
Preparation of cycloheptane ring by nucleophilic cyclopropanation of 1,2-diketones with bis(iodozincio)methane†
Ryosuke
Haraguchi
,
Yoshiaki
Takada
and
Seijiro
Matsubara
*
Department of Material Chemistry, Graduate School of Engineering, Kyoto University, Kyotodaigaku-Katsura, Nishikyo, Kyoto 615-8510, Japan. E-mail: matsubara.seijiro.2e@kyoto-u.ac.jp; Fax: +81(75)3832438
First published on 20th October 2014
Abstract
The nucleophilic cyclopropanation of hexa-1,5-diene-3,4-diones with bis(iodozincio)methane afforded the Zn alkoxides of cis-dialkenylcyclopropane-1,2-diols stereoselectively. The subsequent oxy-Cope rearrangement afforded the corresponding Zn alkoxides of 5,6-dialkylcyclohepta-3,7-diene-1,3-diols.
Introduction
The Cope rearrangement of cis-divinylcyclopropanes has been recognized as an efficient route to synthesize cycloheptane rings. The disadvantageous entropic factor for a seven-membered ring construction is overcome by the close proximity of both ends caused by the rigid configuration of cyclopropane.1 The difficulty in the stereoselective preparation of the cis-isomer of the substrate, however, often makes the transformation less valuable. Although some practical methods have been developed for the preparation of the cis-isomers,2 most of the methods afforded the trans-isomers, which required a temperature of >100 °C to perform the Cope rearrangement.3 Thus, a direct route to synthesize cis-isomers stereoselectively is desirable in order to construct cycloheptane rings easily. During our studies on bis(iodozincio)methane (1),4 we found that the nucleophilic cyclopropanation of 1,2-diketones afforded cis-cyclopropane-1,2-diols stereoselectively.5 The mechanism of the reaction was elucidated by a computational method; the cis-selectivity was attributed to the face-to-face coordination of 1 with the diketones.6 We envisioned that the reaction of 1,6-dialkylhexa-1,5-diene-3,4-diones 2 with 1 would afford the Zn alkoxides of cis-divinylcyclopropane-1,2-diols 4, via face-to-face coordination 3, thus facilitating the oxy-Cope rearrangement of 4 to 5, with additional acceleration by the alkoxide groups (Scheme 1).7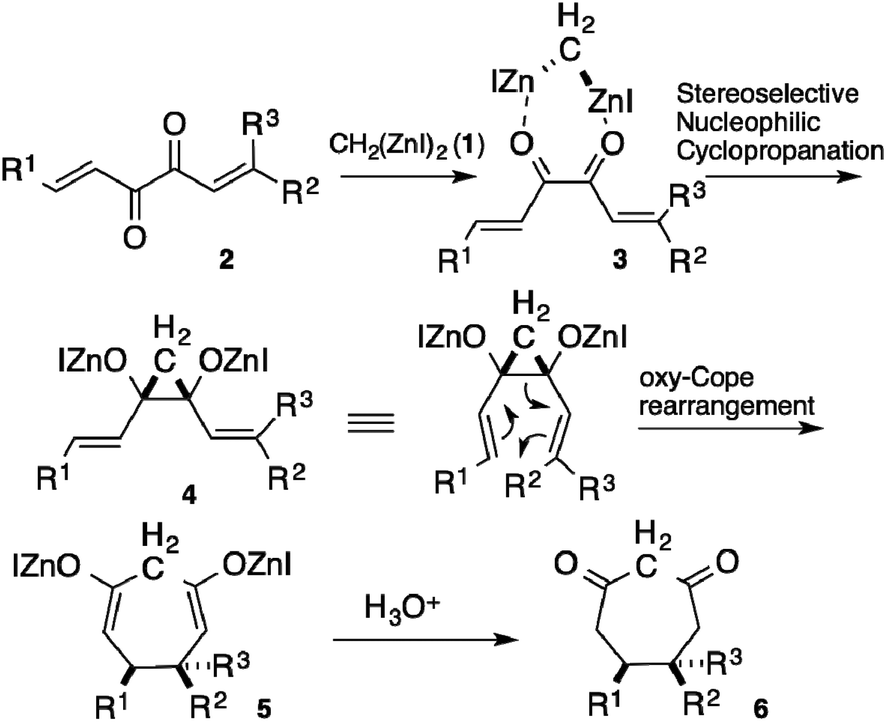 | ||
| Scheme 1 Synthesis of cycloheptane ring 5 by nucleophilic cyclopropanation of 1,2-diketone 2. | ||
Results and discussion
As a one-pot reaction
The reaction of (1E,5E)-1,6-diphenylhexa-1,5-diene-3,4-dione (R1, R2 = Ph, R3 = H, 2a) with dizinc 1 at −20 °C, however, afforded a complex mixture, even though it contained a small amount of the desired cycloheptane-1,3-dione 6a after the hydrolysis. The main byproduct was the adduct of an enolate 5a with the substrate 2a. This result indicates that the first reaction, i.e., the cyclopropanation of 2a with 1 should be completed before the start of Cope rearrangement to prevent side reactions of the rearranged product 5 with substrate 2. For this purpose, we reacted diketone 2 with 1 at the lower temperatures, which do not allow Cope-rearrangement, for an appropriate period, until the complete conversion of 2; the resulting mixture was warmed up to promote the subsequent Cope-rearrangement. In fact, the reaction of 2a with 1 for 3 h at −78 °C, followed by warming up the resulting mixture to 25 °C afforded the seven-membered ring 6a in 78% yield.8 Moreover, instead of the simple heating to 25 °C, the addition of THF (25 °C) to the reaction mixture afforded 6a in 84% yield, because the dilution suppressed the intermolecular side reactions without affecting the rate of the intramolecular rearrangement reaction. Some examples of the preparation of cycloheptane-1,3-diones are shown in Table 1. Various cycloheptane-1,3-diones substituted with two cis-aryl groups 6 were prepared and isolated in good yields (Table 1, entries 1–4). The presence of an electron-withdrawing group on the benzene ring resulted in a low yield (entry 5). The presence of a bulky group such as 1-naphthyl also resulted in a low yield (entry 7). The presence of alkyl groups as the substituents (R1, R2, and R3) did not hinder the reaction (Table 1, entries 8–11). These transformations were stereospecific. As shown in entries 8 and 9, the cis- and trans-isomers were obtained specifically depending on the E,Z-configuration of the substrate.|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 | 6 (yield %)b,c | |||
| R1 | R2 | R3 | |||
| a The reaction was performed with the following scale: 1 (1.2 mmol, 0.35 M THF solution), 2 (1.0 mmol, 0.35 M in THF). After 3 h at −78 °C, 10 mL of THF (25 °C) were added in one portion. b Isolated yields. c The diastereomer was not detected. | |||||
| 1 | 2a | Ph | Ph | H | 6a (84%) |
| 2 | 2b | 4-Me–C6H4 | 4-Me–C6H4 | H | 6b (93%) |
| 3 | 2c | 4-MeO–C6H4 | 4-MeO–C6H4 | H | 6c (98%) |
| 4 | 2d | 4-t-Bu-C6H4 | 4-t-Bu-C6H4 | H | 6d (96%) |
| 5 | 2e | 4-F–C6H4 | 4-F–C6H4 | H | 6e (47%) |
| 6 | 2f | 2-Furyl | 2-Furyl | H | 6f (78%) |
| 7 | 2g | 1-Naphthyl | 1-Naphthyl | H | 6g (41%) |
| 8 | 2h | Me | Me | H | 6h (99%) |
| 9 | 2i | Me | H | Me | 6i (65%) |
| 10 | 2j | Me | Ph | H | 6j (88%) |
| 11 | 2k | Me | Me | Me | 6k (86%) |
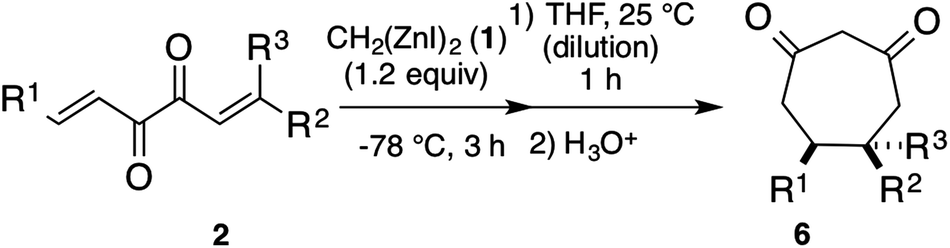
The Zn-enolate intermediate 5 in Scheme 1 was able to be trapped with chlorotrimethylsilane (TMSCl) or acetic anhydride (Ac2O). As shown in Scheme 2, after the treatment of 2h with dizinc 1 at −78 °C for 3 h and at 25 °C for 1 h with additional THF, TMSCl was added. The corresponding silyl enol ether 7 was obtained in 96% isolated yield. Instead of TMSCl, the addition of Ac2O afforded the corresponding enol acetate 8 in 82% isolated yield.
 | ||
| Scheme 2 Trapping of Zn-enolate intermediate from 2h with electrophiles. | ||
In a microflow reactor
This [6 + 1] transformation contains two reactions, that is, the nucleophilic cyclopropanation of 1,2-diketone and the oxy-Cope rearrangement of cis-1,2-divinylcyclopropane-1,2-diol. As the activation energy for the first step is smaller than the second one, the first reaction can be completed before the second reaction proceeds at −78 °C. Otherwise, the formed zinc enolate 5 reacts with the diketone 2. Therefore, careful temperature control made the entire transformation proceed reasonably to obtain the 7-membered product in good yields. In other words, the two reactions were separated by the reaction temperature.Instead of temperature control, it is possible to separate the two reactions with space. The microflow system (space integration)9 may improve the problem arising from a premature start of the second reaction, because it can supply a minimum amount of the substrate to be consumed at the micromixer spontaneously.10 Thus, as shown in Fig. 1, we constructed a microflow system consisting of two T-shaped SUS micromixers (M1 and M2, Φ = 0.5 mm) and SUS microtube reactors (R1, Φ = 1.0 mm).11 A THF solution of 1 (0.16 M, 3.92 mL min−1) and a THF or CH2Cl2 solution of 1,2-diketone (0.09 M, 3.92 mL min−1) were introduced by a syringe pump; after passage through reactor R1, the enolate 5 and the excess amount of 1 were quenched with methanol in M2. The residence time was optimized by varying the length of the microtube reactor (see the ESI†). In the flow system, a residence time of 6 seconds (1 m length, Φ = 1.0 mm, SUS microtube reactor (R1)) afforded the products in good yields continuously. In this case, the residence time of the reaction mixture of 1 and 2 in R1 was 6 s. The period was calculated from the flow rate (3.92 × 2 mL min−1) and the inner volume of R1. The results are summarized in Table 2.
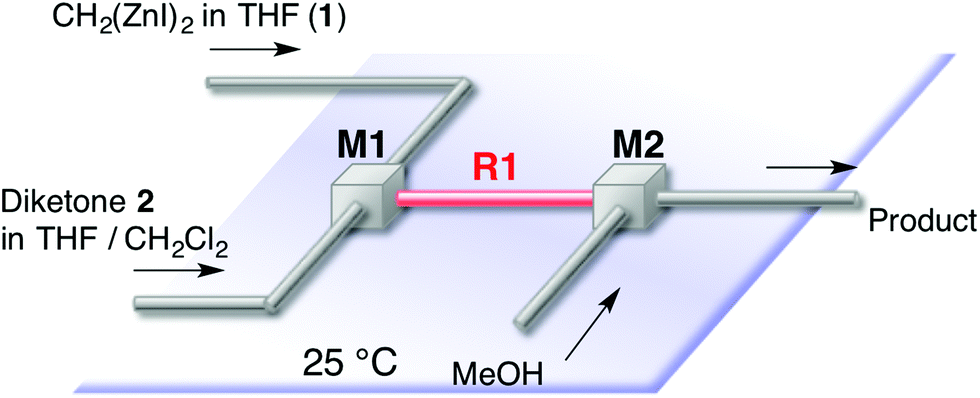 | ||
| Fig. 1 Microflow system for the preparation of cycloheptane-1,3-dione. | ||
| Entry | 2 | 6 (yield %)b,c | |||
|---|---|---|---|---|---|
| R1 | R2 | R3 | |||
| a The reaction was performed using the microflow system shown in Fig. 1: T-shaped SUS micromixer: M1 (inner diameter: 0.5 mm) and M2 (inner diameter: 0.5 mm), SUS microtube reactor: R1 (Φ) = 1.0 mm, length = 1 m), a solution of 1: 3.92 mL min−1, 0.16 M; a solution of 2 in CH2Cl2: 3.92 mL min−1, 0.09 M; methanol: 7.25 mL min−1. b THF was used as the solvent for the solution of 2 instead of CH2Cl2. c The diastereomer was not observed. | |||||
| 1 | 2a | Ph | Ph | H | 6a (>99%) |
| 2 | 2b | 4-Me–C6H4 | 4-Me–C6H4 | H | 6b (>99%) |
| 3 | 2c | 4-MeO–C6H4 | 4-MeO–C6H4 | H | 6c (92%) |
| 4 | 2d | 4-t-Bu-C6H4 | 4-t-Bu-C6H4 | H | 6d (81%) |
| 5b | 2e | 4-F–C6H4 | 4-F–C6H4 | H | 6e (82%) |
| 6b | 2f | 2-Furyl | 2-Furyl | H | 6f (77%) |
| 7 | 2h | Me | Me | H | 6h (70%) |
As shown in Table 2, the products were obtained in reasonable yields at 25 °C for 6 s continuously. Except entries 5 and 6, dichloromethane was used as the solvent to prepare the solution of diketones 2, because the corresponding diketones except 2e and 2f were not very soluble in THF. Notably, the microflow system allowed us to use dichloromethane as a cosolvent for the reactions using a fairly basic dizinc reagent 1. Moreover, dichloromethane is difficult to use as a cosolvent in a batch reaction, because the monomeric structure of dizinc 1 in THF is changed into the polymethylenezinc form through the Schlenk equilibrium by the addition of any other less polar solvent such as dichloromethane. The polymeric structure often loses nucleophilicity.12
In the microflow system shown in Fig. 1, Zn-enolate 5 and an excess amount of dizinc 1 was protonated with methanol in M2. Subsequently, instead of protonation, the resulting reaction mixture viaR1 was introduced into a THF solution of ketones 9a–c as shown in Scheme 3. Although dienolate 5 was treated with an excess amount of ketone, 5 reacted with only one molar equivalent of ketone to afford the corresponding aldol adducts 10a–c diastereoselectively.13
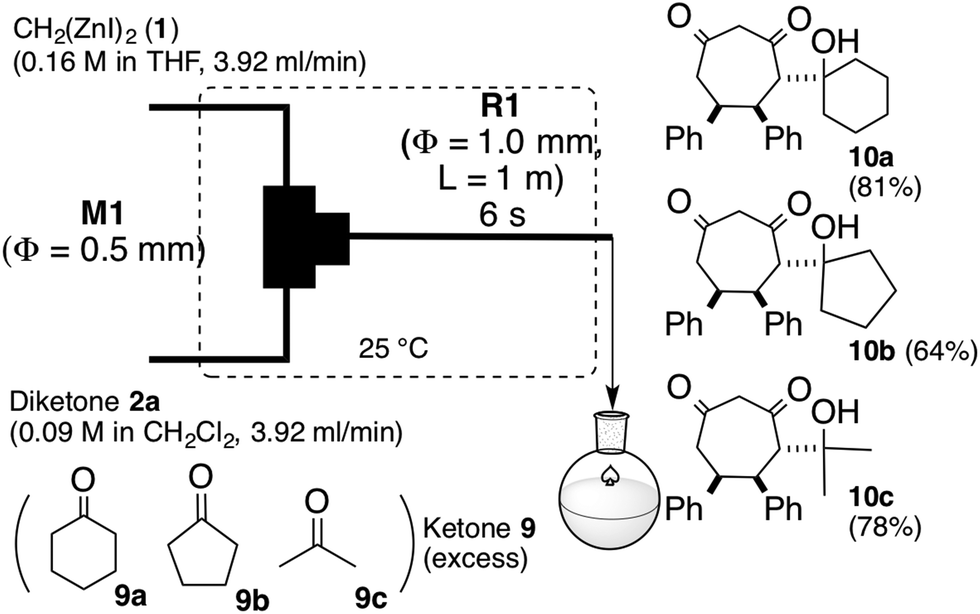 | ||
| Scheme 3 Reaction of zinc-enolate intermediate 5a with ketones 9. | ||
Conclusion
In conclusion, the reaction of bis(iodozincio)methane 1 with divinyl-1,2-diketones 2 afforded cycloheptane-1,3-diones 6 efficiently via a reactive cis-divinylcyclopropane derivative as the key intermediate. Bis(iodozincio)methane was found to be a unique reagent for performing a nucleophilic cyclopropanation reaction with vicinal electrophiles such as 1,2-diketone, and affords reactive cyclopropanol derivatives efficiently.14 Although classical batch reactions required careful temperature control to suppress side-reactions of the product with the starting substrate, the microflow system removed the reactive product from the reaction site continuously, thus improving the yield of the desired product.Experimental section
Nuclear magnetic resonance spectra were taken on Varian UNITY INOVA 500 (1H, 500 MHz; 13C, 125.7 MHz) spectrometer using tetramethylsilane for 1H NMR as an internal standard (δ = 0 ppm), and CDCl3 for 13C NMR as an internal standard (δ = 77.0 ppm). 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constants (Hz), and integration. High-resolution mass spectra were obtained with a Thermo Fisher SCIENTIFIC EXACTIVE (ESI, APCI). Infrared (IR) spectra were determined on a SHIMADZU FTIR-8200PC spectrometer. Melting points were determined using a YANAKO MP-500D. TLC analyses were performed by means of Merck Kieselgel 60 F254 (0.25 mm) Plates. Visualization was accomplished with UV light (254 nm) and an aqueous vanillin solution followed by heating. Flash column chromatography was carried out using Kanto Chemical silica gel (spherical, 40–100 μm).Unless otherwise noted, commercially available reagents were used without purification. Tetrahydrofuran, Dehydrated stabilizer free – Super – was purchased from Kanto Chemical Co., stored under argon, and used as is. Zinc powder was used after washing with 10% HCl according to the reported procedure.15
Preparation of bis(iodozincio)methane (1)
A mixture of pure zinc dust (150 mmol), diiodomethane (1.0 mmol), and PbCl2 (0.005 mmol) in THF (5.0 mL) was sonicated for 1 h in an ultrasonic cleaning bath under Ar. When pyrometallurgy zinc dust was used instead of pure zinc, it was not necessary to add PbCl2. Both pure zinc and pyrometallurgy zinc are commercially available. Diiodomethane (50 mmol) in THF (45 mL) was added dropwise to the mixture over 30 min at 0 °C with vigorous stirring. The mixture was then stirred for 4 h at 25 °C. After the stirring was stopped, the reaction vessel was allowed to stand undisturbed for several hours. Excess zinc was separated by sedimentation. 1H NMR spectra of the obtained supernatant showed a broad singlet at −1.2 ppm at 0 °C, which corresponded to the methylene proton of 1. The supernatant was used in further reactions as a solution of 1 in THF (0.1–0.5 M). The concentration of 1 was estimated by 1H NMR analysis using 2,2,3,3-tetramethylbutane as an internal standard. Bis(iodozincio)methane in THF can be kept for at least a month in a sealed reaction vessel.(1E,5E)-1,6-Diphenylhexa-1,5-diene-3,4-dione (2a) (CAS RN [126201-33-0])
Yellow solid. m.p. 163.2–164.6 °C. 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 16.5 Hz, 2H), 7.69–7.64 (m, 4H), 7.48 (d, J = 16.5 Hz, 2H), 7.46–7.41 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 189.1, 147.8, 134.5, 131.3, 129.04, 128.95, 119.7. IR (KBr) 1706, 1669, 1607, 1595, 1574, 1449, 1032, 1001, 988, 755, 722, 698, 688, 557, 434 cm−1.(1E,5E)-1,6-Bis(4-methylphenyl)hexa-1,5-diene-3,4-dione (2b) (CAS RN [263249-11-2])
Yellow solid. m.p. 182.5–185.3 °C. 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 16.5 Hz, 2H), 7.55 (d, J = 8.0 Hz, 4H), 7.41 (d, J = 16.0 Hz, 2H), 7.32 (d, J = 8.0 Hz, 4H), 2.40 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 189.5, 147.9, 142.1, 131.8, 129.8, 129.0, 118.9, 21.6. IR (KBr) 1669, 1595, 1560, 1512, 1307, 1328, 1295, 1183, 993, 806, 684, 428 cm−1.(1E,5E)-1,6-Bis(4-methoxyphenyl)hexa-1,5-diene-3,4-dione (2c) (CAS RN [263249-10-1])
Orange solid. m.p. 167.0–171.6 °C. 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 16.0 Hz, 2H), 7.62 (dt, J = 9.0, 2.5 Hz, 4H), 7.33 (d, J = 16.0 Hz, 2H), 6.94 (dt, J = 9.0, 2.5 Hz, 4H), 3.86 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 189.6, 162.4, 147.5, 130.9, 127.3, 117.7, 114.5, 55.5. IR (KBr) 1680, 1671, 1593, 1568, 1509, 1421, 1282, 1253, 1175, 1030, 996, 816, 790, 552 cm−1.(1E,5E)-1,6-Bis(4-tert-butylphenyl)hexa-1,5-diene-3,4-dione (2d)
Yellow solid. m.p. 164.8–166.2 °C. 1H NMR (500 MHz, CDCl3) δ 7.85 (d, J = 16.0 Hz, 2H), 7.62–7.58 (m, 4H), 7.46–7.43 (m, 4H), 7.43 (d, J = 16.0 Hz, 2H), 1.34 (s, 18H). 13C NMR (125 MHz, CDCl3) δ 189.5, 155.2, 147.8, 131.8, 128.9, 126.1, 119.1, 35.1, 31.1. IR (KBr) 2960, 1669, 1592, 1560, 993, 822, 746, 646 cm−1. HRMS Calcd for C26H30O2: M+ 374.2246. Found: m/z 374.2248.(1E,5E)-1,6-Bis(4-fluorophenyl)hexa-1,5-diene-3,4-dione (2e)
Yellow solid. m.p. 184.7–187.0 °C. 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 16.5 Hz, 2H), 7.68–7.64 (m, 4H), 7.43 (d, J = 16.5 Hz, 2H), 7.15–7.10 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 188.6, 164.6 (d, J = 253.4 Hz), 146.4, 131.0 (d, J = 8.7 Hz), 130.7, 119.2, 116.3 (d, J = 22.1 Hz). IR (KBr) 1675, 1617, 1599, 1589, 1507, 1245, 1161, 995, 815, 695, 523, 440 cm−1. HRMS Calcd for C18H12F2O2: M+ 298.0805. Found: m/z 298.0805.(1E,5E)-1,6-Difurylhexa-1,5-diene-3,4-dione (2f) (CAS RN [1190209-38-1])
Brown solid. m.p. 156.3–158.9 °C. 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 15.5 Hz, 2H), 7.57 (ddd, J = 1.5, 0.5, 0.5 Hz, 2H), 7.31 (d, J = 15.5 Hz, 2H), 6.80 (ddd, J = 3.5, 0.5, 0.5 Hz, 2H), 6.53 (dd, J = 3.5, 1.5 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 188.6, 151.5, 146.0, 133.0, 117.6, 117.4, 113.0. IR (KBr) 1666, 1592, 1548, 1267, 999, 750, 641 cm−1.(1E,5E)-1,6-Di(naphthalen-1-yl)hexa-1,5-diene-3,4-dione (2g) (CAS RN [1192343-59-1])
Orange solid. m.p. 184.3–185.7 °C. 1H NMR (500 MHz, CDCl3) d 8.82 (d, J = 16.0 Hz, 2H), 8.29 (d, J = 8.5 Hz, 2H), 8.01 (d, J = 6.5 Hz, 2H), 7.97 (d, J = 8.5 Hz, 2H), 7.93–7.89 (m, 2H), 7.67 (d, J = 16.0 Hz, 2H), 7.65–7.61 (m, 2H), 7.59–7.53 (m, 4H). 13C NMR (125 MHz, CDCl3) d 188.8, 144.4, 133.8, 131.85, 131.79, 131.57, 128.9, 127.3, 126.4, 125.8, 125.5, 123.2, 121.9. IR (KBr) 1669, 1594, 1585, 1569, 1347, 998, 982, 799, 770, 689, 592 cm−1.(2E,6E)-Octa-2,6-diene-4,5-dione (2h) (CAS RN [55409-19-3])
Yellow solid. m.p. 43.6–46.0 °C. 1H NMR (500 MHz, CDCl3) δ 7.09 (dq, J = 7.0, 19.5 Hz, 2H), 6.61 (dq, J = 16.0, 1.5 Hz, 2H), 1.98 (dd, J = 7.0, 1.5 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 190.5, 149.5, 126.5, 19.0; IR (KBr) 1670, 1598, 1437, 1284, 1024, 993, 966, 895, 738, 684 cm−1.(2Z,6E)-Octa-2,6-diene-4,5-dione (2i)
Orange liquid. Rf: 0.61 (hexane–EtOAc = 3/1). 1H NMR (500 MHz, CDCl3) δ 7.15 (dq, J = 16.0, 7.0 Hz, 1H), 6.82 (dq, J = 11.5, 1.5 Hz, 1H), 6.77 (dq, J = 16.0, 2.0 Hz, 1H), 6.60 (dq, J = 15.5, 7.5 Hz, 1H), 2.21 (dd, J = 7.5, 1.5 Hz, 3H), 1.99 (dd, J = 7.0, 2.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 190.2, 188.9, 149.4, 149.0, 125.1, 122.0, 19.0, 16.6. IR (neat) 2983, 2941, 1717, 1447. 1377, 1260, 1046, 971 cm−1. HRMS Calcd for C8H10O2: M+ 138.0681. Found: m/z 138.0681.(1E,5E)-1-Phenylhepta-1,5-diene-3,4-dione (2j)
Orange solid. m.p. 68.5–71.8 °C. 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 16.0 Hz, 1H), 7.64–7.61 (m, 2H), 7.46–7.39 (m, 3H), 7.33 (d, J = 16.0 Hz, 1H), 7.19 (dq, J = 15.5, 6.5 Hz, 1H), 6.77 (dq, J = 15.5, 1.5 Hz, 1H), 2.01 (dd, J = 6.5, 1.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 189.8, 189.6, 149.6, 147.7, 134.3, 131.3, 129.0, 128.9, 126.0, 120.1, 19.1. IR (KBr) 2968, 2936, 1689, 1597, 1449, 1206, 974, 698 cm−1. HRMS Calcd for C13H12O2: M+ 200.0837. Found: m/z 200.0842.(E)-2-Methylocta-2,6-diene-4,5-dione (2k)
Orange liquid. Rf 0.71 (hexane–EtOAc = 3/1). 1H NMR (500 MHz, CDCl3) δ 7.13 (dq, J = 16.0, 7.0 Hz, 1H), 6.78 (dq, J = 16.0, 1.5 Hz, 1H), 6.72 (qq, J = 1.5, 1.0 Hz, 1H), 2.26 (d, J = 1.0 Hz, 3H), 2.01 (d, J = 1.5 Hz, 3H), 1.98 (dd, J = 7.0, 1.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 189.7, 189.2, 163.1, 148.6, 125.3, 118.1, 28.5, 21.6, 19.0. IR (neat) 2977, 2941, 2915, 1674, 1619, 1442, 1377, 1312, 1010, 972, 743 cm−1. HRMS Calcd for C9H12O2: M+ 152.0837. Found: m/z 152.0836.Preparation of (5R*,6S*)-5,6-diphenylcycloheptane-1,3-dione (6a) (CAS RN [222629-92-7]) (general procedure for one-pot synthesis of 6)
To a solution of diketone 2a (1.0 mmol) in THF (5.0 mL), dizinc 1 (1.2 mmol, 0.35 M in THF) was added dropwise at −78 °C under Ar. After being stirred for 3 h at −78 °C, the mixture was diluted with THF (10 mL, 25 °C). The resulting mixture was stirred at 25 °C for 1 h, and poured into sat. NH4Claq. (10 mL). The resulting mixture was extracted with ethyl acetate. The combined organic layers were washed with brine and dried over sodium sulfate. Purification by silica gel column chromatography (hexane–ethyl acetate) gave the title compound in 84% yield (233 mg).White solid. m.p. 106.7–112.8 °C. 1H NMR (500 MHz, CDCl3) δ 7.18–7.10 (m, 6H), 6.74–6.69 (m, 4H), 3.95 (d, J = 17.5 Hz, 1H), 3.74–3.67 (m, 2H), 3.57 (d, J = 17.5 Hz, 1H), 3.22 (dd, J = 15.5, 11.0 Hz, 2H), 2.79 (dd, J = 15.5, 5.0 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 204.5, 139.3, 128.2, 128.1, 127.1, 58.2, 46.2, 45.8. IR (KBr) 3060, 3030, 2969, 2889, 1710, 1600, 1491, 1452, 1383, 1284, 1255, 1240, 1220, 1133, 1076, 806, 755, 738, 712, 537, 462 cm−1.
(5R*,6S*)-5,6-Bis(4-methylphenyl)cycloheptane-1,3-dione (6b)
Yellow solid. m.p.129.2–131.3 °C. 1H NMR (500 MHz, CDCl3) δ 6.45 (d, J = 8.0 Hz, 4H), 6.62 (d, J = 8.0 Hz, 4H), 3.95 (d, J = 17.0 Hz, 1H), 3.69–3.62 (m, 2H), 3.55 (d, J = 17.0 Hz, 1H), 3.17 (dd, J = 15.5, 11.0 Hz, 2H), 2.76 (dd, J = 15.0, 5.0 Hz, 2H), 2.27 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 204.7, 136.6, 136.3, 128.8, 128.2, 58.3, 46.1, 45.8, 20.9. IR (KBr) 3024, 2954, 1715, 1513, 1248, 1196, 1019, 806, 480 cm−1. HRMS Calcd for C21H22O2: M+ 306.1620. Found: m/z 306.1625.(5R*,6S*)-5,6-Bis(4-methoxyphenyl)cycloheptane-1,3-dione (6c)
Yellow solid. m.p. 105.1–106.2 °C. 1H NMR (500 MHz, CDCl3) δ 6.71–6.67 (m, 4H), 6.66–6.62 (m, 4H), 3.93 (d, J = 17.0 Hz, 1H), 3.75 (s, 6H), 3.65–3.59 (m, 1H), 3.54 (d, J = 17.0 Hz, 1H), 3.14 (d, J = 15.0, 11.0 Hz, 2H), 2.75 (dd, J = 15.0, 5.5 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 204.7, 158.4, 131.4, 129.2, 113.4, 58.2, 55.1, 46.1, 45.5. IR (KBr) 2958, 1697, 1611, 1511, 1250, 1178, 1028, 839, 779, 541 cm−1. HRMS Calcd for C21H22O4: M+ 338.1518. Found: m/z 338.1521.(5R*,6S*)-5,6-Bis(4-fluorophenyl)cycloheptane-1,3-dione (6d)
White solid. m.p. 165.7–169.8 °C. 1H NMR (500 MHz, CDCl3) δ 6.88–6.83 (m, 4H), 6.71–6.66 (m, 4H), 3.92 (d, J = 17.0 Hz, 1H), 3.70–3.63 (m, 2H), 3.57 (d, J = 17.0 Hz, 1H), 3.14 (dd, J = 15.0, 11.0 Hz, 2H), 2.77 (dd, J = 15.0, 5.0 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 204.0, 161.9 (d, J = 246.6 Hz), 134.8, 129.7 (d, J = 8.2 Hz), 115.4 (d, J = 21.1 Hz), 58.1, 45.8, 45.5. IR (KBr) 1722, 1696, 1603, 1511, 1224, 1159, 843, 812, 789, 514 cm−1. HRMS Calcd for C19H16F2O2: M+ 314.1118. Found: m/z 314.1115.(5R*,6S*)-5,6-Di(naphthalen-1-yl)cycloheptane-1,3-dione (6e)
White solid. m.p. 67.5–69.8 °C. 1H NMR (500 MHz, CDCl3) δ 7.69–7.65 (m, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 7.33–7.29 (m, 2H), 7.11–7.03 (m, 4H), 6.80–6.75 (m, 2H), 4.88–4.81 (m, 2H), 4.15 (d, J = 17.5 Hz, 1H), 3.73 (d, J = 17.5 Hz, 1H), 3.52 (dd, J = 15.5, 11.5 Hz, 2H), 2.89 (dd, J = 15.5, 5.0 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 205.0, 135.0, 133.5, 131.9, 128.7, 127.6, 125.9, 125.3, 124.51, 124.45, 121.9, 58.5, 46.3, 38.2. IR (KBr) 3050, 2926, 2364, 2343, 1696, 1598, 1507, 1396, 1259, 778 cm−1. HRMS Calcd for C27H22O2: M+ 378.1620. Found: m/z 378.1621.(5R*,6S*)-5,6-Di(furan-2-yl)cycloheptane-1,3-dione (6f)
Yellow solid. m.p. 85.5–87.6 °C. 1H NMR (500 MHz, CDCl3) δ 7.31 (dd, J = 2.0, 1.0 Hz, 2H), 6.23 (dd, J = 3.0, 2.0 Hz, 2H), 5.84 (ddd, J = 3.0, 1.0, 1.0 Hz, 2H), 3.93–3.88 (m, 2H), 3.89 (d, J = 15.0 Hz, 1H), 3.72 (d, J = 15.0 Hz, 1H), 3.05 (dd, J = 15.0, 9.5 Hz, 2H), 2.91 (dd, J = 15.0, 4.5 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 201.8, 153.4, 141.9, 110.2, 106.9, 59.9, 45.5, 38.9. IR (KBr) 3150, 3119, 1719, 1503, 1206, 1006, 942, 748 cm−1. HRMS Calcd for C15H14O4: M+ 258.0892. Found: m/z 258.0891.(5R*,6S*)-5,6-Bis(4-tert-butylphenyl)cycloheptane-1,3-dione (6g)
White solid. m.p. 125.1–126.8 °C. 1H NMR (500 MHz, CDCl3) δ 7.13–7.09 (m, 4H), 6.64–6.60 (m, 4H), 3.95 (d, J = 17.0 Hz, 1H), 3.69–3.63 (m, 2H), 3.56 (d, J = 17.0 Hz, 1H), 3.19 (dd, J = 15.0, 11.0 Hz, 2H), 2.78 (dd, J = 15.0, 4.5 Hz, 2H), 1.25 (s, 18H). 13C NMR (125 MHz, CDCl3) δ 204.7, 150.0, 136.4, 127.8, 124.8, 58.4, 45.9, 45.7, 34.3, 31.3. IR (KBr) 2961, 2360, 1730, 1700, 1509, 853 cm−1. HRMS Calcd for C27H34O2: M+ 390.2559. Found: m/z 390.2552.(5R*,6S*)-5,6-Dimethylcycloheptane-1,3-dione (6h)
Colorless liquid. Rf 0.33 (hexane–EtOAc = 3/1). 1H NMR (500 MHz, CDCl3) δ 3.62 (d, J = 15.5 Hz, 1H), 3.35 (d, J = 15.5 Hz, 1H), 2.51 (dd, J = 14.0, 4.5 Hz, 2H), 2.47 (dd, J = 14.0, 8.5 Hz, 2H), 2.33–2.24 (m, 2H), 0.97 (d, J = 6.5 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 204.4, 59.7, 48.7, 34.9, 16.5. IR (neat) 2964, 2364, 1701, 1254, 1199, 1099 cm−1. HRMS Calcd for C9H14O2: M+ 154.0994. Found: m/z 154.0998.(5S*,6S*)-5,6-Dimethylcycloheptane-1,3-dione (6i)
Colorless liquid. Rf 0.29 (hexane–EtOAc = 3/1). 1H NMR (500 MHz, CDCl3) δ 3.43 (s, 2H), 2.59 (dd, J = 14.0, 3.0 Hz, 2H), 2.44 (dd, J = 14.0, 8.5 Hz, 2H), 1.87–1.77 (m, 2H), 1.09 (d, J = 6.5 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 205.4, 59.1, 50.0, 38.0, 20.7. IR (neat) 2963, 2360, 1698, 1611, 1459, 1388, 1252 cm−1. HRMS Calcd for C9H14O2: M+ 154.0994. Found: m/z 154.0990.(5S*,6R*)-5-Methyl-6-phenylcycloheptane-1,3-dione (6j)
Colorless liquid. Rf 0.28 (hexane–EtOAc = 3/1). 1H NMR (500 MHz, CDCl3) δ 7.35–7.31 (m, 2H), 7.28–7.23 (m, 1H), 7.15–7.11 (m, 2H), 3.72 (d, J = 17.0 Hz, 1H), 3.51 (ddd, J = 12.5, 4.0, 4.0 Hz, 1H), 3.49 (d, J = 17.0 Hz, 1H), 3.17 (dd, J = 14.0, 12.5 Hz, 1H), 2.75 (dd, J = 13.5, 4.5 Hz, 1H), 2.72 (dd, J = 14.0, 4.0 Hz, 1H), 2.56 (dd, J = 13.5, 9.0 Hz, 1H), 2.52–2.44 (m, 1H), 0.81 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 204.9, 204.3, 141.6, 128.6, 127.6, 127.0, 59.0, 49.9, 45.9, 45.9, 44.3, 35.5. IR (neat) 2963, 2360, 1716, 1695, 1452, 1387, 1257, 1202, 1149, 744, 703 cm−1. HRMS Calcd for C14H16O2: M+ 216.1150. Found: m/z 216.1145.5,5,6-Trimethylcycloheptane-1,3-dione (6k)
Colorless liquid. Rf 0.36 (hexane–EtOAc = 3/1). 1H NMR (500 MHz, CDCl3) δ 3.47 (d, J = 17.5 Hz, 1H), 3.33 (d, J = 17.5 Hz, 1H), 2.60 (dd, J = 16.0, 11.5 Hz, 1H), 2.51 (d, J = 12.0 Hz, 1H), 2.44 (dd, J = 16.0, 3.0 Hz, 1H), 2.32 (d, J = 12.0 Hz, 1H), 2.03 (ddq, J = 11.5, 7.0, 3.0 Hz, 1H), 1.09 (s, 3H), 1.01 (d, J = 7.0 Hz, 3H), 0.94 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 206.3, 204.3, 58.5, 56.3, 47.7, 39.7, 37.3, 28.5, 22.3, 16.5. IR (neat) 2966, 2362, 1695, 1469, 1395, 1304, 1267, 1196, 1135 cm−1. HRMS Calcd for C10H16O2: M+ 168.1150. Found: m/z 168.1147.Preparation of ((5R*,6S*)-5,6-dimethylcyclohepta-3,7-diene-1,3-diyl)bis(oxy)bis(trimethylsilane) (7)
To a solution of diketone 2a (1.0 mmol) in THF (1.0 mL), dizinc 1 (1.2 mmol, 0.35 M in THF) was added dropwise at −78 °C under Ar. After being stirred for 3 h at −78 °C, the mixture was diluted with THF (10 mL, room temperature). The resulting mixture was stirred at 25 °C for 1 h. To the resulting mixture, chlorotrimethylsilane (2.4 mmol) was added at 0 °C. The resulting mixture was stirred for 1 h at 25 °C, and poured into sat. NH4Claq. (10 mL). The resulting mixture was extracted with ethyl acetate. The combined organic layers were washed with brine and dried over sodium sulfate. Purification by silica gel column chromatography (hexane–ethyl acetate) gave the title compound in 96% yield. Instead of treatment of the reaction mixture with chlorotrimethylsilane, an addition of acetic anhydride gave 8.Colorless liquid. Rf 0.56 (hexane). 1H NMR (500 MHz, CDCl3) δ 4.76 (dd, J = 6.0, 2.0 Hz, 2H), 3.28 (dtt, J = 19.5, 2.0,2.0 Hz, 1H), 2.44 (d, J = 19.5 Hz, 1H), 2.42–2.35 (m, 2H), 0.948 (d, J = 7.0 Hz, 6H), 0.18 (s, 18H). 13C NMR (125 MHz, CDCl3) δ 146.8, 113.7, 40.6, 34.6, 17.5, 0.2. IR (neat) 2961, 2874, 1669, 1253, 1198, 1173, 1147, 962, 846, 752 cm−1. HRMS Calcd for C15H30O2Si2: M+ 298.1784. Found: m/z 298.1782.
(5R*,6S*)-5,6-Dimethylcyclohepta-3,7-diene-1,3-diyl diacetate (8)
Colorless liquid. Rf 0.53 (hexane–EtOAc = 3/1). 1H NMR (500 MHz, CDCl3) δ 5.30 (dd, J = 6.5, 2.0 Hz, 2H), 3.74 (dtt, J = 19.5, 2.0, 2.0 Hz, 1H), 2.65–2.55 (m, 2H), 2.55 (d, J = 19.5 Hz, 1H), 2.01 (s, 6H), 1.04 (d, J = 7.0 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 169.5, 143.7, 123.0, 35.1, 34.2, 20.9, 16.7. IR (neat) 2967, 2876, 1755, 1693, 1370, 1225, 1134, 1092, 1061, 1020, 900 cm−1. HRMS Calcd for C13H18O4: M+ 238.1205. Found: m/z 238.1215.Reaction in a microreactor
Stainless steel (SUS304) T-shaped micromixer with an inner diameter of 0.5 mm was manufactured by Sanko Seiki Co., Inc. Stainless steel (SUS316) microtube reactor with an inner diameter of 1.0 mm was purchased from GL Sciences and was cut to the appropriate length (R1 = 1 m). The micromixers and microtube reactors were connected with stainless steel fittings (GL Sciences, 1/16 OUW) to construct the flow microreactor in the laboratory. The flow microreactor was dipped in the bath to control the temperature. Solutions were continuously introduced to the flow microreactor using syringe pumps, Harvard Model 11. After a steady state was reached, the product solution was collected for 90 s. When the collection time was longer, the product solution could be obtained in a preparative scale. As shown in Fig. 1, a flow microreactor consisting of two T-shaped micromixers (M1 and M2, Φ = 0.5 mm) and one microtube reactor (R1, Φ = 1.0 mm) was used. To M1, bis(iodozincio)methane (1, 0.16 M solution in THF) was introduced by a syringe pump with a rate of 3.92 mL min−1. The syringe pump and M1 were connected with a Teflon tube (Φ = 1.0 mm). To M1, a solution of 2 (0.09 M solution in dichloromethane for 2a–d,h and in THF for 2e,f) was introduced by a syringe pump with a rate of 3.92 mL min−1. The syringe pump and M1 were also connected with a Teflon tube (Φ = 1.0 mm). The mixture was passed through R1 (Φ = 1.0 mm) and was mixed with methanol (7.84 mL min−1) in M2. The resulting mixture was poured into 1 M HClaq.. The product was extracted with ethyl acetate. The combined organic layers were washed with brine and dried over sodium sulfate. Purification by silica gel column chromatography (hexane–ethyl acetate) gave the cycloheptane derivatives 6.Instead of quenching, the product flow from R1 was poured into a solution of a ketone (3.0 mmol) in THF (10 mL) at 25 °C. The addition from R1 was continued for 3 min (the amount of the substrate was 1.06 mmol (0.09 M × 3.92 mL min−1 × 3 min)). After the addition, the resulting mixture was stirred for 30 min, and poured into sat. NH4Claq. (10 mL). The resulting mixture was extracted with ethyl acetate. The combined organic layers were washed with brine and dried over sodium sulfate. Purification by silica gel column chromatography (hexane–ethyl acetate) gave the adduct 10.
Preparation of (5S*,6S*)-4-(1-hydroxycyclohexyl)-5,6-diphenylcycloheptane-1,3-dione (10a)
Yellow solid. m.p. 175.2–175.8 °C. 1H NMR (CDCl3) δ 7.11–7.19 (m, 6H), 6.70 (d, J = 6.5 Hz, 2H), 6.64–6.65 (m, 2H), 3.81 (m, 4H), 3.60 (d, J = 17.5 Hz, 1H), 3.37 (dd, J = 9.0, 2.0 Hz, 1H), 3.11 (dd, J = 17.5, 15.0 Hz, 1H), 2.61 (dd, J = 17.5, 4.0 Hz 1H), 1.63–1.72 (m, 2H) 1.24–1.46 (m, 4H), 1.15 (d, J = 16 Hz, 1H), 0.98–1.07 (m, 2H) 0.47 (t, 11.5 Hz 1H). 13C NMR (CDCl3) δ 211.3, 205.3, 140.6, 137.7, 129.5, 128.0, 127.9, 127.8, 127.0, 126.8, 77.2, 73.8, 60.9, 49.3, 43.8, 43.8, 37.5, 35.9, 25.4, 21.8, 21.1. IR (KBr) 3510.6, 2960.9, 2938.7, 2852.8, 1692.6, 1495.9, 1457.3, 1398.5, 1380.1, 1359.9, 1282.7, 1175.7, 1156.4, 1139.0, 1076.3, 982.8, 845.8, 765.8, 708.9. cm−1. HRMS (ESI) Calcd for C25H28O3Cl: [M + Cl]−, 411.1721. Found: m/z 411.1736.(5S*,6S*)-4-(1-Hydroxycyclopentyl)-5,6-diphenylcycloheptane-1,3-dione (10b)
Yellow solid. m.p. 185.0–185.5 °C. 1H NMR (CDCl3) δ 7.10–7.16 (m, 6H), 6.73 (d, J = 6.5 Hz, 2H), 6.66–6.64 (m, 2H), 3.89 (dd, J = 9.5, 4.0 Hz, 1H), 3.77–3.84 (m, 2H), 3.59 (dd, J = 17.5, 1.0 Hz, 1H), 3.46 (d, J = 2.5 Hz, 1H), 3.28 (dd, J = 9.0, 2.0 Hz, 1H) 3.21 (ddd, J = 17.5, 15.5, 1.0 Hz, 1H), 2.64 (ddd, J = 17.5, 4.0, 1.0 Hz, 1H), 1.84–1.89 (m, 1H) 1.71–1.78 (m, 1H) 1.24–1.59 (m, 4H) 0.93–0.97 (m, 1H) 0.41–0.47 (m, 1H). 13C NMR (CDCl3) δ 210.5, 205.6, 140.6, 137.6, 129.6, 128.0, 127.9, 127.8, 127.0, 127.0, 82.7, 77.2, 61.7, 59.5, 50.4, 43.9, 41.0, 38.3, 23.4, 22.3. IR (KBr) 3525.1, 2974.4, 2924.2, 2869.2, 1714.8, 1693.6, 1493.9, 1456.3, 1382.1, 1266.3, 1456.3, 1382.1, 1266.3, 1232.6, 1139.0, 1096.6, 1003.0, 767.7, 708.9. cm−1. HRMS (ESI) Calcd for C24H26O3Cl: [M + Cl]−, 397.1565. Found: m/z 397.1579.(5S*,6S*)-4-(2-Hydroxypropan-2-yl)-5,6-diphenylcycloheptane-1,3-dione (10c)
Yellow solid. m.p. 111.2–112.0 °C. 1H NMR (CDCl3) δ 7.11–7.18 (m, 6H), 6.67 (d, J = 7.0 Hz, 2H), 6.65–6.63 (m, 2H), 3.86–3.80 (m, 3H), 3.75 (dd, J = 9.0, 3.5 Hz, 1H), 3.61 (dd, J = 18.0, 1.0 Hz, 1H), 3.35 (dd, J = 9.0, 2.0 Hz, 1H), 3.14 (ddd, J = 18.0, 14.0, 1.0 Hz, 1H), 2.62 (ddd, J = 18.0, 3.5, 1.0 Hz, 1H) 1.20 (s, 3H) 0.64 (s, 3H). 13C NMR (CDCl3) δ 210.9, 205.3, 140.5, 137.7, 129.4, 128.0, 128.0, 127.9, 127.1, 126.9, 72.4, 61.2, 60.4, 50.2, 43.9, 43.8, 30.2, 28.5. IR (KBr) 3500.6, 2974.4, 1721.5, 1689.7, 1495.9, 1455.4, 1392.7, 1380.1, 1362.8, 1235.5, 1197.9, 1158.3, 1136.1, 1095.6, 1078.3, 958.7, 767.7, 704.1. cm−1. HRMS (ESI) Calcd for C22H24O3Cl: [M + Cl]−, 371.1408. Found: m/z 371.1422.Notes and references
- H. M. L. Davies, Tetrahedron, 1993, 49, 5203 CrossRef CAS; D. A. Foley and A. R. Maguire, Tetrahedron, 2010, 66, 1131 CrossRef PubMed; G. A. Molander, Acc. Chem. Res., 1998, 31, 603 CrossRef; Y. Ni and J. Montgomery, J. Am. Chem. Soc., 2004, 126, 11162 CrossRef PubMed.
- H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed; K. Takeda, M. Takeda, A. Nakajima and E. Yoshii, J. Am. Chem. Soc., 1995, 117, 6400 CrossRef; V. Vuligonda, M. E. Garst and R. A. S. Chandraratna, Bioorg. Med. Chem. Lett., 1999, 9, 589 CrossRef; N. J. Wallock and W. A. Donaldson, Org. Lett., 2005, 7, 2047 CrossRef PubMed; S. Pantke-Böcker, G. Pohnert, I. Fischer-Lui, W. Boland and A. F. Peters, Tetrahedron, 1991, 51, 7927 CrossRef; J. P. Olson and H. M. L. Davies, Org. Lett., 2008, 10, 573 CrossRef PubMed.
- P. A. Wender and M. P. Filosa, J. Org. Chem., 1976, 41, 3490 CrossRef CAS; H. M. L. Davies, T. J. Clark and H. D. Smith, J. Org. Chem., 1991, 56, 3817 CrossRef; H. M. Davies and D. B. Doan, J. Org. Chem., 1999, 64, 8501 CrossRef; J. E. Baldwin and K. E. Gilbert, J. Am. Chem. Soc., 1976, 98, 8283 CrossRef.
- M. Sada, S. Komagawa, M. Uchiyama, M. Kobata, T. Mizuno, K. Utimoto, K. Oshima and S. Matsubara, J. Am. Chem. Soc., 2010, 132, 17452 CrossRef CAS PubMed; M. Sada and S. Matsubara, J. Am. Chem. Soc., 2010, 132, 432 CrossRef PubMed; R. Haraguchi and S. Matsubara, Org. Lett., 2013, 15, 3378 CrossRef PubMed.
- K. Ukai, K. Oshima and S. Matsubara, J. Am. Chem. Soc., 2000, 122, 12047 CrossRef CAS; K. Nomura, K. Asano, T. Kurahashi and S. Matsubara, Heterocycles, 2008, 76, 1381 CrossRef PubMed.
- S. Matsubara, K. Ukai, H. Fushimi, Y. Yokota, H. Yoshino, K. Oshima, K. Omoto, A. Ogawa, Y. Hioki and H. Fujimoto, Tetrahedron, 2002, 58, 8255 CrossRef CAS.
- D. A. Evans and A. M. Golob, J. Am. Chem. Soc., 1975, 97, 4765 CrossRef CAS.
- Y. Takada, K. Nomura and S. Matsubara, Org. Lett., 2010, 12, 5204 CrossRef CAS PubMed.
- S. Suga, D. Yamada and J. Yoshida, Chem. Lett., 2010, 39, 404 CrossRef CAS; J. Yoshida, K. Saito, T. Nokami and A. Nagaki, Synlett, 2011, 1189 CrossRef PubMed.
- R. Haraguchi, Y. Takada and S. Matsubara, Chem. Lett., 2012, 41, 628 CrossRef CAS.
- H. Wakami and J. Yoshida, Org. Proc. Res. Dev., 2005, 9, 787 CrossRef CAS.
- S. Matsubara, H. Yoshino, Y. Yamamoto, K. Oshima, H. Matsuoka, K. Matsumoto, K. Ishikawa and E. Matsubara, J. Organomet. Chem., 2005, 690, 5546 CrossRef CAS PubMed.
- The structure of 10a was determined by single crystal X-ray analysis (see ref. 10). The structures of the other products 10b,c were determined by analogy to the structure of 10a.
- K. Nomura and S. Matsubara, Chem. – Eur. J., 2010, 16, 703 CrossRef CAS PubMed; K. Nomura and S. Matsubara, Chem. – Asian J., 2010, 5, 147 CrossRef PubMed.
- L. F. Fieser and M. Fieser, Reagents for Organic Synthesis, Wiley, New York, 1967, vol. 1, p. 1276 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob01474j |
| This journal is © The Royal Society of Chemistry 2015 |