
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced formation of stable Ag-nanoparticles from a ternary ligand-DNA-Ag+ complex†‡
Daria V.
Berdnikova
,
Heiko
Ihmels
*,
Holger
Schönherr
,
Marc
Steuber
and
Daniel
Wesner
Center of Micro and Nanochemistry and Engineering; Department Chemie-Biologie, Universität Siegen, Adolf-Reichwein-Str. 2, 57068 Siegen, Germany. E-mail: ihmels@chemie.uni-siegen.de
First published on 12th February 2015
Abstract
The combination of (i) the light-harvesting nature and excited-state redox reactivity of a cationic DNA intercalator, (ii) a conjugated Ag+-binding crown ether, and (iii) the stabilizing effect of DNA on AgNPs in one integral ternary complex enables the mild photoinduced formation of Ag nanoparticles.
Metal nanoparticles (NPs) constitute an important class of materials with promising chemical and optical properties that may be employed in chemical synthesis,1 materials science,2 and diagnostic3 or therapeutic medicine.4 As a consequence, the efficient and reliable synthesis of stable metal NPs of desired size and shape is an essential key discipline in this research area.5 In this context, silver nanoparticles (AgNPs) are attractive targets considering their potential antibacterial, antibiotic, and antiviral activity.6 Among the many synthetic routes to metal NPs5 the photochemically induced reduction of Ag+ offers the particular advantage of spatial and temporal control of this reaction.7 Hence, it has been demonstrated that photochemically generated α-amino- or α-hydroxyalkyl (ketyl) radicals have the propensity to reduce Ag+ to Ag0. The latter forms AgNPs in the presence of an appropriate stabilizing additive.7 In addition, it has been shown that organic dyes such as bis-styryl dyes,8 cyanine dyes,9 or thionine10 may also serve as photosensitizers for the synthesis of AgNPs. Notably, DNA may act as a photosensitizer and a stabilizing reagent at the same time. Thus, AgNPs are formed upon direct irradiation of a mixture of Ag+ and DNA.11 However, the latter approach requires a rather unpractical excitation at λex < 300 nm. Furthermore, the covalent attachment of a Cy5 dye to DNA and irradiation in the presence of Ag+ at 647 nm was shown to induce the formation of DNA-capped AgNPs.12 But in the latter case intense laser light was applied with irradiation times up to 60 min.
Herein we present our attempts to explore milder reaction conditions for the photosensitized formation of AgNPs, because UV light or highly intense laser light, as applied in the above-mentioned methods, may lead to secondary photoreactions or photoinduced degradation of other components in the reaction mixture. Thus, we proposed that high-energy light may be avoided with a photosensitizer or photocatalyst that exhibits an appropriately red-shifted absorption and that may generate a reducing intermediate in the course of the photoreaction, as shown in the photoinduced AgNP formation in the presence of a Cy5 and DNA.12 However, other than in the latter experiment, an ideal set-up employs a photocatalyst that binds to DNA and Ag+ at the same time to establish and guarantee a close proximity of all reactive components.
Considering these basic requirements we identified the benzo[b]quinolizinium-crown ether conjugate 1a13 as a potential “mediator” of photoinduced formation of AgNPs. The parent 9-aminobenzo[b]quinolizinium and derivatives thereof are DNA intercalators with a long-wavelength absorption between 380 and 500 nm.14 In addition, the azadithiacrown ether binds Ag+ ions even in water with high affinity,15 and we have demonstrated that the attachment of this crown ether to the benzo[b]quinolizinium enables the delivery of Hg2+ ions to double-stranded (ds) DNA.16 Based on the established photophysical properties of the benzo[b]quinolizinium ion,17 we expected an initial photoinduced electron transfer reaction between the excited, DNA-bound ligand 1a and the DNA upon irradiation of the ternary complex of 1a, DNA and Ag+ to give an oxidized DNA base such as guanine, e.g. dG˙+, and the reduced charge neutral quinolizinyl radical 1ared (Scheme 1),17d that represents formally an α-aminoalkyl radical. Because of the complexation to the crown-ether substituent the Ag+ is in close proximity to 1red and may be readily reduced to Ag0 (red arrow in Scheme 1).18 With a large ligand-DNA ratio and high concentration of Ag+ this photoreaction is proposed to lead to the formation of AgNPs that are in turn directly stabilized by the nearby nucleic acid. A similar method has been demonstrated with polymer nanocomposites that contain Ag+ and a photoreducing dye;8 however, in the latter case the redox cascade is different, because the excited dye reduces the Ag+ ions directly with subsequent electron transfer from a sacrificial donor. Herein, we present the proof of principle of the varied approach described above; and we demonstrate that this method may be employed for the mild and efficient synthesis of stable AgNPs with reproducible size.
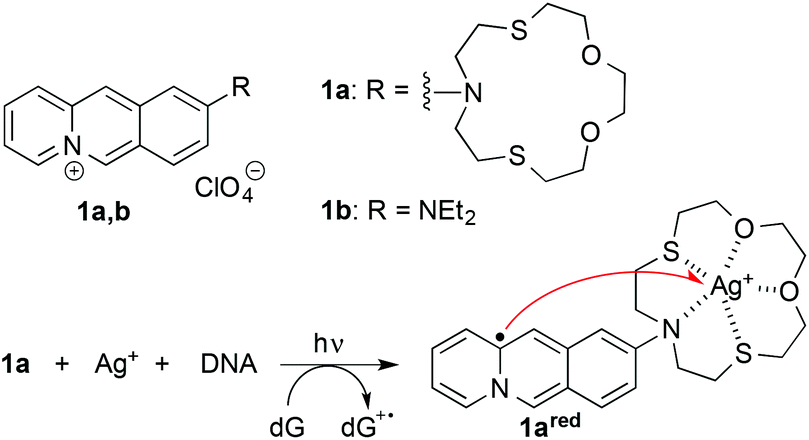 | ||
| Scheme 1 Photoinduced reduction of Ag+ by DNA-bound benzoquinolizinium 1a. | ||
The interactions of substrate 1a with Ag+ or DNA were determined by spectrophotometric titration in aqueous solution (Fig. 1A). Such as already observed for complexes with Hg2+,13 the addition of Ag+ to 1a led to a decrease of the absorbance and a slight blue shift (Δλ = 4 nm) of the long-wavelength maximum. The latter denotes the reduced electron-donating properties of the amino substituent upon complexation of Ag+.13 Analysis of the photometric titration data revealed a binding constant of Kb = 1.0 × 106 M−1. The addition of calf thymus (ct) DNA to 1a in aqueous buffer solution resulted in a decrease of the absorbance and a red shifted long-wavelength absorbance maximum (Δλ = 9 nm), that is similar to the results of resembling DNA-intercalating 9-aminobenzo[b]quinolizinium derivatives.14b A Scatchard analysis of the titration data gave a binding constant of K = 1.2 × 106 M−1 and a binding site size n = 1.1 (Fig. 1B).
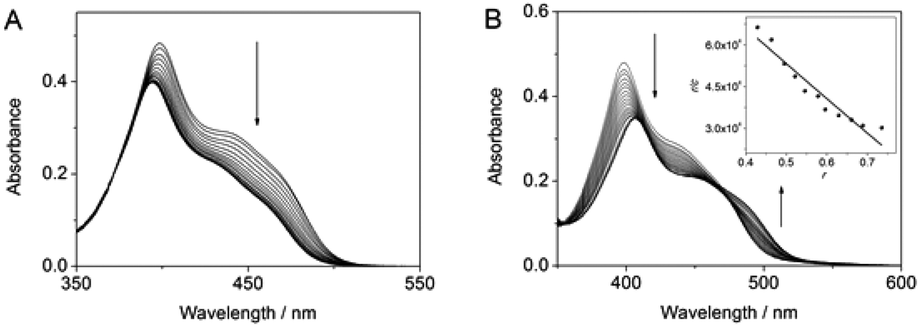 | ||
| Fig. 1 (A) Spectrophotometric titration of AgClO4 to 1a in water, c1 = 30 μM, cAg = 0–0.28 mM; T = 20 °C. (B) Spectrophotometric titration of ct DNA to 1 in HEPES (pH 7.5), c1 = 40 μM, cDNA = 0–0.25 mM, T = 20 °C. Inset: Scatchard plot, r/c vs. r; r = ligand-to-DNA ratio, the line indicates the best fit of the experimental data to the theoretical model. | ||
The association of DNA with 1a and Ag+ was also examined by circular dichroism (CD) spectroscopy (Fig. 2B). Thus, the addition of the ligand 1a to ct DNA resulted in the formation of bisignate induced circular dichroism (ICD) signal in the absorption range of the ligand (300–500 nm), along with increase of the CD band at ca. 275 nm, where both DNA and the ligand absorb. This development of CD spectra is characteristic of DNA-intercalating benzo[b]quinolizinium derivatives.19 As shown already for the interaction of Ag+ with double- and single stranded DNA,20 the addition of Ag+ to ct DNA resulted in a slightly increased absorption band with a small red shift of the maximum. In contrast, the characteristic CD signature of ds DNA with a maximum at 275 nm and a minimum at 245 nm disappears. At the same time, a CD band with a weak maximum at 295 nm and an intense negative band with a minimum at 265 nm develops, indicating the structural changes of the DNA upon Ag+ complexation.20 In the presence of Ag+ and ligand 1a the CD bands of the DNA resemble the ones of the DNA-Ag+ complex, but they are less intense. In addition, relatively strong ICD bands are formed in the absorption range of the ligand. These data provide sufficient evidence for the formation of a ternary complex between DNA, Ag+, and the ligand 1a.
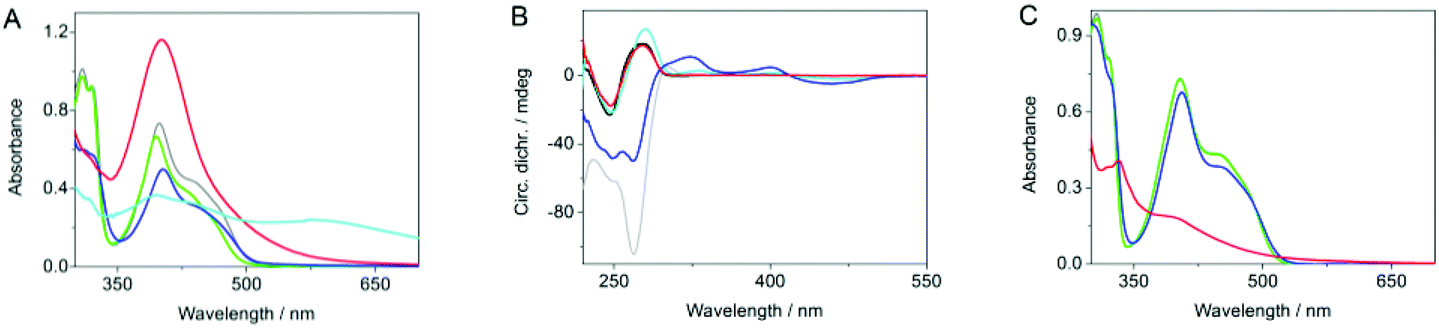 | ||
| Fig. 2 (A) Absorption spectra of 1a (grey), 1a-Ag+ (c1 = 50 μM, cAgClO4 = 0.1 mM) before (green) and after irradiation (cyan, λex = 420 nm, t = 1 h), and the ternary system 1a-Ag+-ct DNA (c1a = 50 μM, cAgClO4 = 0.1 mM, cDNA = 120 μg ml−1) before (blue) and after irradiation (red; λex = 420 nm, t = 1 h). (B) Circular dichroism spectra of ct DNA (black), ct DNA-1a (cyan; c1 = 50 μM, cDNA = 120 μg ml−1), ct DNA-Ag+ (grey; cDNA = 120 μg ml−1, cAgClO4 = 0.1 mM), and the ternary system 1a-Ag+-ct DNA (c1a = 50 μM, cAgClO4 = 0.1 mM, cDNA = 120 μg ml−1) before (blue) and after irradiation (red; λex = 420 nm, t = 1 h). (C) Absorption spectra of 1b (grey), 1b-Ag+ (green), and the ternary system 1b-Ag+-ct DNA (c1b = 50 μM, cAgClO4 = 0.1 mM, cDNA = 120 μg ml−1) before (blue) and after irradiation (red; λex = 420 nm, t = 1 h). | ||
A mixture of the ligand 1a, of Ag+ (2 molar equiv.), and ct DNA was irradiated with λex = 420 nm, i.e. an excitation wavelength where only the ligand 1a absorbs. After 1 h of irradiation, AgNPs were formed, as clearly indicated by the development of the characteristic surface plasmon band (SPB) centered at 401 nm. A similar result was obtained with circular DNA, namely plasmid PBR322; however in the latter case an additional shoulder was observed at ca. 490 nm along with the main SPB at 401 nm (cf. ESI†). The absorption spectrum did not change, except for a slight decrease of the SPB absorbance, with longer irradiation times (6.5 h). This decrease originates from the disappearance of the residual ligand absorption in the spectral region of 350–500 nm due to complete photodegradation of 1a upon prolonged irradiation. The DNA-stabilized AgNPs are stable in aqueous solution, as indicated by the maintenance of the SPB of the photolysate over several weeks without change of the absorption maximum position and intensity (cf. ESI†). Most notably, the CD spectrum of the photolysate, i.e. after the completion of the AgNPs formation, resembles the characteristic CD bands of a regular ds DNA. This observation indicates consumption of the DNA-bound Ag+ ions during the reaction and resulting reorganization to the regular double-helical DNA structure. Control experiments were conducted to show that the formation of nanoparticles actually originates from the ternary complex of DNA, Ag+ ions and 1a. For that purpose, the irradiation of the derivative 1b,21 that does not contain the crown-ether unit, in the presence of Ag+ and DNA under identical conditions did not lead to the formation of AgNPs according to photometric analysis of the photolysate. Instead, the characteristic absorption band of derivative 1b disappears, and a less intense broad band developed that may indicate a photodimerization of 1b.22 This result shows that the crown-ether unit is essential for the photoinduced formation of the AgNPs, presumably because it provides a close proximity between Ag+ and the absorbing chromophore required for the initiation of the reduction of Ag+ to Ag0. In another control experiment, irradiation of a solution of the quinolizinium-crown ether conjugate 1a and Ag+ ions in the absence of DNA resulted in the reduction of Ag+ as shown by the formation of a very broad absorption from 350 to 800 nm, that is typical for colloidal silver solutions with a wide particle size/shape distribution.23 Apparently, the DNA is not necessarily required to induce the reduction process. But in the absence of the nucleic acid, the growing AgNPs are not stabilized and, therefore, undergo fast aggregation and precipitation.
The size and shape of the NPs were analyzed by SEM and TEM imaging showing round-shaped particles with a diameter of 11.8 ± 3.7 nm and 11.8 ± 3.6 nm (mean ± standard deviation, SD) for NPs synthesized on ct DNA and plasmid DNA pBR322, respectively (Fig. 3 and ESI†). This observation indicates that the type of the DNA does not have significant influence on the dimensions of the individual NPs. For AgNPs generated with plasmid DNA the fraction of agglomerated particles on the surface is higher, but this effect might also be influenced by the drying process of the droplet containing the AgNPs. In TEM images a corona was observed around the AgNPs that indicates the presence of material of lower electron density, which is assigned to the DNA (cf. ESI†). The surface elemental composition as well as the DNA corona, that covers the AgNPs, were unambiguously confirmed by XPS analysis (cf. ESI†). In addition, the presence of Ag was confirmed.
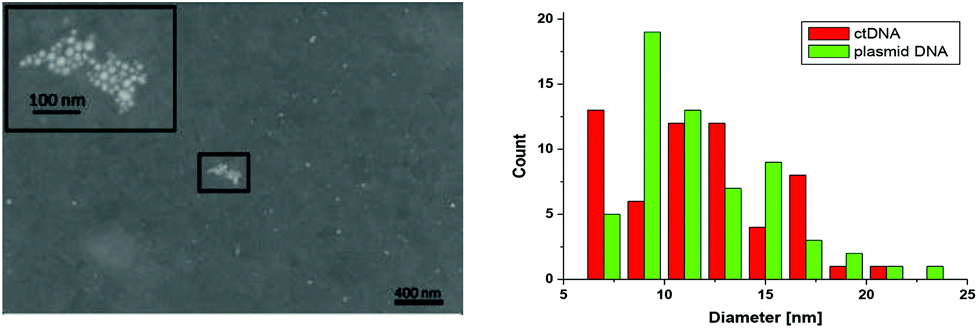 | ||
| Fig. 3 Left: SEM images showing silver NPs, which were prepared from irradiation of 1, Ag+ and ct DNA, on template-stripped Au. Right: diameters of the round-shaped AgNPs as obtained from irradiation of 1, Ag+ and ct DNA or plasmid pBR322. | ||
Since only the electrically conductive AgNPs are visible in the SEM micrographs, AFM imaging was used to investigate the NPs together with the DNA. The DNA is electrostatically bound at the surface and the remaining sample droplet was removed, so that the NPs present are assumed to be immobilized through the DNA they are associated with. At a DNA concentration of 120 μg ml−1 the whole mica surface is covered by DNA with the NPs distributed on the surface (Fig. 4A and C). The height of the particles of 17.4 ± 10.3 nm (mean ± SD) is slightly larger than the diameter measured with SEM, which may be explained by the different interaction forces between the AFM-tip and the surface (DNA vs. AgNP).24 At lower DNA concentrations of 2.5 μg ml−1 individual DNA molecules can be differentiated and the NPs are closely associated with those strands (Fig. 4B).
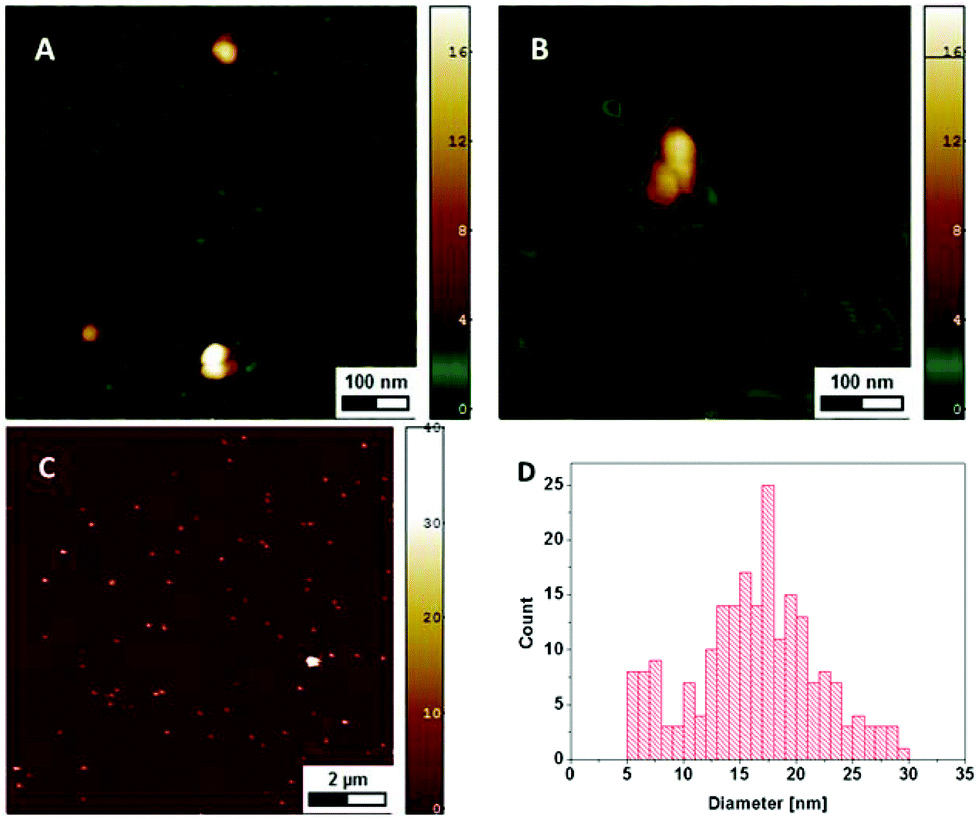 | ||
| Fig. 4 AFM height images showing AgNPs and DNA on mica at a DNA concentration of 120 μg ml−1 (A, C) and 2.5 μg ml−1 (B) The diameter of the NPs is given by the measured height (D). | ||
All experimental results point to a photoinduced formation of AgNPs that is triggered through the light absorption by the benzo[b]quinolizinium chromophore. It is assumed that the ligand 1a has essentially the same reduction potential in the excited state as the parent 9-aminobenzo[b]quinolizinium (Ered* = 1.9 V vs. NHE)17d such that a photoinduced electron transfer between the excited intercalator and the DNA bases25 takes place with formation of the quinolizinyl radical 1ared (Scheme 1), as has been shown already for benzo[b]quinolizinium derivatives.17d In a subsequent electron transfer reaction, the intermediate 1ared reduces the Ag+ ion to Ag0 and the benzo[b]quinolizinium 1a is regained. The Ag0 is then ejected from the crown ether unit and may be substituted with another DNA-bound Ag+ ion, so that the process can be reiterated. The immediate availability of another Ag+ ion, that is associated directly with DNA, is clearly indicated by the CD spectroscopic analysis (Fig. 2C).20 It has been demonstrated already that nucleic acids may serve as a template and appropriate stabilizing component during the formation of AgNPs.11,20b,26 Therefore it is proposed that the AgNPs are readily formed along the DNA backbone once the critical concentration of Ag atoms is reached.7 Apparently, the critical phase of the AgNP formation is the sequence of electron transfer reactions directly after excitation of the benzo[b]quinolizinium ligand. Although it has been observed that in the absence of a secondary reaction pathways for the quinolizinyl radical 1ared an efficient electron back transfer to the oxidized nucleic base takes place,17d it has also been shown that a competing electron transfer from a DNA-bound molecule to an adjacent electron acceptor may constitute a competing secondary reaction.18 At the same time, the directly oxidized DNA base will induce a charge migration through the DNA in a multistep hopping mechanism, followed by hydrolysis, and eventually leading to an oxidized DNA base, e.g. 8-oxo-7,8-dihydroguanine, according to established mechanisms.27 The reduction of Ag+ complexed to 1a in the absence of DNA may be the result of an initial intra- or intermolecular photoinduced electron transfer from the electron-donating crown ether to the excited quinolizinium, that also generates – although to lesser extent due to a fast back electron transfer – the reduced benzoquinolizinium. The latter subsequently reduces the Ag+ ions.
It should be noted that there is already one reported case of photoinduced AgNP formation in the presence of a dye, Cy5, and DNA.12 In the latter case, however, a completely different mechanism is proposed, namely the initial photoinduced oxidation of Cy5 and subsequent reduction of Ag+ by the oxidized excited Cy5; and the fate of the oxidized dye is not addressed. Most notably, this particular reaction is suggested to occur in a complex between Cy5 and Ag+, whereupon the DNA is not involved in the formation, but just in the stabilization of the AgNPs after their formation. In the case presented herein, a different mechanism takes place. Firstly, the benzo[b]quinolizinium is reduced in its excited state by initial electron transfer reaction with the DNA.17d Secondly, we have unambiguously demonstrated that the ternary complex between 1a, Ag+ and DNA is a general requirement for the photoinduced formation of the AgNPs.
Conclusions
Overall we have shown that a ternary complex of an intercalator-crown ether conjugate, DNA, and Ag+ provides all requirements to accomplish the photoinduced formation of AgNPs, namely a photosensitizer as light-harvesting and reducing component, a conjugated crown ether that provides close vicinity of Ag+ ions to the in situ formed, short-lived reductant, and the DNA as stabilizing reagent for the AgNPs. Most notably, the AgNPs made by this method are stable in solution for a relatively long period of time and the size is highly reproducible. The mechanism is not yet proven, but at least supported by literature data and precedences, so that further detailed photophysical measurements are necessary. Nevertheless, these results are a proof of principle for milder variant of photoinduced formation of DNA-stabilized AgNPs that may be employed complementary to the established method that requires significantly more excitation energy to initiate the process. Several modifications of this approach may be proposed that enable a rather modular approach to metal NP synthesis. Thus with a variation of the ligand-receptor unit, the metal ion, or the host system different types of nanoparticles may be available or – with different chromophores – the irradiation conditions may be changed to other, ideally red-shifted excitation wavelengths. It is also anticipated that the same effect may be accomplished with other ligand-crown ether conjugates that bind selectively to non-canonical DNA forms such as quadruplex or triplex DNA, which may even lead to the formation of AgNPs with different shape and size, as has been demonstrated recently for chemically generated AgNPs in the presence of different DNA forms.28 Moreover, this method may be applied for the targeted deposition of AgNPs on DNA-functionalized surfaces.Acknowledgements
Generous support by the Deutsche Forschungsgemeinschaft is gratefully acknowledged. Marc Steuber, Daniel Wesner and Holger Schönherr acknowledge financial support from the European Research Council (ERC starting grant to HS, ERC grant agreement no. 279202). The authors gratefully acknowledge Prof. Dr. Xin Jiang (Department of Mechanical Engineering, University of Siegen, Germany) for access to the FESEM. We thank Ms Sarah Kölsch for the preparation of AgNPs for TEM and XPS experiments and Ms Inga Lilge for conducting the XPS experiments.Notes and references
- (a) B. S. Takale, M. Bao and Y. Yamamoto, Org. Biomol. Chem., 2014, 12, 2005–2027 RSC; (b) Metal Nanoparticles for Catalysis: Advances and Applications, ed. Franklin Tao, The Royal Society of Chemistry, Cambridge, 2014 Search PubMed; (c) B. R. Cuenya, Acc. Chem. Res., 2013, 46, 1682–1691 CrossRef PubMed.
- (a) Z. Lu and Y. Yin, Chem. Soc. Rev., 2012, 41, 6874–6887 RSC; (b) M. Oyama, X. M. Chen and X. Chem, Anal. Sci., 2014, 30, 529–538 CrossRef CAS PubMed; (c) W. Y. Ko and K. J. Lin, J. Nanomater., 2013, 505292 Search PubMed.
- (a) M. Perfézou, A. Turner and A. Merkoçi, Chem. Soc. Rev., 2012, 41, 2606–2622 RSC; (b) M. Swierczewska, G. Liu, S. Lee and X. Chen, Chem. Soc. Rev., 2012, 41, 2641–2655 RSC.
- (a) E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC; (b) R. R. Arvizo, S. Bhattacharyya, R. A. Kudgus, K. Giri, R. Bhattacharya and P. Mukherjee, Chem. Soc. Rev., 2012, 41, 2943–2970 RSC.
- (a) A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS PubMed; (b) M.-A. Neouze, J. Mater. Sci., 2013, 48, 7321–7349 CrossRef CAS; (c) Z. Pengxiang, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef; (d) Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed; (e) J. A. Dahl, B. L. S. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228 CrossRef CAS PubMed; (f) I. Willner, R. Baron and B. Willner, Adv. Mater., 2006, 18, 1109 CrossRef CAS.
- (a) S. Galdiero, A. Falanga, M. Vitiello, M. Cantisani, V. Marra and M. Galdiero, Molecules, 2011, 16, 8894–8918 CrossRef CAS PubMed; (b) A. J. Huh and Y. J. Kwon, J. Controlled Release, 2011, 156, 128–145 CrossRef CAS PubMed; (c) M. K. Rai, S. D. Deshmukh, A. P. Ingle and A. K. Gade, J. Appl. Microbiol., 2012, 112, 841–852 CrossRef CAS PubMed; (d) A. D. Politano, K. T. Campbell, L. H. Rosenberger and R. G. Sawyer, Surg. Infect., 2013, 14, 8–20 CrossRef PubMed.
- (a) M. Sakamoto, M. Fujistuka and T. Majima, J. Photochem. Photobiol., C, 2009, 10, 33–56 CrossRef CAS; (b) M. Sakamoto and T. Majima, Bull. Soc. Chem. Jpn., 2012, 83, 1133–1154 CrossRef; (c) K. G. Stamplecoskie and J. C. Scaiano, Photochem. Photobiol., 2012, 88, 762–768 CrossRef CAS PubMed; (d) J. C. Scaiano, K. G. Stamplecoskie and G. L. Hallett-Tapley, Chem. Commun., 2012, 48, 4798–4808 RSC.
- F. Stellacci, C. A. Bauer, T. Meyer-Friedrichsen, W. Wenseleers, V. Alain, S. M. Kuebler, S. J. K. Pond, Y. Zhang, S. R. Marder and J. W. Perry, Adv. Mater., 2002, 14, 194–198 CrossRef CAS.
- D. M. Eisele, H. V. Berlepsch, C. Bottcher, K. J. Stevenson, D. A. V. Bout, S. Kirstein and J. P. Rabe, J. Am. Chem. Soc., 2010, 132, 2104–2105 CrossRef CAS PubMed.
- P. K. Sudeep and P. V. Kamat, Chem. Mater., 2005, 22, 5404–5410 CrossRef.
- (a) L. Berti, A. Alessandrini and P. Facci, J. Am. Chem. Soc., 2005, 127, 11216–11217 CrossRef CAS PubMed; (b) S. Kundu, Phys. Chem. Chem. Phys., 2013, 15, 14107–14119 RSC.
- V. B. Zon, G. A. Burley and U. Rant, Nanotechnology, 2012, 23, 115607 CrossRef PubMed.
- M. Tian and H. Ihmels, Chem. Commun., 2009, 3175–3177 RSC.
- (a) H. Ihmels, K. Faulhaber, B. Engels and C. Lennartz, Chem. – Eur. J., 2000, 6, 2854–2864 CrossRef CAS PubMed; (b) A. Granzhan, H. Ihmels and G. Viola, J. Am. Chem. Soc., 2007, 129, 1254–1267 CrossRef CAS PubMed.
- (a) M. Schmittel and H. Lin, Inorg. Chem., 2007, 46, 9139 CrossRef CAS PubMed; (b) A. Coskun and E. U. Akkaya, J. Am. Chem. Soc., 2006, 128, 14474 CrossRef CAS PubMed; (c) M. J. Yuan, Y. L. Li, J. B. Li, C. H. Li, X. F. Liu, J. Lv, J. L. Xu, H. B. Liu, S. Wang and D. B. Zhu, Org. Lett., 2007, 9, 2313 CrossRef CAS PubMed; (d) M. Zhu, M. Yuan, X. Liu, J. Xu, J. Lv, C. Huang, H. Liu, Y. Li, S. Wang and D. Zhu, Org. Lett., 2008, 10, 1481 CrossRef CAS PubMed; (e) L. Wang, W.-K. Wong, L. Wu and Z.-Y. Li, Chem. Lett., 2005, 34, 934 CrossRef CAS.
- M. Tian, H. Ihmels and K. Benner, Chem. Commun., 2010, 46, 5719–5721 RSC.
- (a) J. Bendig, J. Wagner, W. Buchwitz and D. Kreysig, Ber. Bunsen-Ges. Phys. Chem., 1981, 85, 437 CrossRef CAS; (b) J. Bendig, S. Helm, D. Kreysig and J. Wilda, J. Prakt. Chem., 1982, 324, 978 CrossRef CAS; (c) R. Mitzner, J. Bendig, R. Ziebig, F. Graichen, D. Kreysig and F. Pragst, J. Prakt. Chem., 1985, 327, 241–250 CrossRef CAS; (d) C. Bohne, K. Faulhaber, B. Giese, A. Häfner, A. Hofmann, H. Ihmels, A.-K. Köhler, S. Perä, F. Schneider and M. A. L. Sheepwash, J. Am. Chem. Soc., 2005, 126, 76–85 CrossRef PubMed.
- For electron transfer cascades with bifunctional DNA, see: (a) J. Joseph, N. V. Eldho and D. Ramaiah, Chem. – Eur. J., 2003, 9, 5926–5935 CrossRef CAS PubMed; (b) T. Zhang, S. Sun, F. Liu, Y. Pang, J. Fan and X. Peng, Phys. Chem. Chem. Phys., 2011, 13, 9789–9795 RSC.
- H. Ihmels, K. Faulhaber, D. Vedaldi, F. Dall'Acqua and G. Viola, Photochem. Photobiol., 2005, 81, 1107–1115 CrossRef CAS PubMed.
- (a) B. Norden, Y. Matsuoka and T. Kurucsev, Biopolymers, 1986, 25, 1531–1545 CrossRef CAS PubMed; (b) J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS PubMed.
- A. Granzhan and H. Ihmels, ARKIVOC, 2007, viii, 136–149 Search PubMed.
- H. Ihmels and J. Luo, J. Photochem. Photobiol., A, 2008, 200, 3–9 CrossRef CAS.
- T. Huang and X.-H. Nancy Xu, J. Mater. Chem., 2010, 20, 9867–9876 RSC.
- S. J. T. van Noort, K. O. van der Werf, B. G. de Grooth, N. F. van Hulst and J. Greve, Ultramicroscopy, 1997, 69, 117–127 CrossRef CAS.
- (a) C. A. M. Seidel, A. Schulz and M. H. M. Sauer, J. Phys. Chem., 1996, 100, 5541 CrossRef CAS; (b) S. Steenken and S. Jovanovic, J. Am. Chem. Soc., 1997, 119, 617 CrossRef CAS.
- G. Shemer, O. Krichevski, G. Markovich, T. Molotsky, I. Lubitz and A. B. Kotlyar, J. Am. Chem. Soc., 2006, 128, 11006 CrossRef CAS PubMed.
- S. Kanvah, J. Joseph, G. B. Schuster, R. N. Barnett, C. L. Cleveland and Z. Landman, Acc. Chem. Rev., 2010, 43, 280–287 CrossRef CAS PubMed.
- J. Wu, L. Huey Tan, K. Hwang, H. Xing, P. Wu, W. Li and Y. Lu, J. Am. Chem. Soc., 2014, 136, 15195–15202 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures. See DOI: 10.1039/c5ob00295h |
| ‡ Author names are in alphabetical order that does not reflect the specific contribution of each author. |
| This journal is © The Royal Society of Chemistry 2015 |