allowing these interactions to be manipulated in a controlled manner.
It is important to note that at both mole fractions of ionic liquid considered, the activation parameters for both the [Bmim][N(CF3SO2)2] 5 and the [Bmim][PF6] 6 cases are the same within uncertainty; this was expected given the relatively similar coordinating abilities of the two anions, and differing order of coordinating ability and Kamlet-Taft β value mentioned earlier for ionic liquids 5 and 6. While the coordinating ability scale allows a qualitative correlation between the activation parameters and the anions’ coordination strength to be seen, consideration of the Kamlet-Taft β parameters allows a quantitative assessment of this correlation to be made (Fig. 6 and 7).
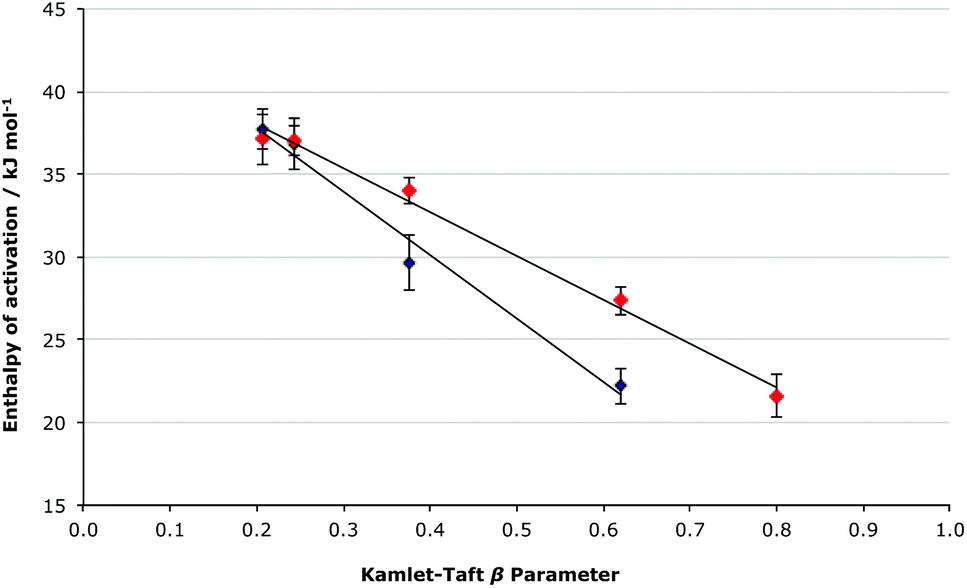 | ||
Fig. 6 The relationship between the enthalpy of activation and the Kamlet-Taft β parameter for the ionic liquids 5–9 (Table 1) at ca. χIL 0.2 ( ) and 0.9 (◆), with the linear correlations having R2 values of 0.992 and 0.983, respectively. ) and 0.9 (◆), with the linear correlations having R2 values of 0.992 and 0.983, respectively. | ||
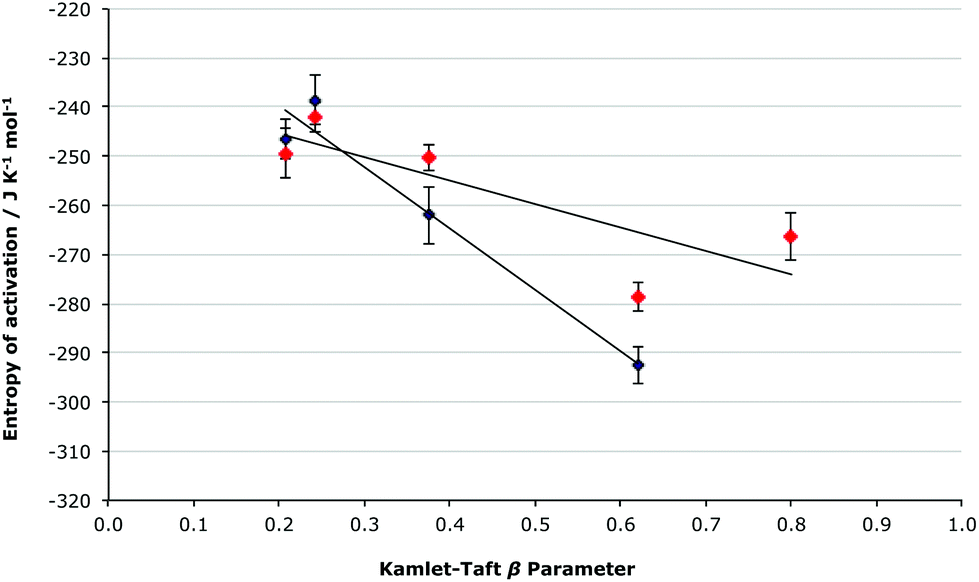 | ||
Fig. 7 The relationship between the entropy of activation and the Kamlet-Taft β parameter for the ionic liquids 5–9 (Table 1) at ca. χIL 0.2 ( ) and 0.9 (◆), with the linear correlations having R2 values of 0.671 and 0.955, respectively. ) and 0.9 (◆), with the linear correlations having R2 values of 0.671 and 0.955, respectively. | ||
Consideration of the relationship between the activation parameters and the hydrogen bond accepting ability of the ionic liquids reinforces the qualitative analysis presented above. There is a linear correlation between the β value and the enthalpy of activation at both χILca. 0.2 and 0.9 (Fig. 6), with both cases having an R2 value greater than 0.98. The entropy of activation also has a linear relationship to the β value at χILca. 0.9, but similar to as was discussed above, at χILca. 0.2 the trend is less pronounced (Fig. 7).
These linear relationships (particularly for the higher mole fraction cases) further demonstrate the validity of the predictions made based on the extent to which the anion will interact with the [Bmim]+ cations. Given these linear relationships seen for the enthalpy and entropy of activation, it is still surprising that there is no systematic change in the rate constant when changing the anion. No correlation is seen between the Gibbs free energy of activation and the Kamlet-Taft β value (see Fig. S3, ESI†). Such a lack of correlation clearly suggests that there is some other factor affecting the delicate balance of the activation parameters, which give the Gibbs free energy of activation.
While the trends presented in Tables 2, 3 and Fig. 6, 7 suggest that the main factor affecting the activation parameters is the competing interactions between the cation, and both the anion and amine 2; more subtle effects due to differing extents of anion–amine and anion–transition state interaction (Fig. 8) may be responsible for the subtle variations in the activation parameters, resulting in the lack of a correlation between the Gibbs free energy of activation and the β parameter, and the unexpected ordering of the rate constants.
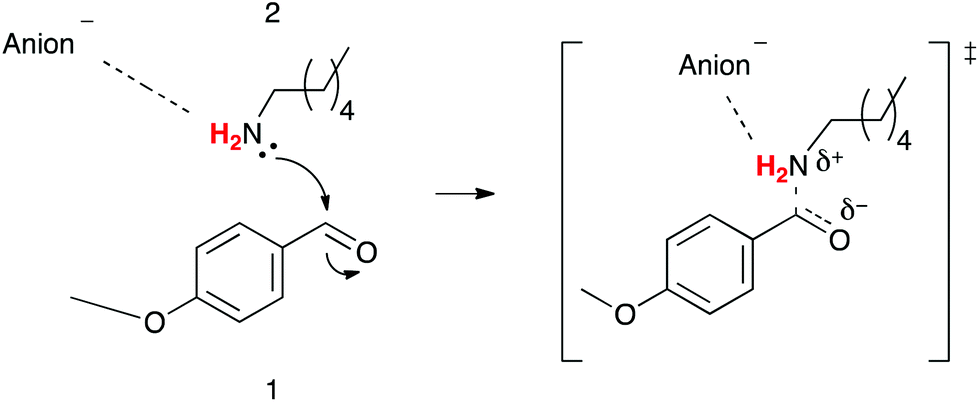 | ||
| Fig. 8 A visualisation of the interactions being discussed: an anion could interact with the amino hydrogen atoms in the nucleophile 2 (red), with the extent of this interaction increasing on moving from starting material (left) to transition state (right) as these hydrogen atoms become more acidic. Note that only the stepwise process is shown for simplicity; the same argument holds for the concerted process. | ||
There are two implications of these proposed interactions. Firstly interaction between the anion and a hydrogen atom on the nitrogen centre of the nucleophile 2 will increase the nucleophilicity of the amine 2 and lower the enthalpic barrier of the reaction. Secondly, on moving to the transition state there will be an increase in the hydrogen bond donor ability of the nucleophile, resulting in an increase in any directional anion–hydrogen interaction that may exist; there is literature precedent for such an interaction existing for a substitution reaction involving primary and secondary amine nucleophiles in ionic liquids.10 The greater ordering of the anion about the transition state relative to the starting material 2, would introduce an entropic cost that could, to a certain extent, offset the opposing effects of the cation–nucleophile 2 interaction. The transition state would also be stabilised by this directional anion–hydrogen interaction, further decreasing the enthalpic barrier for the reaction, resulting in the enthalpic benefit from such an interaction being compounded from both activation of the starting material 2 and stabilisation of the transition state. It is important to note that these effects on the enthalpy and entropy of activation are the opposite to what is observed; this suggests that while such interactions may contribute to the overall outcomes, the cation–nucleophile interaction is dominant.
It would be expected that such interactions involving the anion would be most pronounced for anions that are (i) weakly coordinating, as they will interact less with the cation and be more available to interact with the amine 2; and (ii) good hydrogen-bond acceptors, as they will be better able to interact with the hydrogen atoms of the amine group in the nucleophile 2. Conversely, this effect will be least significant for anions that are highly coordinating and poor hydrogen bond acceptors.
Considering this, the weakly coordinating [N(CF3SO2)2]− and [PF6]− anions, which have relatively high Kamlet-Taft β values of 0.2441,42 and 0.21,41,43 respectively, are likely to be most affected. It could be reasoned that the [N(CF3SO2)2]− anion will interact with the amino hydrogen atoms to a larger extent than [PF6], having a more substantial ‘offsetting’ effect, resulting in the enthalpies and entropies of activation for the [Bmim][N(CF3SO2)2] 5 cases being slightly lower than what would be expected if only considering the extents of cation–nucleophile 2 interaction; thus bringing the activation parameters for these two ionic liquids closer together. This minor competing interaction could also explain the lack of a trend in the rate constants seen across the range of ionic liquids 5–9 and the activation entropies at χILca. 0.2; such an effect could disturb the delicate enthalpy–entropy balance, resulting in activation energies and rate constants that don't uniformly change with increasing cation–nucleophile interaction. Molecular dynamics simulations are underway to further investigate this possible balance of competing interactions.
Discussion of the [Bmim][Br] 10/[Bmim][N(CF3SO2)2] 5 mixture has been left until last; due to its highly coordinating nature the [Br]− anion was of interest to analyse, yet its high melting point required it to be used as a mixture with another ionic liquid. These results were initially quite surprising, as previous studies on a bimolecular substitution reaction24 found the activation parameters for the mixture of bromide 10 in ionic liquid 5 were comparable to those for [Bmim][N(CF3SO2)2] 5. The key difference in this case is that the nucleophile is a hydrogen bond donor; the nucleophile used for the substitution case was not.24 Hence, these results can be rationalised by considering both the discussion above relating to hydrogen bond donation in the transition state; and the importance of preferential interaction of ions previously considered.37–39 The more charge dense [Br]− anion will coordinate to the [Bmim]+ cation to a greater extent than [N(CF3SO2)2]− will; resulting in two different effects. Firstly, a reduction in the extent of cation–nucleophile interaction; in turn reducing the enthalpy and entropy of activation, relative to [Bmim][N(CF3SO2)2] 5. This decrease will be ameliorated by the secondary effect; there will be a larger amount of ‘free’ [N(CF3SO2)2]− able to interact with the transition state, due to the preferential coordination of [Br]− to [Bmim]+, further reducing the enthalpic and entropic effects relative to [Bmim][N(CF3SO2)2] 5. These effects are so substantial that they completely offset the favourable [Bmim]+ – nucleophile interaction, resulting in activation parameters very similar to acetonitrile, with the enthalpy of activation for the mixture of 5/10 and acetonitrile being the same within uncertainty. Interestingly, despite this, the reaction still proceeded much faster in the ionic liquid mixture 5/10 than in acetonitrile; it is the slightly greater entropy of activation that is responsible for this rate enhancement. This further demonstrates the importance of entropic effects in ionic liquids; the reported solvent effects of ionic liquids are dominated by such, and highlights that when using ionic liquids consideration of entropic effects is paramount for these highly ordered solvents.
Conclusions
The effect of a series of ionic liquids on the activation parameters of a simple condensation reaction was successfully predicted based on a developing understanding of how ionic liquids affect reaction outcomes gained from previous studies. This framework was particularly accurate in predicting the systematic changes in the activation parameters when varying the ability of the anion to interact with the cation; with increased anion–cation interaction the extent of cation–nucleophile interaction was systematically decreased and resulted in a predicted decrease in the enthalpic and entropic effects down the series of ionic liquids studied. The validity of these predictions was further reinforced by the linear relationship between both the enthalpy and entropy of activation and the Kamlet-Taft basicity parameter; the more basic the ionic liquid the greater the enthalpy and entropy of activation. This is the first time that such an effect has been effectively forecast, and that such a clear relationship has been demonstrated.As the mole fraction of ionic liquid in the solvent mixture with acetonitrile was increased there was a gradual increase in the rate constant, with the most prominent rate enhancement occurring between χIL 0 and χIL 0.2. This general trend was also in accordance with what was predicted based on previous studies, suggesting that for [Bmim]+-based ionic liquids it may not be essential to use high mole fractions of ionic liquid to achieve significant rate enhancements for bimolecular reactions involving a nitrogen nucleophile.
The changes in the rate constants when varying the anion of the ionic liquid were not as forecast. The importance of the delicate balance of the entropic and enthalpic effects was reinforced, with the variation in the rate constants for the different ionic liquids explained by considering two competing effects; the primary cation–nucleophile interaction, and the more subtle interactions involving the anion of the ionic liquid and components along the reaction coordinate, the importance of which wasn't initially anticipated. For this reaction, involving a nucleophile that is a hydrogen-bond donor, the major effect of the cation–nucleophile interaction on the activation parameters was readily controlled by varying the anion, yet the need to consider the more subtle interaction between the anion and the amino hydrogen atoms was realised, especially when attempting to rationalise the rate constants in each of the ionic liquids considered.
Overall, the effectiveness of the predictive model highlights that there is the opportunity to tailor ionic liquids to introduce specific microscopic interactions that alter reaction outcomes in a desired fashion, which is of interest to synthetic chemists looking for solvent control of reaction outcome.
Acknowledgements
STK acknowledges the support of the Australian government through the receipt of an Australian Postgraduate Award. JBH acknowledges financial support from the Australian Research Council Discovery Project Funding Scheme (Project DP130102331).Notes and references
- S. Z. El Abedin and F. Endres, Acc. Chem. Res., 2007, 40, 1106–1113 CrossRef CAS PubMed.
- K. R. Seddon, Kinet. Catal., 1996, 37, 693–697 CAS.
- C. L. Hussey, Pure Appl. Chem., 1988, 60, 1763–1772 CrossRef CAS.
- M. J. Earle, J. M. S. S. Esperanca, M. A. Gilea, J. N. Canongia Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- B. Wu, W. Liu, Y. Zhang and H. Wang, Chem. – Eur. J., 2009, 15, 1804–1810 CrossRef CAS PubMed.
- H. M. Yau, A. G. Howe, J. M. Hook, A. K. Croft and J. B. Harper, Org. Biomol. Chem., 2009, 7, 3572–3575 CAS.
- S. T. Keaveney and J. B. Harper, RSC Adv., 2013, 3, 15698–15704 RSC.
- T. Fischer, A. Sethi, T. Welton and J. Woolf, Tetrahedron Lett., 1999, 40, 793–796 CrossRef CAS.
- H. Yanai, H. Ogura and T. Taguchi, Org. Biomol. Chem., 2009, 7, 3657–3659 CAS.
- L. Crowhurst, N. L. Lancaster, J. M. Pérez Arlandis and T. Welton, J. Am. Chem. Soc., 2004, 126, 11549–11555 CrossRef CAS PubMed.
- J. P. Hallett, C. L. Liotta, G. Ranieri and T. Welton, J. Org. Chem., 2009, 74, 1864–1868 CrossRef CAS PubMed.
- C. Chiappe, D. Capraro, V. Conte and D. Pieraccini, Org. Lett., 2001, 3, 1061–1063 CrossRef CAS PubMed.
- C. Chiappe, V. Conte and D. Pieraccini, Eur. J. Org. Chem., 2002, 2831–2837 CrossRef CAS.
- F. D'Anna, V. Frenna, R. Noto, V. Pace and D. Spinelli, J. Org. Chem., 2005, 70, 2828–2831 CrossRef PubMed.
- F. D'Anna, V. Frenna, R. Noto, V. Pace and D. Spinelli, J. Org. Chem., 2006, 71, 9637–9642 CrossRef PubMed.
- C. E. Rosella and J. B. Harper, Tetrahedron Lett., 2009, 50, 992–994 CrossRef CAS PubMed.
- H. M. Yau, S. J. Chan, S. George, J. Hook, A. Croft and J. Harper, Molecules, 2009, 14, 2521–2534 CrossRef CAS PubMed.
- H. M. Yau, S. T. Keaveney, B. J. Butler, E. E. L. Tanner, M. S. Guerry, S. R. D. George, M. H. Dunn, A. K. Croft and J. B. Harper, Pure Appl. Chem., 2013, 85, 1979–1990 CrossRef CAS.
- E. E. L. Tanner, R. R. Hawker, H. M. Yau, A. K. Croft and J. B. Harper, Org. Biomol. Chem., 2013, 11, 7516–7521 CAS.
- B. Y. W. Man, J. M. Hook and J. B. Harper, Tetrahedron Lett., 2005, 46, 7641–7645 CrossRef CAS PubMed.
- H. M. Yau, S. A. Barnes, J. M. Hook, T. G. A. Youngs, A. K. Croft and J. B. Harper, Chem. Commun., 2008, 3576–3578 RSC.
- S. G. Jones, H. M. Yau, E. Davies, J. M. Hook, T. G. A. Youngs, J. B. Harper and A. K. Croft, Phys. Chem. Chem. Phys., 2010, 12, 1873–1878 RSC.
- H. M. Yau, A. K. Croft and J. B. Harper, Faraday Discuss., 2012, 154, 365–371 RSC.
- S. T. Keaveney, D. V. Francis, W. Cao, R. S. Haines and J. B. Harper, Aust. J. Chem., 2014, 68, 31–35 CrossRef.
- E. E. L. Tanner, H. M. Yau, R. R. Hawker, A. K. Croft and J. B. Harper, Org. Biomol. Chem., 2013, 11, 6170–6175 CAS.
- S. T. Keaveney, R. S. Haines and J. B. Harper, in Encyclopedia of Physical Organic Chemistry, ed. U. Wille, Wiley, in press Search PubMed.
- S. T. Keaveney, K. Schaffarczyk McHale, R. S. Haines and J. B. Harper, Org. Biomol. Chem., 2014, 12, 7092–7099 CAS.
- J. March and M. Smith, Advanced Organic Chemistry: Reactions, Mechanisms and Structure, Wiley, 6th edn, 2007 Search PubMed.
- S. Steines, P. Wasserscheid and B. Drießen-Hölscher, J. Prakt. Chem., 2000, 342, 348–354 CrossRef CAS.
- L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192–5200 RSC.
- H. Srour, H. Rouault, C. C. Santini and Y. Chauvin, Green Chem., 2013, 15, 1341–1347 RSC.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS PubMed.
- For discussion see: M. Ciaccia and S. Di Stefano, Org. Biomol. Chem., 2005, 13, 646–654 Search PubMed , and references cited therein.
- N. E. Hall and B. J. Smith, J. Phys. Chem. A, 1998, 102, 4930–4938 CrossRef CAS.
- M. Ciaccia, R. Cacciapaglia, P. Mencarelli, L. Mandolini and S. Di Stefano, Chem. Sci., 2013, 4, 2253–2261 RSC.
- S. T. Keaveney, R. H. Haines and J. B. Harper, unpublished results.
- R. Bini, C. Chiappe, E. Marmugi and D. Pieraccini, Chem. Commun., 2006, 897–899 RSC.
- R. Bini, O. Bortolini, C. Chiappe, D. Pieraccini and T. Siciliano, J. Phys. Chem. B, 2007, 111, 598–604 CrossRef CAS PubMed.
- A. Aggarwal, N. L. Lancaster, A. R. Sethi and T. Welton, Green Chem., 2002, 4, 517–520 RSC.
- P. G. Jessop, D. A. Jessop, D. Fu and L. Phan, Green Chem., 2012, 14, 1245–1259 RSC.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790–2794 RSC.
- S. Coleman, R. Byrne, S. Minkovska and D. Diamond, Phys. Chem. Chem. Phys., 2009, 11, 5608–5614 RSC.
- A. Jeličić, N. García, H.-G. Löhmannsröben and S. Beuermann, Macromolecules, 2009, 42, 8801–8808 CrossRef.
- Q. Chen, A. Xu, Z. Li, J. Wang and S. Zhang, Green Chem., 2011, 13, 3446–3452 RSC.
- M. A. Ab Rani, A. Brant, L. Crowhurst, A. Dolan, M. Lui, N. H. Hassan, J. P. Hallett, P. A. Hunt, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Welton and R. Wilding, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- H. Ohno and Y. Fukaya, Chem. Lett., 2009, 38, 2–7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Determination of order of reaction with respect to the nucleophile 2; mole fraction dependent rate constant data on which the plots in Fig. 2 and 3 are based; temperature dependent kinetic data on which Fig. 4 and 5 are based and from which the activation parameters shown in Tables 2–3 are derived; summaries of activation parameter data presented; plot of the Gibbs free energy of activation against the Kamlet-Taft β parameter for the ionic liquids 5–9. See DOI: 10.1039/c4ob02482f |
| This journal is © The Royal Society of Chemistry 2015 |