
An enantioselective total synthesis of Sch-725674†
Kota
Ramakrishna
and
Krishna P.
Kaliappan
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai-400076, India. E-mail: kpk@chem.iitb.ac.in
First published on 30th October 2014
Abstract
An enantioselective total synthesis of Sch-725674, a unique 14-membered macrolactone, has been accomplished in 13 steps. The step-wise dithiane alkylation served as a strategic step to assemble the upper and lower fragments of the molecule, whereas cross metathesis reaction, Yamaguchi macrolactonization and a substrate controlled stereoselective reduction are used as key steps to complete the total synthesis.
Introduction
Macrolactones, an important subclass of natural products, are often used as a template for designing new drugs owing to their conformational flexibility towards various biological macromolecules.1 Sch-725674 (1), a 14-membered macrolactone, isolated by Yang and co-workers from the culture of Aspergillus sp., has been shown to exhibit an antifungal activity against Saccharomyces cerevisiae and Candida albicans with MICs 8 and 32 μg mL−1 respectively.2 The 14-membered macrocyclic mono-lactone skeletons without additional methyl group substitution on the ring are very rare and Sch-725674 (1) represents one such example in this class (Fig. 1).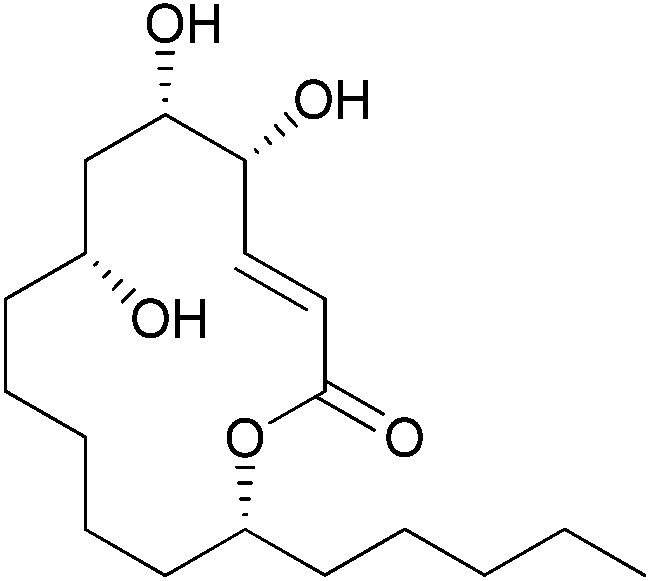 | ||
| Fig. 1 Sch-725674 (1). | ||
The presence of an unusual n-pentyl substitution on the macrolactone ring along with the multiple sites of oxygenation makes Sch-725674 (1) a distinct and attractive synthetic target. The first total synthesis of Sch-725674 (1) with the complete stereoisomer library has been reported3 by the Curran's group using fluorous mixture synthesis. During the preparation of our manuscript, Prasad and co-workers reported4 the second total synthesis of 1 using a RCM approach.
The 1,3-dithiane has been utilized as an acyl anion equivalent in organic synthesis ever since its discovery by Seebach and Corey.5 In 1994, the pioneering study by Tietze on the alkylation of lithiated-1,3-dithianes led to the inception of linchpin coupling,6 whose synthetic utility has been significantly demonstrated by Smith et al. with their key scientific finding that tertiary amines such as HMPA and DMPU are capable of inducing a solvent controlled Brook rearrangement on lithiated-1,3-dithianes.7a Furthermore, Smith and co-workers introduced the concept of multicomponent linchpin coupling, popularly known as anion relay chemistry and the efficiency of this methodology has been enormously utilized in the total synthesis of various complex natural products thereby rendering the dithiane chemistry as an important tool for C–C bond formation in organic synthesis.7
Results and discussion
Our long-standing interest in synthesizing biologically active macrolides8 prompted us to devise a concise synthetic strategy toward Sch-725674 (1) using the linchpin coupling strategy. As shown in Scheme 1, we envisaged that an oxidative cleavage of dithiane in macrolactone 2 followed by a stereoselective reduction would install the C-7 hydroxyl center in 1. The 14-membered macrolactone 2 was thought to be obtained from triol 3, which in turn could be synthesized from the linchpin product 4. The linchpin coupling of TBS-dithiane 5 with epoxide 6 and alkyl iodide 7 was envisaged as a key strategy to secure 4. The alkyl iodide 7 could be synthesized over a few steps from the enantio-enriched (S)-epichlorohydrin, whereas the kinetic resolution of racemic divinylcarbinol followed by protection of the resulting hydroxyl as its TBS ether would readily give the other linchpin partner epoxide 6.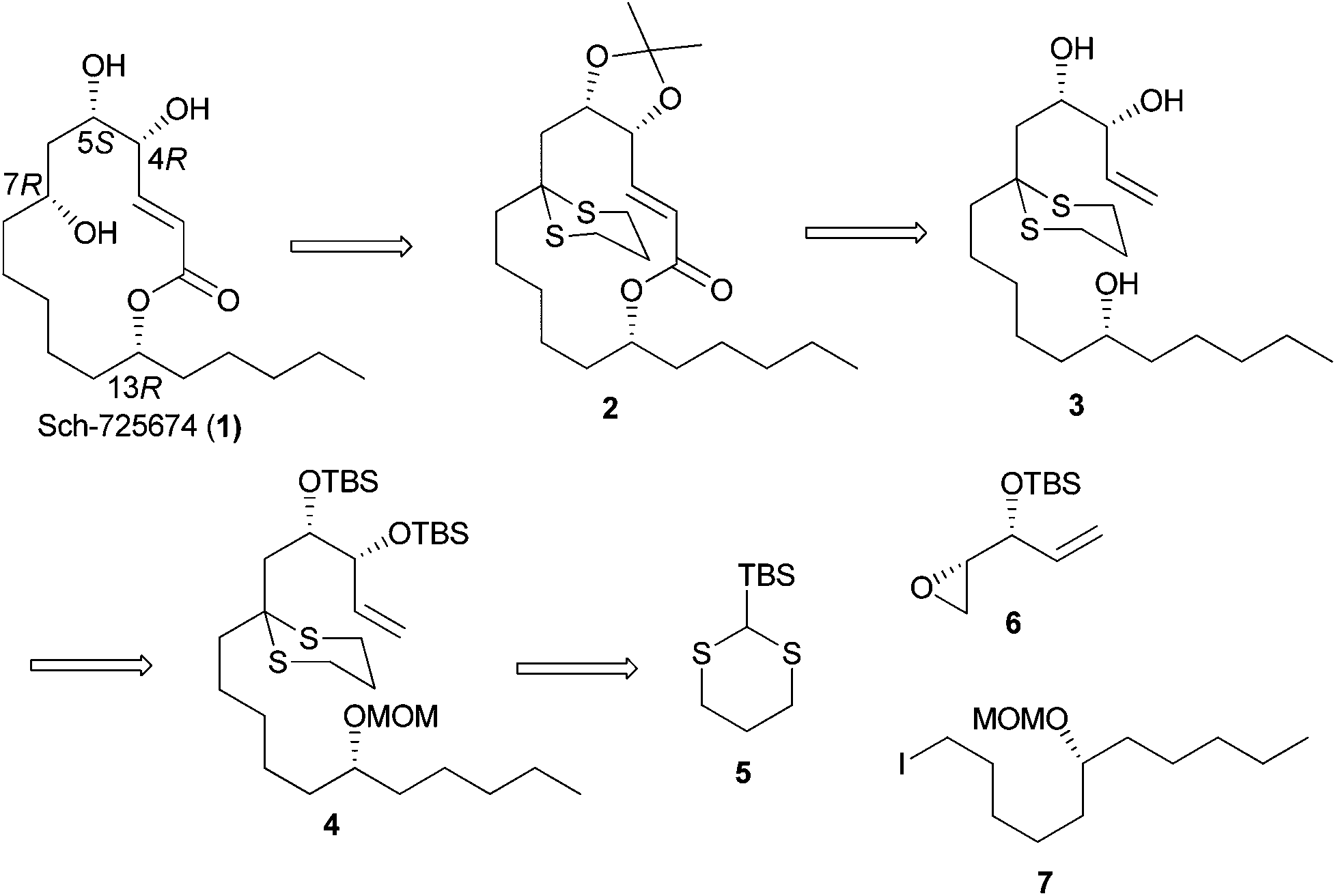 | ||
| Scheme 1 Retrosynthetic analysis of Sch-725674 (1). | ||
Thus, our synthetic journey towards 1 started with the preparation of linchpin partners’ epoxide 6 and iodide 7. The free hydroxyl group of the known compound 9, obtained from 8 in 3 steps,9 was protected as its MOM ether 10 using MOMCl and DIPEA.10 The hydroboration–oxidation sequence on alkene 10 using either the commercially available BH3·SMe2 or in situ generated BH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 afforded the alcohol 11 in a moderate yield of 60%, which in turn was transformed into iodide 7 using PPh3 and I2 (Scheme 2).
11 afforded the alcohol 11 in a moderate yield of 60%, which in turn was transformed into iodide 7 using PPh3 and I2 (Scheme 2).
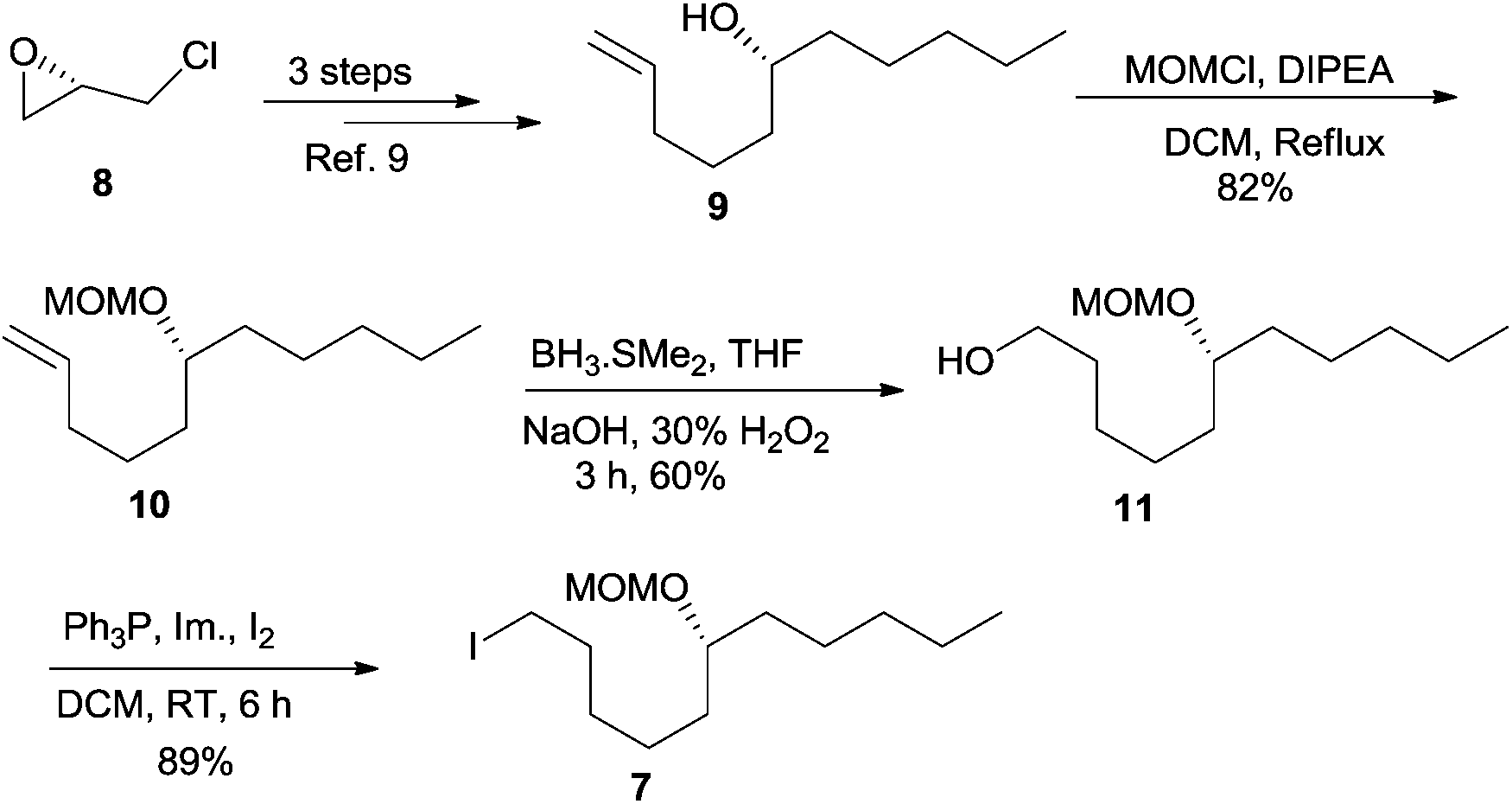 | ||
| Scheme 2 Synthesis of iodide 7. | ||
The kinetic resolution12 of divinyl carbinol 12 resulted in an enantio-enriched epoxy alcohol 13 and the protection of the free hydroxyl group as TBS ether gave the known epoxide 6 in good yield (Scheme 3).13
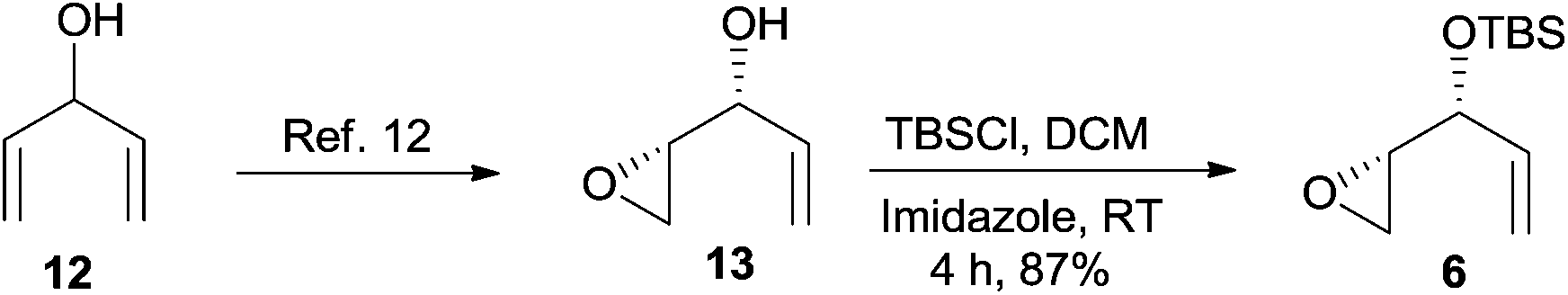 | ||
| Scheme 3 Synthesis of epoxide 6. | ||
With both the linchpin partners in hand, the stage was now set for carrying out the key linchpin coupling. However, all our initial attempts to effect the linchpin coupling with TBS-dithiane 5 under conventional conditions did not succeed and we could only recover the unreacted starting materials. After extensive experimentation, we were able to synthesize the Brook rearranged product 15 by performing lithiation on an azeotroped TBS-dithiane 5 using t-BuLi as a base at −40 °C. The resulting dithiane anion was deaggregated by the addition of HMPA followed by treatment with epoxide 6 in Et2O and HMPA (5:1) at −40 °C over a period of 1 h to give 15 in 68% yield (Scheme 4).
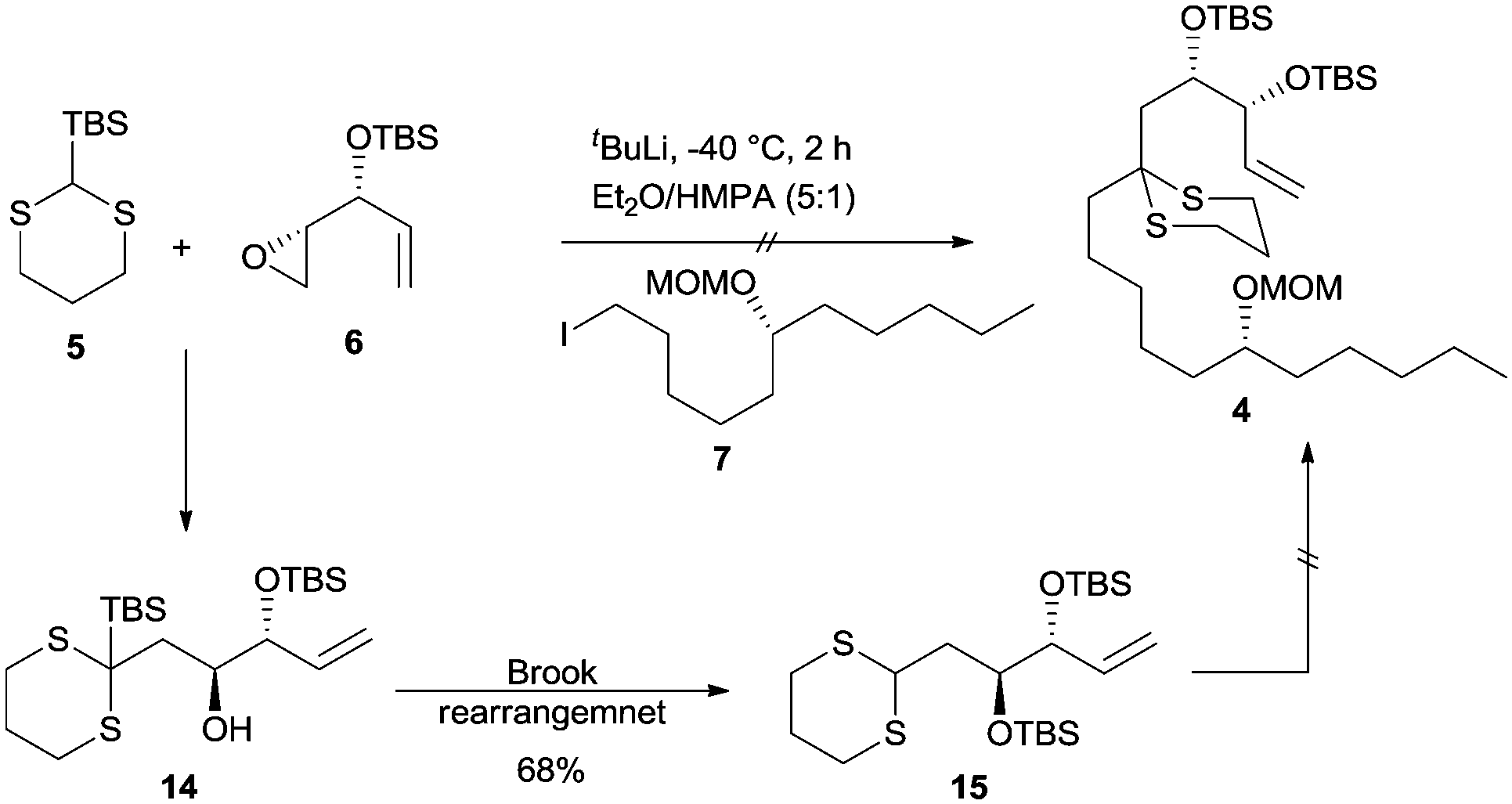 | ||
| Scheme 4 Attempted linchpin coupling. | ||
Having failed to perform the three-component linchpin coupling, we opted for a step-wise alkylation on dithiane 15. Unfortunately, our attempts to synthesize linchpin product 4 by the lithiation of dithiane 15 using t-BuLi as a base in THF and Et2O solvents over a range of temperatures with HMPA as an additive in different concentrations also failed and resulted in the recovery of unreacted dithiane 15 and iodide 7. Based on the literature, reported by Smith and co-workers in their synthesis of (+)-calyculin A and (−)-calyculin B,14 the difficulty in the lithiation of dithiane 15 may be attributed to the fact that the presence of a terminal double bond in 15 would indeed decrease the acidity of the dithiane proton. A possible explanation accounted by Smith et al. suggests that the interaction between the alkene π-system and the unfilled C–S σ* orbital would considerably decrease the acidity of the dithiane proton by assuming two equilibrated conformations 16 and 17 (Fig. 2). This rationale was further supported by another example as recently reported by Brimble and co-workers.15
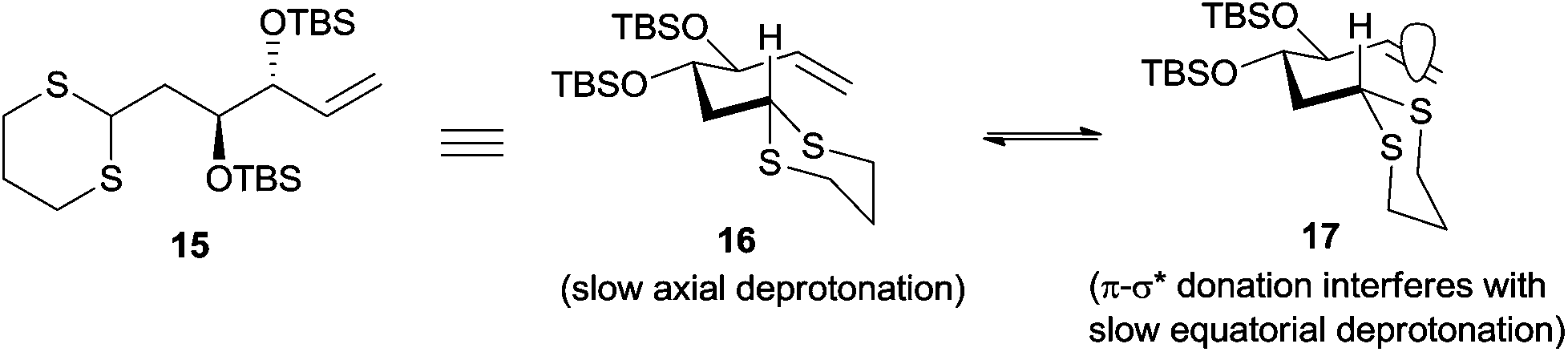 | ||
| Fig. 2 Conformations of dithiane 15. | ||
After the unsuccessful attempts in achieving the three component linchpin coupling, we resorted to a step-wise alkylation with 1,3-dithiane 18 using iodide 7 as the first electrophile and epoxide 6 as the second electrophile. Accordingly, lithiation of 1,3-dithiane 18 was performed with t-BuLi as a base at −78 °C and after adding HMPA, the anion was quenched with iodide 7 to afford the monoalkylated product 19 in an excellent yield of 86%. The lithiated dithiane 19 on treatment with epoxide 6 also proceeded smoothly to result in dithiane dialkylated product 20 in 72% yield. Removal of the MOM group in 20 proved to be quite cumbersome and after considerable efforts, a concomitant deprotection of MOM ether and TBS ether of 20 was smoothly effected by utilizing 7.5 eq. of PPTS in MeOH at reflux over a period of 32 h that resulted in triol 3 in 60% yield.16 The cross metathesis reaction between triol 3 and methyl acrylate using G-II in refluxing DCM for 48 h furnished the E-methyl ester 21 (1H NMR analysis) in 74% yield.17 The high catalytic loading ensured the complete conversion of the starting material as both the reactant and the product appear at the same Rf as in the TLC pattern (Scheme 5).
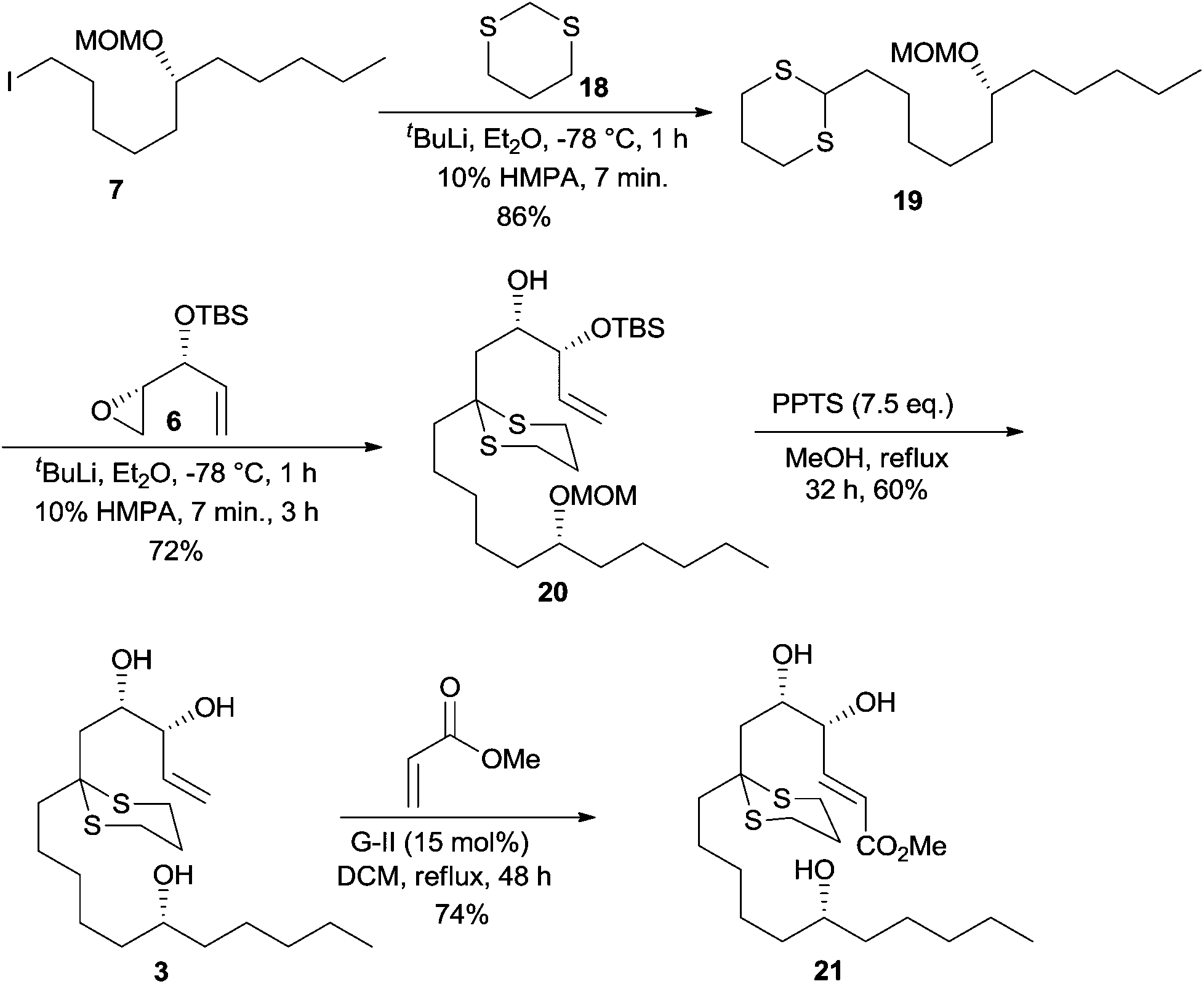 | ||
| Scheme 5 Synthesis of the CM product 21. | ||
Having synthesized the methyl ester 21 in good quantity, the vicinal 1,2-diol in 21 was then protected as acetonide 22 using 2,2-DMP and a catalytic amount of PPTS. The hydrolysis of methyl ester 22 followed by the Yamaguchi macrolactonization of the resulting seco-acid 23 with 2,4,6-trichlorobenzoyl chloride and DMAP in refluxing toluene gave the macrolactone 2 in 64% yield over two steps.18 The deprotection of dithiane ketal was effected using Stork's reagent19 to obtain ketoester 24, albeit in a moderate yield of 59%. The substrate controlled stereoselective reduction of 24 using NaBH4 proceeded with complete diastereoselectivity to give alcohol 25 (1H NMR analysis) in 73% yield. The removal of the acetonide was then easily accomplished by treating alcohol 25 with CSA to afford the natural product whose spectral data were identical in all respects to the reported data (Scheme 6)3,4 and thus completed an enantioselective total synthesis of Sch-725674 (1).
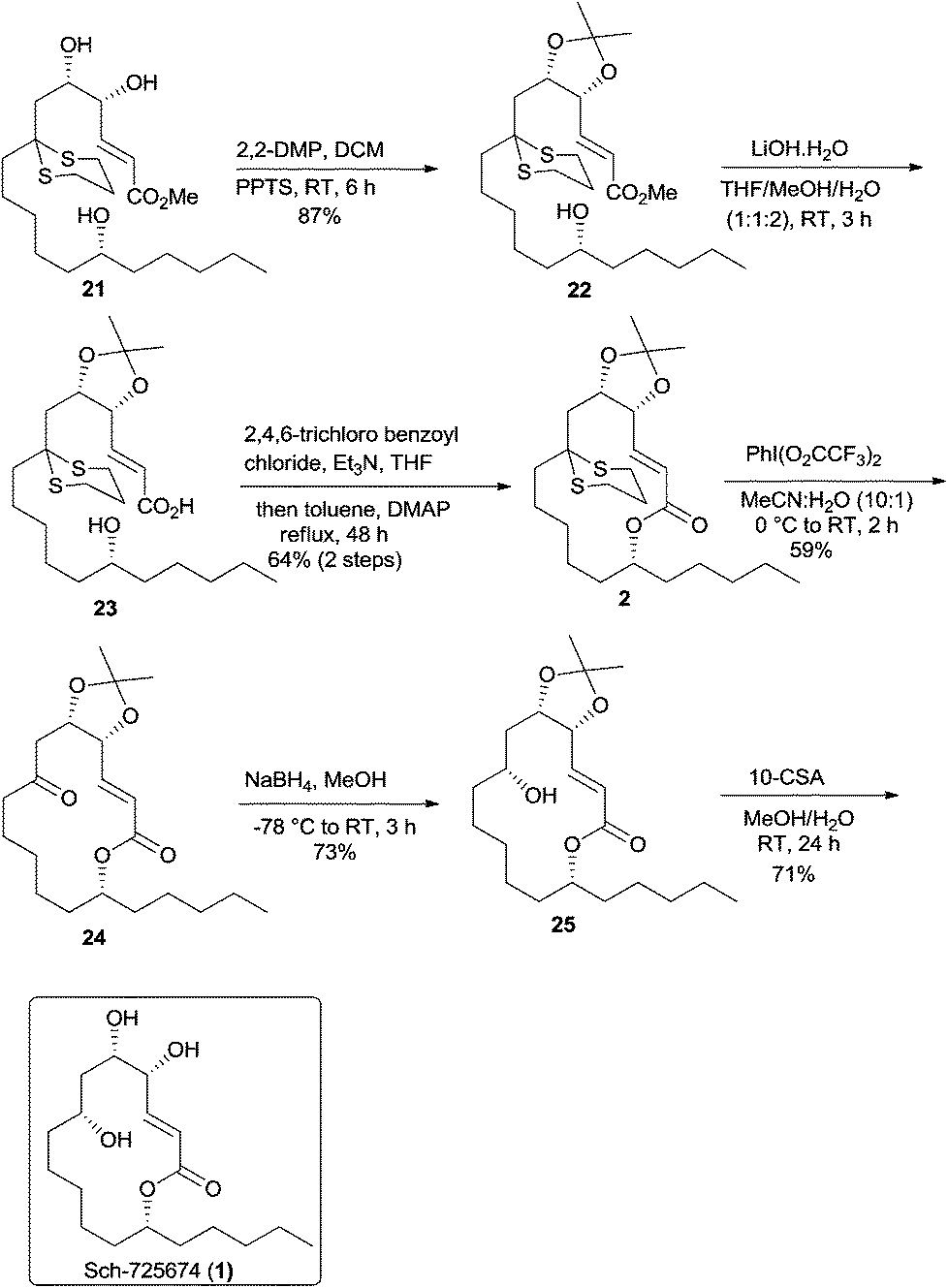 | ||
| Scheme 6 Total synthesis of Sch-725674 (1). | ||
Conclusion
In summary, we have accomplished an enantioselective total synthesis of Sch-725674 (1) in 13 steps from the known alcohol 9. The salient features of our strategy include dithiane alkylation, cross metathesis, Yamaguchi macrolactonization and a substrate controlled stereoselective reduction as key steps.Experimental section
General methods
Unless and otherwise noted, all starting materials and reagents were obtained from commercial suppliers and used after further purification. Tetrahydrofuran was distilled from sodium benzophenone ketyl and toluene from sodium. Dichloromethane, hexanes and HMPA were freshly distilled from calcium hydride. All solvents for routine isolation of products and chromatography were of reagent grade and glass distilled. Reaction flasks were dried in an oven at 100 °C for 12 h. Air and moisture sensitive reactions were performed under an argon/UHP nitrogen atmosphere. Chromatography was performed using silica gel (100–200 mesh, Aceme) with indicated solvents. All reactions were monitored by thin-layer chromatography carried out on 0.25 mm E. Merck silica plates (60F-254) using UV light as the visualizing agent, and charring solution (prepared by dropwise addition of conc. H2SO4 (5 mL) to a solution of phosphomolybdic acid (1 g) and ceric sulphate (2 g) in water (95 mL)), an alkaline KMnO4 solution (prepared by dissolving KMnO4 (2 g) and NaHCO3 (4 g) in water (100 mL)), and heat as developing agents. Optical rotation was recorded on an Autopol IV automatic polarimeter. IR spectra were recorded on a Thermo Nicolet Avater 320 FT-IR and a Nicolet Impact 400 machine. Mass spectra were recorded on a Waters Micromass-Q-Tof micro™ (YA105) spectrometer. 1H and 13C NMR spectra were recorded either on a Bruker AVANCE 400 MHz or a Bruker AVANCE HD 500 MHz. NMR data are of the order of chemical shifts, multiplicity (s, singlet; br s, broad singlet; d, doublet; t, triplet; q, quartet; qn, quintet; m, multiplet), coupling constant in hertz (Hz) and number of protons.:1:2) was added LiOH·H2O (116 mg, 2.76 mmol, 5 eq.) in one portion at 0 °C and stirred at RT for 3 h. MeOH and THF were removed in vacuo and extracted an aqueous layer with Et2O. The aqueous layer was acidified with a 10% aq. citric acid solution (∼5 mL) at 0 °C and extracted with EtOAc (2 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to get the crude acid 23, which was used in the next step without any further purification. Rf = 0.1 (40% EtOAc in hexanes); [α]20D +19.2 (c 0.32, CHCl3); IR (neat): ν = 3019, 2933, 2859, 1701, 1661, 1424, 1216, 1046, 929, 669 cm−1; 1H NMR (400 MHz, CDCl3): δ 6.95 (dd, J = 15.6, 6.4 Hz, 1H), 6.08 (dd, J = 15.6, 1.3 Hz, 1H), 4.71 (td, J = 6.4, 1.3 Hz, 1H), 4.61 (td, J = 7.1, 3.3 Hz, 1H), 3.60–3.58 (m, 1H), 2.83–2.71 (m, 4H), 2.50–2.20 (brm, 5H), 2.13 (dd, J = 15.3, 3.4 Hz, 2H), 2.00 (dd, J = 15.3, 7.1 Hz, 1H), 1.96–1.89 (m, 3H), 1.51 (s, 3H), 1.43–1.41 (m, 5H), 1.38 (s, 3H), 1.35–1.24 (m, 6H), 0.88 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 169.1, 146.2, 122.9, 109.1, 77.8, 75.3, 72.3, 52.6, 39.4, 38.5, 37.5, 37.4, 32.1, 29.9, 28.1, 26.4, 26.3, 25.7, 25.6, 25.5, 25.3, 24.4, 22.8, 14.2; HRMS (ESI): m/z calcd for C24H42O5S2Na (M + Na)+: 497.2371; observed: 497.2392.
:1) was added PhI(O2CCF3)2 (236 mg, 0.55 mmol, 2.5 eq.) at 0 °C and stirred at RT for 2 h. The reaction mixture was diluted with EtOAc (5 mL) and washed successively with water (5 mL) and brine (5 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to get the crude product, which upon purification by silica gel column chromatography with 8% EtOAc in hexanes as an eluent afforded the ketoester 24 (43 mg) as a colorless liquid in 59% yield. Rf = 0.20 (10% EtOAc in hexanes); [α]20D −20.14 (c 0.42, CHCl3); IR (neat): ν = 2931, 2860, 1717, 1658, 1460, 1378, 1300, 1271, 1244, 1222, 1162, 1112, 1039, 984, 874, 798 cm−1; 1H NMR (400 MHz, CDCl3): δ 6.61 (dd, J = 15.6, 6.7 Hz, 1H), 6.08 (dd, J = 15.6, 1.0 Hz, 1H), 5.01–4.94 (m, 1H), 4.83 (td, J = 6.7, 1.0 Hz, 1H), 4.77–4.72 (m, 1H), 2.86 (dd, J = 18.9, 10.9 Hz, 1H), 2.68 (dd, J = 18.9, 2.6 Hz, 1H), 2.49–2.42 (m, 1H), 2.19–2.13 (m, 1H), 1.70–1.58 (m, 4H), 1.49 (s, 3H), 1.47–1.39 (m, 2H), 1.36 (s, 3H), 1.32–1.20 (m, 10H), 0.87 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 208.9, 165.8, 140.6, 125.1, 108.9, 76.7, 75.9, 74.3, 45.9, 41.8, 34.5, 31.9, 31.8, 28.8, 28.0, 25.5, 25.3, 24.5, 24.2, 22.7, 14.2; HRMS (ESI): m/z calcd for C21H34O5Na (M + Na)+: 389.2298; observed: 389.2302.
:1) was added 10-CSA (16 mg, 0.06 mmol, 2 eq.) in one portion at RT and stirred for 24 h. The reaction mixture was diluted with EtOAc (5 mL) and washed successively with sat. aq. NaHCO3 (5 mL) and brine (5 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to get the crude product, which upon purification by silica gel column chromatography with 50% EtOAc in hexanes as an eluent afforded Sch-725674 (1) (7 mg) as an amorphous white solid in 71% yield. Rf = 0.10 (50% EtOAc in hexanes). M.P: 179–185 °C; [α]20D +4.91 (c 0.26, CHCl3)*; IR (KBr): ν = 3454, 3267, 2929, 2859, 1713, 1460, 1273, 1043, 892 cm−1; 1H NMR (400 MHz, MeOD): δ 6.87 (dd, J = 15.8, 6.1 Hz, 1H), 6.08 (dd, J = 15.8, 1.2 Hz, 1H), 4.98–4.94 (m, 1H), 4.51–4.46 (m, 1H), 3.99 (qn, J = 6.1 Hz, 1H), 3.86–3.83 (m, 1H), 1.84 (dt, J = 14.7, 6.0 Hz, 1H), 1.73–1.51 (m, 5H), 1.39–1.25 (m, 11H), 1.23–1.13 (m, 3H), 0.90 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, MeOD): δ 168.4, 149.3, 123.1, 77.7, 76.0, 72.8, 69.5, 38.3, 36.8, 36.5, 34.1, 32.9, 29.5, 27.0, 26.4, 25.7, 23.7, 14.5; HRMS (ESI): m/z calcd for C18H33O5 (M + H)+: 329.2328; observed: 329.2323. *Reported in the literature [α]25D +5.15 (c 0.27, CHCl3).
Acknowledgements
The financial support from DST, New Delhi is gratefully acknowledged. KR thanks the Council of Scientific and Industrial Research for a fellowship. IIT Bombay is thanked for providing the necessary infrastructure and high end equipment facilities.Notes and references
- (a) S. Õmura, in Macrolide Antibiotics: Chemistry, Biology and Practice, ed. S. Õmura, Academic Press, Orlando, 1984, p. 509 Search PubMed; (b) I. Paterson and M. M. Mansuri, Tetrahedron, 1985, 41, 3569 CrossRef CAS; (c) N. Tadashi, in Macrolide Antibiotics: Chemistry, Biology and Practice, ed. S. Õmura, Academic Press, San Diego, 2002, 2nd edn, p. 181 Search PubMed.
- S. W. Yang, T. M. Chan, J. Terracciano, D. Loebenberg, M. Patel and M. Chu, J. Antibiot., 2005, 58, 535 CrossRef CAS PubMed.
- J. D. Moretti, X. Wang and D. P. Curran, J. Am. Chem. Soc., 2012, 134, 7963 CrossRef CAS PubMed.
- (a) A. K. Bali, S. K. Sunnam and K. R. Prasad, Org. Lett., 2014, 16, 4001 CrossRef CAS PubMed; (b) S. K. Sunnam and K. R. Prasad, Tetrahedron, 2014, 70, 2096 CrossRef CAS PubMed.
- (a) E. J. Corey and D. Seebach, Angew. Chem., Int. Ed. Engl., 1965, 4, 1075 CrossRef CAS; (b) B.-T. Gröbel and D. Seebach, Synthesis, 1977, 357 CrossRef.
- L. F. Tietze, H. Geissler, J. A. Gewert and U. Jakobi, Synlett, 1994, 511 CrossRef CAS PubMed.
- (a) A. B. Smith III, S. M. Condon and J. A. McCauley, Acc. Chem. Res., 1998, 31, 35 CrossRef; (b) A. B. Smith III and C. M. Adams, Acc. Chem. Res., 2004, 37, 365 CrossRef PubMed; (c) A. F. Petri, A. Bayer and M. E. Maier, Angew. Chem., Int. Ed., 2004, 43, 5821 CrossRef CAS PubMed.
- (a) K. P. Kaliappan and D. Si, Synlett, 2009, 2441 CrossRef CAS PubMed; (b) P. Gowrisankar, S. A. Pujari and K. P. Kaliappan, Chem. – Eur. J., 2010, 16, 5858 CrossRef CAS PubMed; (c) D. Si, N. M. Sekar and K. P. Kaliappan, Org. Biomol. Chem., 2011, 9, 6988 RSC; (d) D. Si and K. P. Kaliappan, Synlett, 2012, 2822 CAS.
- P. Kubizna, I. Špánik, J. Kožíšek and P. Szolcsányi, Tetrahedron, 2010, 66, 2351 CrossRef CAS PubMed.
- H. Takahata, Y. Yotsui and T. Momose, Tetrahedron, 1998, 54, 13505 CrossRef CAS.
- A. S. B. Prasad, J. V. B. Kanth and M. Periasamy, Tetrahedron, 1992, 48, 4623 CrossRef CAS.
- A. Romero and C.-H. Wong, J. Org. Chem., 2000, 65, 8264 CrossRef CAS PubMed.
- K. J. Quinn, L. Islamaj, S. M. Couvertier, K. E. Shanley and B. L. Mackinson, Eur. J. Org. Chem., 2010, 5943 CrossRef CAS.
- A. B. Smith III, G. K. Friestad, J. Barbosa, E. Bertounesque, K. G. Hull, M. Iwashima, Y. Qiu, B. A. Salvatore, P. G. Spoors and J. J.-W. Duan, J. Am. Chem. Soc., 1999, 121, 10468 CrossRef.
- J. L.-Y. Chen and M. A. Brimble, J. Org. Chem., 2011, 76, 9417 CrossRef CAS PubMed.
- A. K. Ghosh and Y. Wang, J. Am. Chem. Soc., 2000, 122, 11027 CrossRef CAS.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360 CrossRef CAS PubMed.
- (a) J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989 CrossRef CAS; (b) For a review on macrolactonizations in the total synthesis of natural products see: A. Parenty, X. Moreau, G. Niel and J.-M. Campagne, Chem. Rev., 2013, 113, PR1 CrossRef CAS PubMed.
- G. Stork and K. Zhao, Tetrahedron Lett., 1989, 30, 287 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02136c |
| This journal is © The Royal Society of Chemistry 2015 |