
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systematic methodology for the development of biocatalytic hydrogen-borrowing cascades: application to the synthesis of chiral α-substituted carboxylic acids from α-substituted α,β-unsaturated aldehydes†
Tanja
Knaus‡
 a,
Francesco G.
Mutti‡
b,
Luke D.
Humphreys
c,
Nicholas J.
Turner
b and
Nigel S.
Scrutton
*a
a,
Francesco G.
Mutti‡
b,
Luke D.
Humphreys
c,
Nicholas J.
Turner
b and
Nigel S.
Scrutton
*a
aManchester Institute of Biotechnology, Faculty of Life Sciences, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: nigel.scrutton@manchester.ac.uk
bManchester Institute of Biotechnology, School of Chemistry, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK
cGSK Medicines Research Centre, Gunnel's Wood Road, Stevenage, Herts, SG1 2NY, UK
First published on 29th October 2014
Abstract
Ene-reductases (ERs) are flavin dependent enzymes that catalyze the asymmetric reduction of activated carbon–carbon double bonds. In particular, α,β-unsaturated carbonyl compounds (e.g. enals and enones) as well as nitroalkenes are rapidly reduced. Conversely, α,β-unsaturated esters are poorly accepted substrates whereas free carboxylic acids are not converted at all. The only exceptions are α,β-unsaturated diacids, diesters as well as esters bearing an electron-withdrawing group in α- or β-position. Here, we present an alternative approach that has a general applicability for directly obtaining diverse chiral α-substituted carboxylic acids. This approach combines two enzyme classes, namely ERs and aldehyde dehydrogenases (Ald-DHs), in a concurrent reductive-oxidative biocatalytic cascade. This strategy has several advantages as the starting material is an α-substituted α,β-unsaturated aldehyde, a class of compounds extremely reactive for the reduction of the alkene moiety. Furthermore no external hydride source from a sacrificial substrate (e.g. glucose, formate) is required since the hydride for the first reductive step is liberated in the second oxidative step. Such a process is defined as a hydrogen-borrowing cascade. This methodology has wide applicability as it was successfully applied to the synthesis of chiral substituted hydrocinnamic acids, aliphatic acids, heterocycles and even acetylated amino acids with elevated yield, chemo- and stereo-selectivity. A systematic methodology for optimizing the hydrogen-borrowing two-enzyme synthesis of α-chiral substituted carboxylic acids was developed. This systematic methodology has general applicability for the development of diverse hydrogen-borrowing processes that possess the highest atom efficiency and the lowest environmental impact.
Introduction
Nowadays, there is an urgent demand for new chemical reactions and processes that possess an elevated atom efficiency as well as a low environmental impact.1–3 Multi-step chemical reactions using enzymes one pot allow this goal to be achieved as intermediate isolation and purification steps are avoided and energy consumption is minimized.4,5 The major challenge is to perform cascade reactions wherein an oxidative and a reductive step are running simultaneously without any compartmentalization.6 One of the early examples of two-step concurrent oxidative-reductive enzymatic cascades was the deracemization and the stereoinversion of secondary alcohols using stereocomplementary alcohol dehydrogenases (ADHs).7 However, this cascade was operated by four enzymes constituting two redox independent steps (i.e. non-interconnected); as a consequence, redox equivalents were supplied at the expense of formate and molecular oxygen as sacrificial co-substrates, hence generating hydrogen peroxide and carbon dioxide as waste. A similar concept was applied to the two-step oxidative-reductive combination of an enzyme with an artificial metal-enzyme or a metal catalyst.8,9 In contrast, a two-step redox self-sufficient biocatalytic network has been recently presented for the amination of primary alcohols.10 In this case, the redox equivalents liberated in the first oxidative step were consumed in the second reductive step in the form of NAD(P)H. As the cofactor acts as a shuttle of hydride within the catalytic cycle, such a process is also defined as a hydrogen-borrowing cascade. Another example is biocatalytic redox isomerisation of allylic alcohols.11 Recently, a hydrogen-borrowing biocatalytic synthesis of α-substituted carboxylic acids from α-substituted α,β-unsaturated aldehydes was presented.12 However, the two-step cascade was run using crude preparations of the enzymes (cell extracts) and thereby efficient internal recycling of the nicotinamide cofactor was not demonstrated as other enzymes may contribute to the overall process. Furthermore, a general strategy for setting up a hydrogen-borrowing biocatalytic cascade has not been presented until to date. Thus, in this work, we decided to study a systematic approach for developing a successful biocatalytic hydrogen-borrowing cascade. The asymmetric hydrogen-borrowing biocatalytic synthesis of α-substituted carboxylic acids from α-substituted α,β-unsaturated aldehydes was chosen as the case study. The development of such a cascade is not trivial since the enzymes involved in the reductive and in the oxidative steps have to operate concomitantly with high activity and stability, and under the same reaction conditions (T, pH, type of cofactor, type of buffer, cosolvent, etc.). Moreover, enzyme concentrations and kinetics have to be thoroughly studied in order to maximize chemo- and stereoselectivity.Ene-reductases (ERs) from the “Old Yellow Enzyme Family” (OYEs) are flavin dependent enzymes that, in the last decade, have been applied extensively in biocatalysis.13–16 These enzymes catalyze the asymmetric reduction of carbon–carbon double bonds that are activated by conjugation with an electron-withdrawing substituent. In particular, α,β-unsaturated carbonylic compounds (e.g. enals and enones) as well as nitroalkenes are rapidly reduced by the ERs, affording quantitative yields in most cases.17,18,19–22 In contrast, alkenes bearing a single conjugated ester moiety or a free carboxylic group are poorly reduced or not converted at all.23–25 Reduction of acid derivatives is restricted to only a few families such as α,β-unsaturated, 1–2 substituted diesters or diacids (e.g. fumarate, maleate, citraconate, mesaconate) or β-cyano-, β-alkoxy- and β-aryloxy-α,β-unsaturated esters.26–30 More recently, it has been shown that an electron-withdrawing halogen group in the α-position can significantly increase the reactivity of α,β-unsaturated esters towards bio-reduction.31–33 The requirement of an additional activating group, significantly narrows the substrate scope for the biocatalytic asymmetric reduction of α,β-unsaturated esters, whereas the same reaction on α,β-unsaturated acids is completely inapplicable. This is a potential limitation given that most developed synthetic routes to pharmaceuticals and agrochemicals require optically active α-substituted carboxylic acids as intermediates. Therefore, synthetic routes involving an ER would require a further hydrolytic step to obtain the carboxylic moiety from the related ester. Crucially, the direct reduction of activated α,β-unsaturated esters requires an external source of hydride that is provided by the NAD(P)H cofactor. Most of the recent publications report the reduction of activated α,β-unsaturated esters using suprastoichiometric NAD(P)H or catalytic NAD(P)H in presence of glucose as the sacrificial substrate for cofactor regeneration. Nevertheless, the use of catalytic amounts of cofactor with GDH/glucose, somehow, worsened conversion and stereoselectivity. Additionally, the asymmetric carbon–carbon double bond reduction of some classes of α,β-unsaturated esters is not applicable. For instance, α-substituted cinnamic acid methyl esters bearing an additional α-cyano moiety decomposed or polymerized under the reaction conditions for the enzymatic reduction.33 In this work, we conceived an alternative approach that has general applicability to directly access optically active α-substituted carboxylic acids (Scheme 1). The approach combines two enzyme classes, namely ERs and aldehyde dehydrogenases (Ald-DHs), in a concurrent reductive-oxidative biocatalytic cascade.6
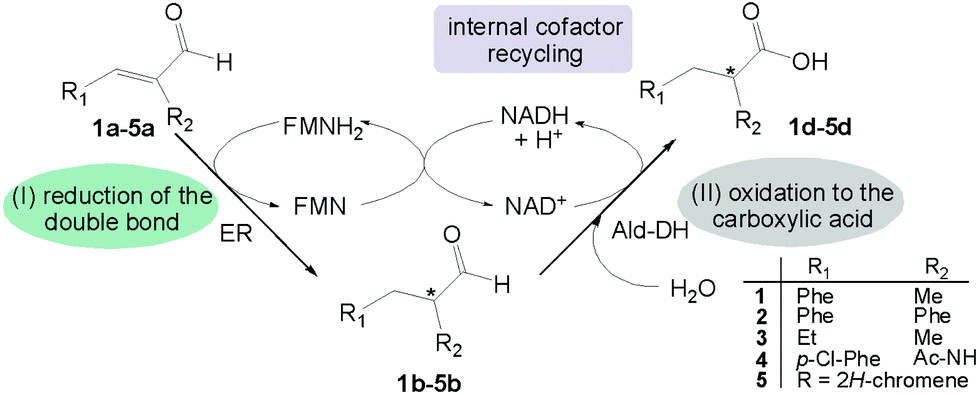 | ||
| Scheme 1 Concurrent redox self-sufficient two enzyme cascade reaction. The redox equivalents required in the first reductive step are provided by the second oxidative step in form of NAD(P)H. | ||
This strategy has several advantages: (I) the starting material is an α-substituted α,β-unsaturated aldehyde, a class of compounds extremely reactive for the carbon–carbon double bond reduction; (II) no external source of hydride from a sacrificial substrate (e.g. glucose, formate) is required; (III) the cascade reaction has the highest atom efficiency as the only additional reagent is water and no waste is produced; (IV) the process takes advantage of the intrinsic stereoselectivity of the ER to enable transfer of the hydride predominantly onto one of the prochiral faces of the alkene moiety.
This challenging two enzyme hydrogen-borrowing cascade was developed following the systematic strategy as depicted in Chart 1. Finally, the potential utility of this cascade sequence was demonstrated in the production of diverse important intermediates for the chemical industry such as chiral substituted hydrocinnamic acids, aliphatic acids, heterocycles and even acetylated amino acids.
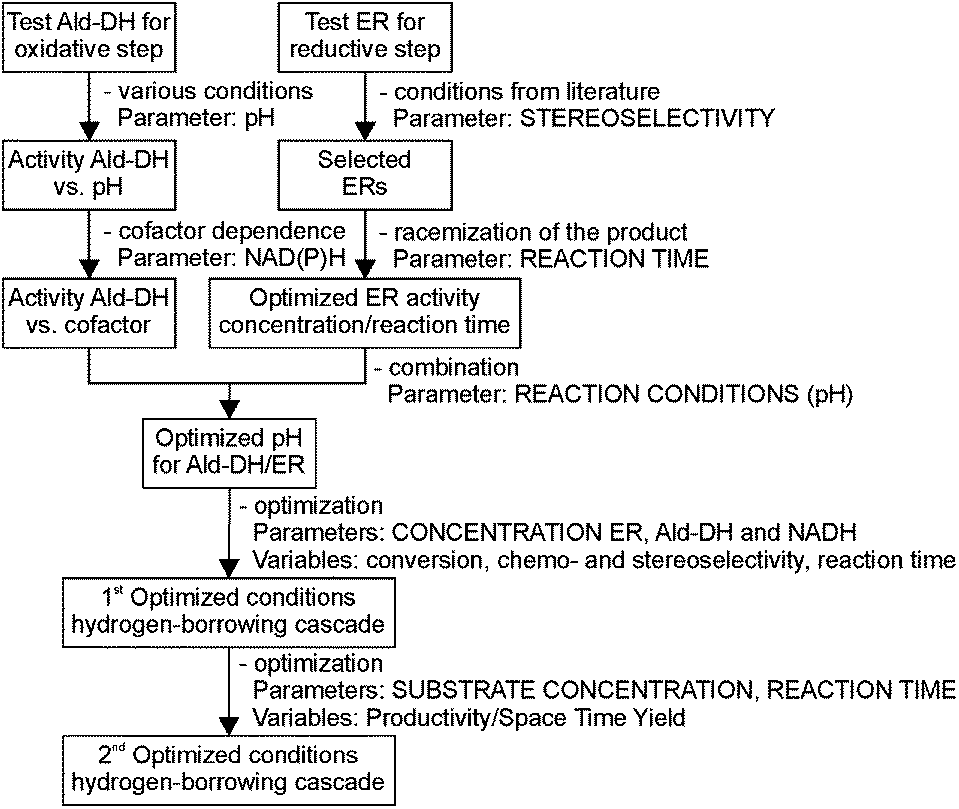 | ||
| Chart 1 Systematic steps for designing a successful hydrogen-borrowing cascade reaction. | ||
Results and discussion
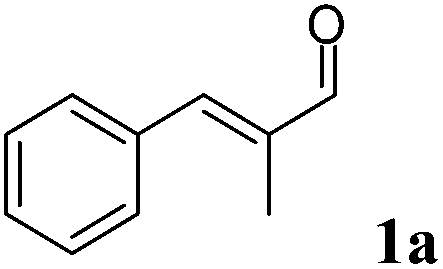
Since the stereogenic centre is introduced into the final product in the first reductive step of the cascade (Scheme 2), we initially tested a wide panel of eleven different ERs that are originated from various sources such as bacteria, yeasts and plants.34–42 The parameter for this initial screening of the ERs was the stereoselectivity for the reduction of the carbon–carbon double bond of the target substrate α-methyl-trans-cinnamaldehyde (1a) (Chart 1). The starting reaction conditions were taken from a survey of the recent literature.21,35,43,44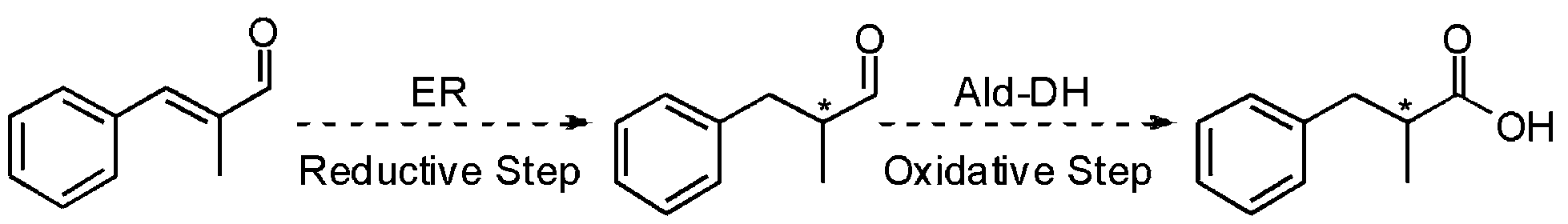 | ||
| Scheme 2 Hydrogen borrowing cascade reaction. | ||
Asymmetric reduction is commonly conducted using NADPH as cofactor that is recycled by glucose dehydrogenase (GDH) at the expense of glucose as the sacrificial co-substrate. Surprisingly, although it is known that α-chiral aldehydes are prone to racemization in aqueous solution,45 the majority of the procedures reported in the literature for this biocatalytic reaction envisage 24 h reaction time. Therefore these reaction conditions and this reaction time were employed in the initial experiment. Quantitative conversion was achieved with the ERs tested, with XenA being the only exception (Table 1). However, the reduced aldehyde (1b) was obtained either in a racemic form or with poor enantiomeric excess (up to 28% (S)). Moreover, a relevant amount of 1b (from 7% to 29%) was further reduced to the corresponding alcohol (1c).
| Entry | ER | Conv.a [%] | 1b [%] | 1c [%] | ee 1bb [%] |
|---|---|---|---|---|---|
| a Achiral GC (DB-Wax). b Chiral HPLC (Chiralsil OJ-H). c Reaction time 24 h. d Reaction time 6 h. Experimental conditions: reaction volume = 1 mL, 50 mM KPi pH 7.0, 30 °C, [ER] = 2 μM, [1a] = 5 mM, [NADPH] = 10 μM, [GDH] = 10 U, [glucose] = 300 mM; extraction with MTBE (2 × 500 μL). | |||||
| 1 | PETNR | >99c | 78 | 22 | rac |
| 2 | TOYE | >99c | 87 | 13 | rac |
| 3 | OYE2 | >99c | 75 | 25 | 28 (S) |
| 4 | OYE3 | >99c | 71 | 29 | 28 (S) |
| 5 | XenA | 66c | 56 | 10 | 18 (R) |
| 6 | XenB | >99c | 82 | 18 | rac |
| 7 | LeOPR1 | >99c | 85 | 15 | rac |
| 8 | NerA | >99c | 79 | 21 | rac |
| 9 | GluOx | >99c | 71 | 29 | 18 (S) |
| 10 | YqjM | >99d | 89 | 11 | rac |
| 11 | MR | >99d | 93 | 7 | rac |
In order to ascertain the origin of the poor enantiomeric excess, biocatalytic reduction was studied in more detail using two selected ERs. OYE2 was chosen since it showed the highest ee (28% (S)) and better chemoselectivity compared to OYE3. YqjM was also selected because it shows high chemoselectivity (89%), although the product was obtained in racemic form. In this experiment, the impact of the reaction time was evaluated on the conversion and ee. Additionally, NADPH was used in stoichiometric amount (i.e. without a recycling system) to avoid any possible cross-activity from GDH. Fig. 1 shows that the reduction of the alkene moiety is complete after two hours using YqjM (Fig. 1A) and in less than 30 min using OYE2 (Fig. 1B). OYE2 performs the reduction of the carbon–carbon double bond with perfect stereoselectivity as witnessed by the ee of >99% after the first few minutes of the reaction. Hence, the origin of the poor enantioselectivity for the reduction with OYE2 stems from the spontaneous chemical racemization of 1b in aqueous buffer. Furthermore, as the reaction is essentially complete after 15 min, prolonging the reaction time is not advantageous as this depletes the ee of 1b. In contrast, the optical purity of 1b was found to be extremely poor at early time points when the reduction was carried out with YqjM.
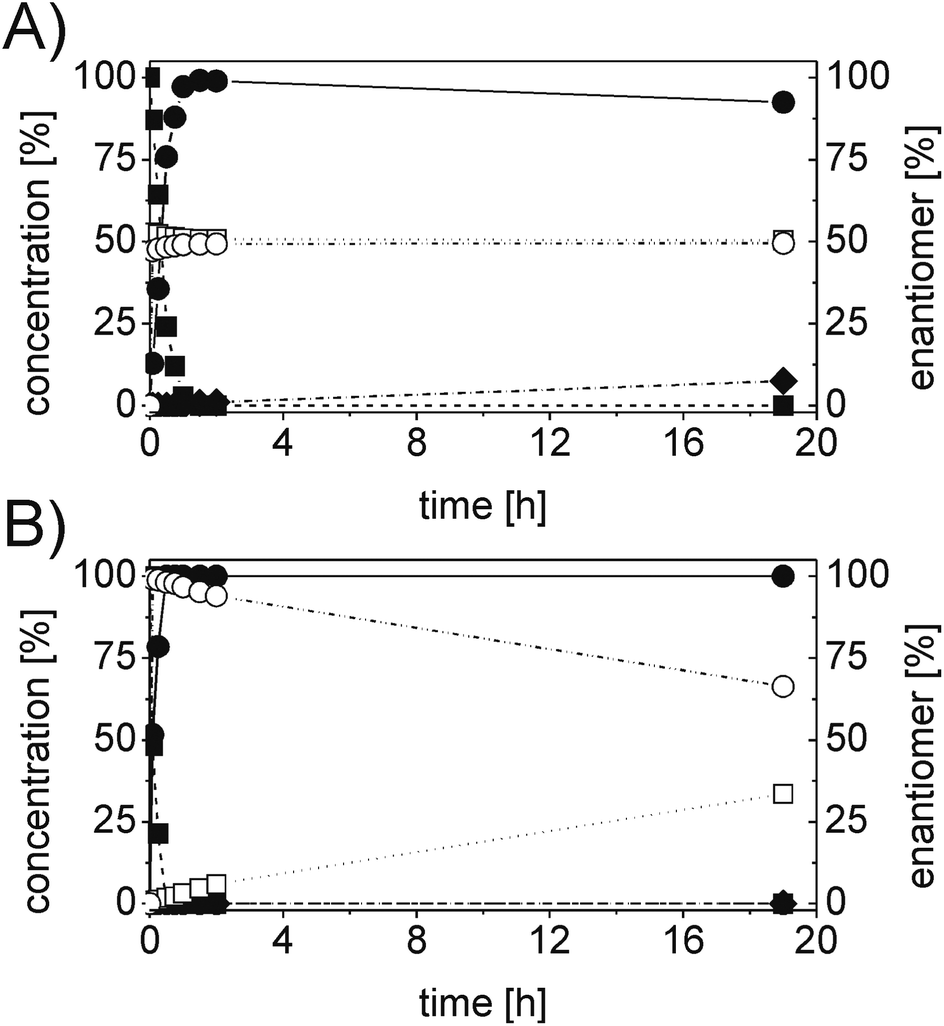 | ||
| Fig. 1 Progress curve for the biotransformation of 1a by (A) YqjM and (B) OYE2. Concentration of α-methyl-trans-cinnamaldehyde 1a (■), α-methylhydro-cinnamaldehyde 1b (●), α-methylhydrocinnamic alcohol 1c (◆) and (R)-α-methyl-hydrocinnamaldehyde (□) and (S)-α-methylhydro-cinnamaldehyde (○). Experimental conditions: reaction volume = 1 mL, 50 mM KPi pH 7.0, 30 °C, [ER] = 2 μM, [1a] = 5 mM, [NADPH] = 11 mM; extraction with MTBE (2 × 500 μL). Conversion measured by achiral GC (DB-Wax); ee measured by chiral HPLC (Chiralsil OJ-H). | ||
In another experiment, the same reaction under the same reaction conditions was repeated using catalytic NADPH and GDH/glucose for cofactor regeneration (Fig. S3 ESI†). In contrast to the experiment using OYE2 with stoichiometric NADPH, the use of catalytic NADPH in presence of GDH led to the formation of the alcohol 1c as side-product in significant amounts (22% after 11 h). Therefore ERs must be in general chemoselective enzymes for the reduction of 1a; further reduction to 1c is attributed to a promiscuous activity of the GDH (a few purified GDHs were tested). The reduction with stoichiometric NADPH and YqjM also led to the formation of a minor quantity of alcohol 1c (8%) after 19 h. However, this is attributed to the presence of some impurities (e.g. alcohol dehydrogenases) in the YqjM protein solution (Fig. S1 ESI†). Therefore, it is important to avoid the use of GDH and employ highly purified ERs to carry out biocatalytic reduction of substrate 1a. Even taking this into account one step reduction does not lead to high conversion and ee, because of spontaneous chemical racemization. The hydrogen-borrowing approach described here also has the advantage of solving this problem, since the aldehyde 1b is promptly oxidized to the corresponding non-enolisable acid 1d.
The second branch of Chart 1 relates to the oxidative step catalyzed by an Ald-DH. The oxidation of 1b to the corresponding carboxylic acid (1d) was performed using three different aldehyde dehydrogenases, namely the Ald-DHs from bovine lens (Ald-DH-BOV),46,47 horse liver (Ald-DH-HL)48,49 and E. coli (Ald-DH-EC).50 The enzymes were tested for the conversion of 1b. The parameters explored experimentally (Chart 1) were reaction pH values (step 1) and the requirement for a cofactor NADP+/NAD+ (step 2). Ald-DH-BOV and Ald-DH-HL were shown to be strictly selective for NAD+; no conversion was observed with NADP+ as cofactor (Table 2, entries 2 and 4; Table S2 ESI† for detailed time study). Conversely, Ald-DH-EC is able to accept both cofactors but the reaction proceeded faster with NAD+ (Table 2, entries 5 and 6). Table 2 clearly shows that the reaction rate for Ald-DH-BOV as well as Ald-DH-HL gradually increases passing from pH 6, 7, 8 and reaching the maximum at pH 9. Moreover, Ald-DH-EC is active at a broader pH range. Also at pH 7 – which is the preferred pH value for ERs – an acceptable reaction rate could be observed.
| Entry | Ald-DH | Cofactor | pH 6 | pH 7 | pH 8 | pH 9 |
|---|---|---|---|---|---|---|
| a n.m. not measurable. Experimental conditions: reaction volume = 1 mL, 50 mM KPi (pH 6.0, 7.0, 8.0) and 50 mM Tris/HCl pH 9.0, 30 °C, reaction time = 23 h, [Ald-DH] = 2 μM, [1b] = 5 mM, [NAD+] = 7 mM; extraction with MTBE (2 × 400 μL), derivatization with (trimethylsilyl)diazomethane to methylester. Conversion measured by achiral GC (DB-Wax). | ||||||
| 1 | BOV | NAD+ | 20% | 52% | 63% | 74% |
| 2 | BOV | NADP+ | n.m. | n.m. | n.m. | n.m. |
| 3 | HL | NAD+ | 7% | 32% | 32% | 51% |
| 4 | HL | NADP+ | n.m. | n.m. | n.m. | n.m. |
| 5 | EC | NAD+ | 7% | >99% | >99% | >99% |
| 6 | EC | NADP+ | 3% | 44% | 66% | 70% |
In the next step (Chart 1) the activity of the ERs was reanalyzed at different pH values and using NADH as cofactor, as this is the preferred cofactor for the Ald-DHs. This step is vital for the set-up of a successful hydrogen-borrowing cascade, as it aims at identifying suitable conditions for the combination of both enzyme classes. The reaction time was set to 1 h to minimize chemical racemization (Table 3). In all the cases at pH 9 no activity for the ERs could be measured. Only at pH 7, using OYE2 was possible to achieve quantitative conversion and a good enantiomeric excess (90% (S); Table 3, entry 3). All the other ERs showed imperfect ee and/or low conversion under these reaction conditions. Unfortunately, none of the ERs tested could provide the (R) enantiomer in high enantioenriched form. As a consequence OYE2 was selected as the best ER and the Ald-DH from E. coli as the best dehydrogenase for performing the cascade reaction.
| Entry | pH 7 | pH 8 | pH 9 | |||
|---|---|---|---|---|---|---|
| ER | Conv.a [%] | eeb [%] | Conv.a [%] | eeb [%] | Conv.a [%] | |
| a Achiral GC (DB-Wax). b Chiral HPLC (Chiralsil OJ-H); n.d. not determined; n.m. not measurable. Experimental conditions: reaction volume = 1 mL, 50 mM KPi (pH 7.0 and 8.0) and 50 mM Tris/HCl pH 9.0, 30 °C, [ER] = 2 μM, [1a] = 5 mM, [NADH] = 10 μM, [GDH] = 10 U, [glucose] = 300 mM; extraction with MTBE (2 × 500 μL). | ||||||
| 1 | PETNR | 2 | n.d. | 2 | n.d. | n.m. |
| 2 | TOYE | 50 | 16 (S) | 32 | rac | n.m. |
| 3 | OYE2 | 99 | 90 (S) | 48 | 83 (S) | n.m. |
| 4 | OYE3 | 15 | 97 (S) | 9 | 95 (S) | n.m. |
| 5 | XenA | 6 | n.d. | 5 | n.d. | n.m. |
| 6 | XenB | 7 | n.d. | 4 | n.d. | n.m. |
| 7 | LeOPR1 | 19 | 19 (R) | 12 | 38 (R) | n.m. |
| 8 | NerA | 46 | 7 (R) | 47 | 14 (R) | n.m. |
| 9 | GluOx | 19 | 50 (S) | 12 | 39 (S) | n.m. |
| 10 | YqjM | 29 | 16 (R) | 32 | 38 (R) | n.m. |
| 11 | MR | 78 | 5 (R) | 71 | 8 (R) | n.m. |
Moving along Chart 1, the further steps are aimed at optimizing the cascade to maximize conversion, chemo- and stereoselectivity and reducing the reaction time. The parameters investigated are the concentrations of the enzymes and of the cofactor. The first combination of both enzymes – ER and Ald-DH – was performed with the optimized conditions identified in the single experiments ([OYE2] = [Ald-DH-EC] = 2 μM, [NADH] = 10 μM). Unfortunately, after 6 h reaction time, the conversion was only 45% (Fig. 2 and Table S3 ESI†).
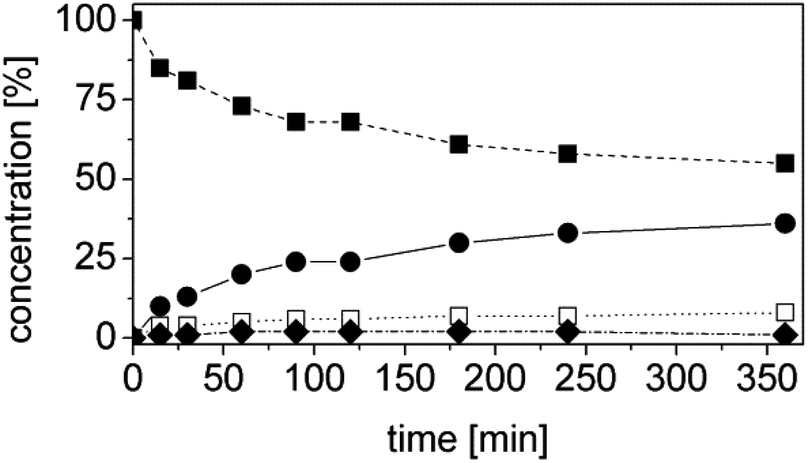 | ||
| Fig. 2 First experiment one-pot two enzyme cascade reaction for the conversion of 1a to 1d. Concentration of α-methylcinnamaldehyde 1a (■), α-methylhydrocinnamic acid 1d (●), α-methylhydrocinnamaldehyde 1b (◆) and α-methylcinnamic acid 1e (□). Experimental conditions: reaction volume = 1 mL, 50 mM KPi pH 7.0, 30 °C, [OYE2] = 2 μM, [Ald-DH-EC] = 2 μM, [1a] = 5 mM, [NADH] = 10 μM; two-step selective extraction with MTBE: (I) under basic conditions (aldehydes) and (II) acidic conditions and derivatization with (trimethylsilyl)diazomethane (acids (ester)); IS = 2-phenylethanol. Conversion was measured by achiral GC (DB-Wax). | ||
Nevertheless we noted that the Ald-DH preferred to oxidize the saturated aldehyde 1b, rather than the unsaturated starting material 1a, as the main product was α-methyl-hydro cinnamic acid (1d). Based on these results, the object parameters (ER, Ald-DH, NAD+ concentration, time) were varied for improving the outcome of the cascade (Table 4). The reaction was stopped after 6 hours. The substrate was kept at 5 mM concentration whereas: (I) the amount of the cofactor was increased and (II) the concentration of the two enzymes was varied to investigate the influence of the ratio [ER]/[Ald-DH]. The latter is a crucial point because the concentrations of the two enzymes have to be carefully balanced. In fact, the Ald-DH must quickly oxidize the saturated aldehyde to avoid racemization of the aldehyde product; also the Ald-DH must oxidize the saturated aldehyde intermediate 1b, rather than the unsaturated starting material 1a.
| Entry | NADH [μM] | OYE2 [μM] | Ald-DH EC [μM] | Conv.a [%] | 1e [%] | 1d [%] | ee (S)-1db [%] |
|---|---|---|---|---|---|---|---|
| a Achiral GC (DB-Wax). b Chiral HPLC (Chiralsil OJ-3); n.d. = not determined. Experimental conditions: reaction volume = 1 mL, 50 mM KPi pH 7.0, 30 °C, [1a] = 5 mM, reaction time = 6 h; two-step selective extraction with MTBE: (I) under basic conditions (aldehydes) and (II) acidic conditions and derivatization with (trimethylsilyl)-diazomethane (acids (ester)); IS = 2-phenylethanol. | |||||||
| 1 | 500 | 2 | 2 | >99 | 2 | 98 | 98 |
| 2 | 500 | 2 | 10 | >99 | 6 | 94 | 98 |
| 3 | 500 | 10 | 2 | >99 | 2 | 98 | 99 |
| 4 | 500 | 10 | 10 | >99 | 4 | 96 | 99 |
| 5 | 250 | 2 | 2 | >99 | 5 | 95 | 98 |
| 6 | 250 | 2 | 10 | >99 | 7 | 93 | 98 |
| 7 | 250 | 10 | 2 | >99 | 4 | 96 | 99 |
| 8 | 250 | 10 | 10 | >99 | 6 | 94 | 99 |
| 9 | 100 | 2 | 2 | 90 | 5 | 82 | 97 |
| 10 | 100 | 2 | 10 | >99 | 6 | 94 | 98 |
| 11 | 100 | 10 | 2 | >99 | 5 | 95 | 99 |
| 12 | 100 | 10 | 10 | >99 | 6 | 94 | 99 |
| 13 | 50 | 10 | 5 | >99 | 5 | 95 | 99 |
| 14 | 25 | 10 | 5 | >99 | 5 | 95 | 99 |
| 15 | 10 | 10 | 5 | >99 | 6 | 94 | 99 |
| 16 | 10 | 2 | 2 | 45 | 8 | 36 | n.d. |
In almost all the cases quantitative conversion was achieved within 6 h reaction time. The only exceptions were when the concentration of both enzymes was reduced to 2 μM, using 10 μM or 100 μM NADH (Table 4, entries 9 and 16). The increased enzyme concentrations (up to 10 μM for the ER and 5 μM for the Ald-DH, respectively) afforded quantitative conversion, albeit only 10 μM of NADH were employed (Table 4, entry 15). The ees were excellent in all the cases, ranging from 97% to 99%. Therefore, the saturated aldehyde intermediate (1b) is quickly oxidized by the Ald-DH. It is also worth noting that very low quantities of the unsaturated carboxylic acid 1e were formed (from 2% to 8%).
At this stage of the work we did not have any information regarding the kinetics of the process. Hence the progress of the reaction was monitored as a function of time. Specifically, the concentrations of the starting material (1a), of the intermediate (1b), of the final product (1d) and of the side product (1e) as well as the ees, were determined as a function of the time (Fig. 3). At this step of our work flow (Chart 1), the chemo- and, in particular, the stereoselectivity are maximized. In fact, the cascade reaction must run the minimum time required to achieve full conversion. Longer reaction times lead to lower ees, due to spontaneous racemization, and diminished productivity. For this experiment, the best conditions from the previous step were taken (Table 4, entry 14).
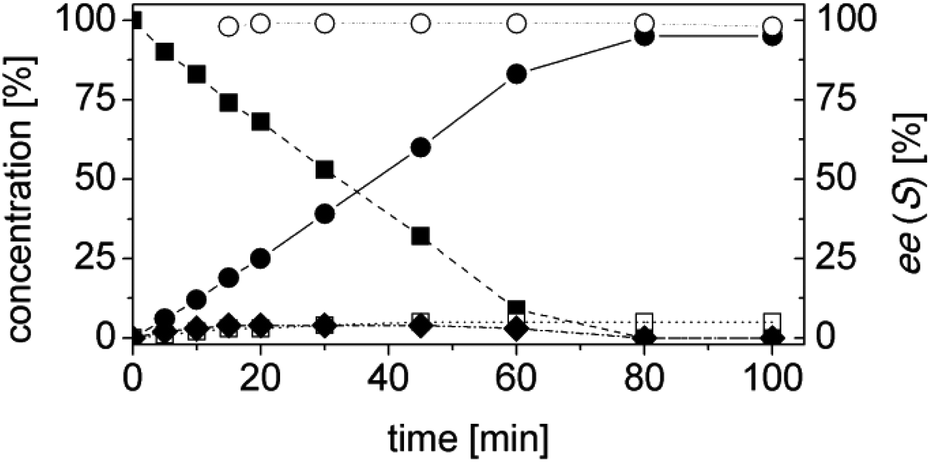 | ||
| Fig. 3 Time study optimized one-pot two enzyme cascade reaction for the conversion of 1a to 1d. Concentration of α-methylcinnamaldehyde 1a (■), α-methylhydrocinnamic acid 1d (●), α-methylcinnamic acid 1e (□), α-methylhydro-cinnamaldehyde 1b (◆) and enantiomeric excess of α-methylhydrocinnamic acid (S)-1d (○). Experimental conditions: reaction volume = 1 mL, 50 mM KPi pH 7.0, 30 °C, [OYE2] = 10 μM, [Ald-DH-EC] = 5 μM, [NADH] = 25 μM, [1a] = 5 mM; two-step selective extraction with MTBE: (I) under basic conditions (aldehydes) and (II) acidic conditions and derivatization with (trimethylsilyl)-diazomethane (acids (ester)); IS = 2-phenylethanol. Conversion was measured by achiral GC (DB-Wax) and ee by chiral HPLC (Chiralsil OJ-3). | ||
The reaction was extremely efficient since it was complete after 90 min despite the low concentrations of the enzymes used (Fig. 3, Table S4 ESI;† [OYE2] = 10 μM, [Ald-DH-EC] = 5 μM). The concentration of the intermediate 1b remained constant with time (ca. 3%). The cinnamic acid by-product 1e was mainly produced at the beginning of the reaction. This is not surprising as the cinnamic aldehyde 1a is present at almost 5 mM concentration, whereas the concentration of the intermediate 1b is almost negligible during the first few minutes of the reaction. However, after having identified the suitable reaction conditions, the concentration of cinnamic acid 1e remained constant during the reaction time and always below 5%. This is an indication that the Ald-DH-EC is significantly more active towards the saturated aldehyde 1b rather than the unsaturated one 1a, which is the crucial point for enabling this hydrogen-borrowing biocatalytic cascade reaction. Moreover, the cascade showed a perfect stereoselectivity (>99% ee).
Productivity is an important factor for a successful process and this objective is considered in the last step of the proposed Chart 1. The studied parameter was the substrate concentration. Applying the optimized reaction conditions from the previous round and increasing the substrate concentration to 10 mM, 97% conversion was still obtained after 6 h reaction time with high chemoselectivity (93%) and excellent stereoselectivity (99%; Table S5 ESI†). Increasing the substrate concentration up to 25 mM required the prolongation of the reaction time up to 24 h and doubling of the amount of enzyme in order to achieve >99% conversion (chemoselectivity = 96%, ee = 96%). However, bio-catalytic reductions with ERs are commonly performed at 5 mM substrate concentration since these enzymes are plagued by substrate and/or product inhibition.51–53 The proposed two-step biocatalytic cascade we have developed here also allows alleviation of this problem because the product of the bioreduction (1b) is immediately removed by the Ald-DH in the second step.
The hydrogen-borrowing biocatalytic cascade was therefore successfully developed for the conversion of the test substrate 1a into 1d with quantitative conversion, improved productivity and excellent chemo- and stereoselectivity. Finally, we wanted to investigate if this strategy for developing a hydrogen-borrowing cascade has a more general validity and it is therefore applicable on a broad range of substrates. Thus, a panel of structurally diverse α,β-unsaturated aldehydes, valuable synthons for the synthesis of chiral active pharmaceutical ingredients, were selected (Fig. 4).
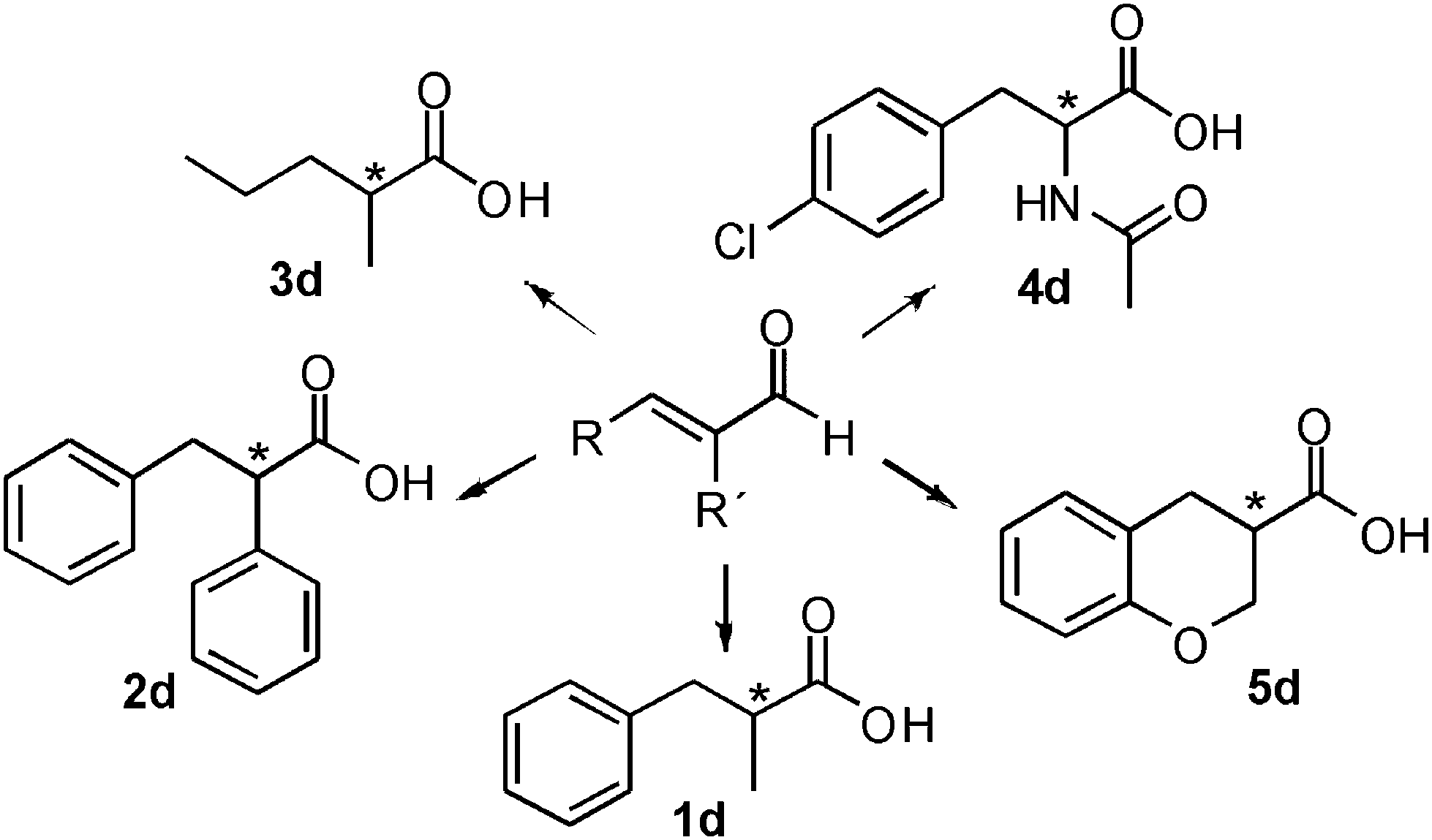 | ||
| Fig. 4 Schematic view of the broad substrate scope applicability of the hydrogen-borrowing cascade combining ERs and Ald-DHs. | ||
For example, enantiopure 1d is a precursor for the production of biological active compounds that possess muscle relaxant activity.54,552d constitutes the main core of drugs possessing antagonist activity for the acetylcholine M1 receptor of human.563d is commonly employed for the production of herbicidals,57 antibacterial58 as well as anticancer compounds (e.g. leukemia, lung and prostate carcinoma).59–61 Optically active compound 5d and derivatives thereof are precursors of enantiopure aminochromane derivatives; these can be obtained through the conversion of the carboxylic moiety into the isocyanate intermediate, followed by Curtius rearrangement. The optically active aminochromanes are key intermediates of many antidepressant drugs such as robalzotan,62 ebalzotan63 as well as DBH inhibitors such as chromanyl-imidazolethiones.64 Finally, our strategy can give access to enantioenriched unnatural amino acids. For instance N-acetylated (S)-para-chloro phenylalanine 4d was obtained. Through the carbon carbon coupling using the chloro group on the aromatic ring, more complex structures possessing various biological activities can be achieved.65–68 (S)-para-chloro phenylalanine derivatives also possess antimycobacterial (e.g. Mycobacterium tuberculosis),69 antifungal70 as well as anticancer activity.71 Additionally, N-derivatized (S)-para-chloro phenylalanine is also used for the production of herbicides.72
For each substrate, the steps described in Chart 1 were applied as described for 1a. For instance, in the case of α-phenylcinnamaldehyde (2a), two ERs, namely PETNR and XenB, showed the highest activity/stereoselectivity for the reduction of the carbon–carbon double bond of the unsaturated aldehyde; the Ald-DH from bovine lens was found to be the most efficient enzyme for the oxidation of the aldehyde intermediate (2b) to the corresponding acid (2d) (Table S6 and S7 ESI†). Fig. 5 depicts the final time studies, after the overall procedure described in Chart 1, for the conversion of 2a into 2d using PETNR and XenB in combination with the aldehyde dehydrogenase from bovine lens (Fig. 5A and B, and Table S8 and S9 ESI† for detailed information). Hence the substrate bearing the bulky phenyl-group in the alpha position was also accepted by PETNR and XenB with high stereoselectivity. In the hydrogen-borrowing cascade reaction using PETNR/Ald-DH-BOV, the conversion reached 92% after 4 h, with 91% chemo-selectivity and a perfect ee (97%). The combination of XenB/Ald-DH-BOV resulted in 91% conversion after 5 h (95% chemoselectivity) and excellent ee of 99%. Further changes on the reaction conditions (e.g. concentration of NADH and AldDH) did not improve the chemoselectivity but had a negative impact on the stereoselectivity (Table S10 ESI†).
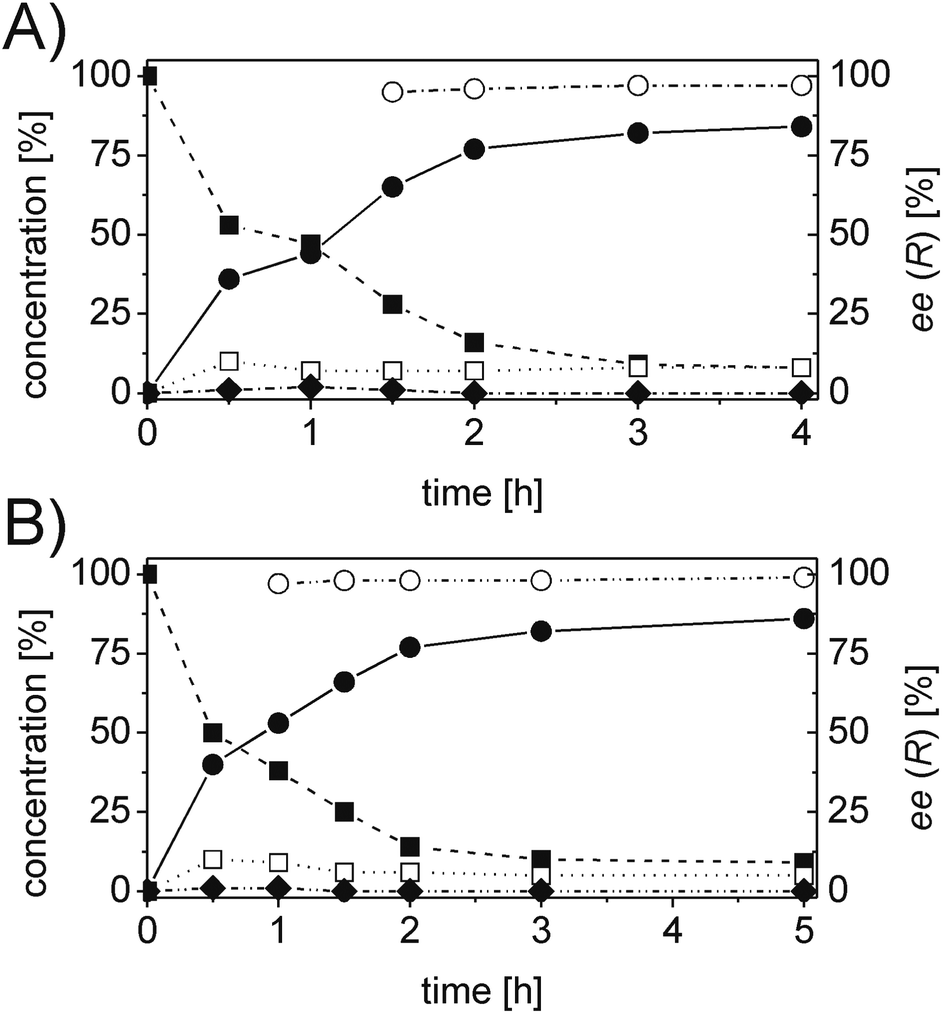 | ||
| Fig. 5 Progress curves for the one-pot two enzyme cascade reaction of 2a by (A) PETNR and Ald-DH-BOV and (B) XenB and Ald-DH-BOV. Concentration of α-phenylcinnamaldehyde 2a (■), α-phenylhydrocinnamic acid 2d (●), α-phenyl-cinnamic acid 2e (□), α-phenylhydrocinnamaldehyde 2b (◆) and enantiomeric excess of α-phenylhydrocinnamic acid (R)-2d (○). Experimental conditions: reaction volume = 1 mL, 50 mM KPi pH 7.0, 30 °C, [PETNR] & [XenB] = 25 μM, [Ald-DH-BOV] = 4 μM, [NADH] = 50 μM, [2a] = 5 mM; two-step selective extraction with MTBE: (I) under basic conditions (aldehydes) and (II) acidic conditions and derivatization with (trimethylsilyl)diazomethane (acids (ester)); IS = 2-phenylethanol. Conversion was measured by achiral GC (DB-Wax) and ee by chiral HPLC (Chiralsil OJ-3). | ||
Using trans-2-methyl-2-pentenal (3a) as substrate, OYE2 and Ald-DH-EC were selected as the best performing ER and Ald-DH, respectively (Table S11 and S12 ESI†). Fig. 6 (Table S13 ESI†) displays the time study for the one-pot concurrent cascade reaction. Full conversion was obtained after 60 min, showing an ee of 98%. Using the Ald-DH from E. coli, 13% of the unsaturated acid side product 3e were formed. Again, any change in the reaction condition resulted in the same chemoselectivity but lower ee (96%). However, the chemoselectivity was improved (only 8–9% of 3c were formed) when the Ald-DH from bovine lens (Ald-DH-BOV) was combined with OYE2 remaining the ee still perfect (>98%, Table S14 ESI†).
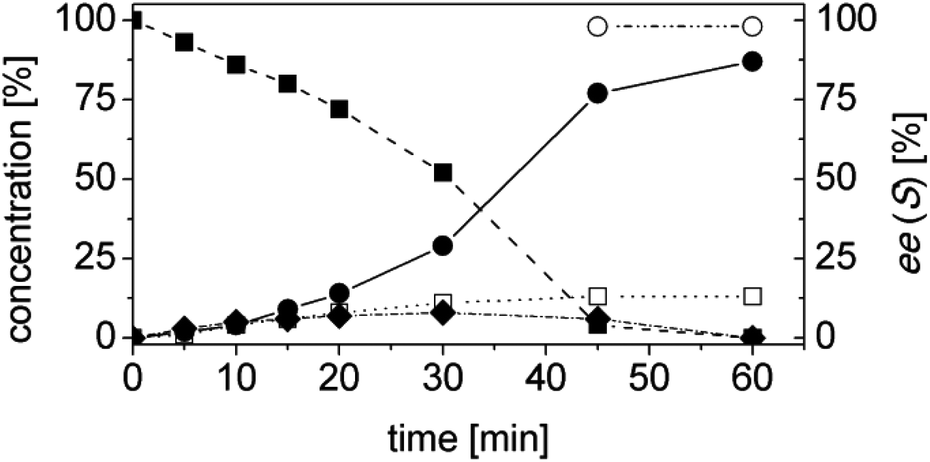 | ||
| Fig. 6 Progress curves for the one-pot two enzyme cascade reaction of 3a by OYE2 and Ald-DH-EC. Concentration of trans-2-methyl-2-pentenal 3a (■), 2-methylpentanoic acid 3d (●), trans-2-methyl-2-pentenoic acid 3e (□), 2-methyl-pentanal 3b (◆) and enantiomeric excess of 2-methylpentanoic acid (S)-3d (○). Experimental conditions: reaction volume = 1 mL, 50 mM KPi pH 7.0, 30 °C, [OYE2] = 10 μM, [Ald-DH-EC] = 3 μM, [NADH] = 25 μM, [3a] = 5 mM; extraction with MTBE (2 × 400 μL) under acidic conditions and derivatization with (trimethylsilyl)-diazomethane to methylester. Conversion was measured by achiral GC (DB1701) and ee by chiral GC (Restek Rt-ßDEXsm). | ||
All three Ald-DHs have been tested for the oxidation of (Z)-N-(1-(4-chlorophenyl)-3-oxoprop-1-en-2-yl)acetamide (4a) as well as the related saturated aldehyde N-(1-(4-chlorophenyl)-3-oxopropan-2-yl)acetamide (4b) to the corresponding carboxylic acids. Two independent experiments showed that the Ald-DHs clearly preferred to oxidize the saturated substrate (4b); after 5 h reaction time as the conversion for 4b was much higher than for 4a (Table S16 and S17 ESI†).
OYE2 and Ald-DH-BOV were combined for performing the cascade reaction starting from 4a, yielding 99% conversion, 95% ee and 81% chemoselectivity after 3 h (Fig. 7, Table S20 ESI†). Increasing the concentration of the Ald-DH from 10 μM to 50 μM slightly increased the chemoselectivity (83%) and the ee value (96%). In general, the ee was never higher than 96% since the few initial minutes of the reaction. Thus, for 4a, the ee is limited by the intrinsic stereoselectivity of the ER (Table S21 ESI†).
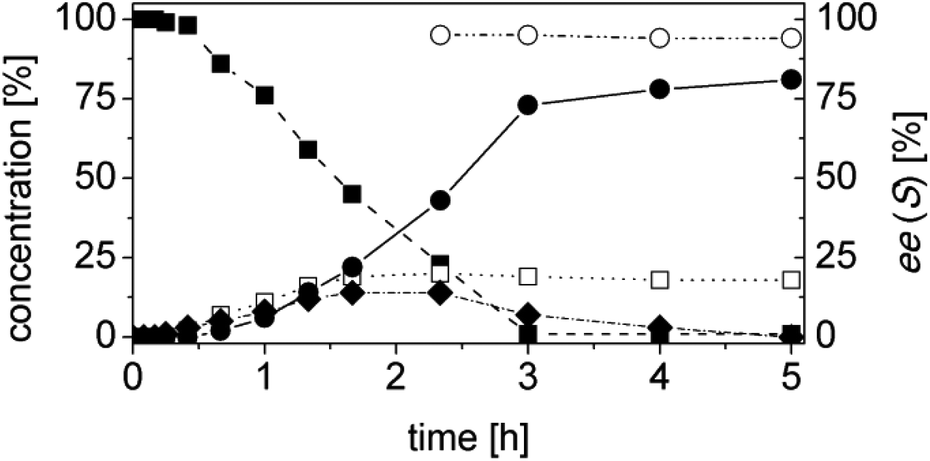 | ||
| Fig. 7 Progress curves for the one-pot two enzyme cascade reaction of 4a by OYE2 and Ald-DH-BOV. Concentration of (Z)-N-(1-(4-chlorophenyl)-3-oxoprop-1-en-2-yl)acetamide 4a (■), 2-acetyl-4-chloro-DL-phenylalanine 4d (●), (Z)-2-acetamido-3-(4-chlorophenyl)acrylic acid 4e (□), N-(1-(4-chlorophenyl)-3-oxopropan-2-yl)acetamide 4b (◆) and enantiomeric excess of 2-acetyl-4-chloro-L-phenyl-alanine (S)-4d (○). Experimental conditions: reaction volume = 1 mL, 50 mM KPi pH 7.0, 30 °C, [OYE2] = 10 μM, [Ald-DH-BOV] = 10 μM, [NADH] = 50 μM, [4a] = 5 mM; extraction under acidic conditions with EtOAc (2 × 400 μL) and derivatization with (trimethylsilyl)diazomethane to methylester. Conversion measured by achiral GC (HP5) and ee value by chiral GC (DEX-CB). | ||
The last substrate investigated was 2H-chromene-3-carbaldehyde (5a). After 2 h reaction time, quantitative conversion was obtained for the one-pot two enzymes cascade reaction using OYE2 or GluOx in combination with Ald-DH-EC resulting in 81% chemoselectivity and 95% ee (S-enantiomer, Fig. 8, Table S25 and S26 ESI†).
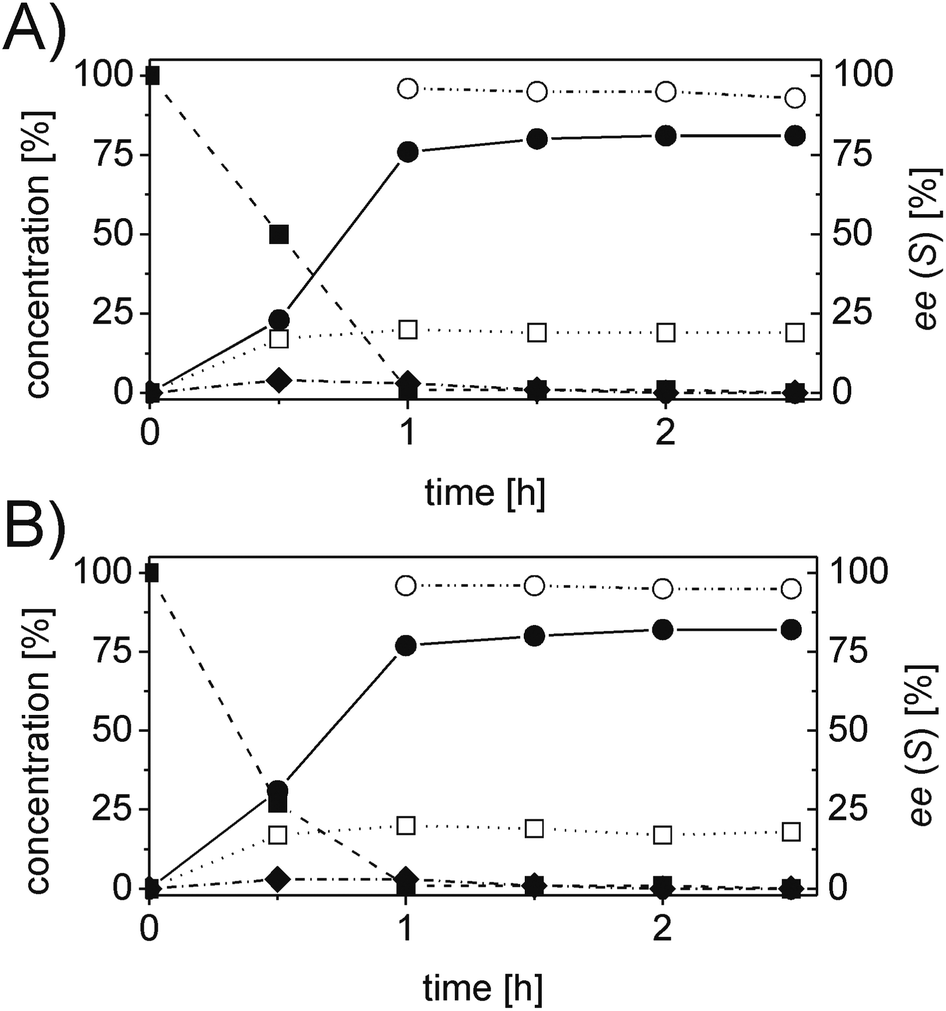 | ||
| Fig. 8 Progress curves for the one-pot two enzyme cascade reaction of 5a by (A) OYE2 and Ald-DH-EC and (B) GluOx and Ald-DH-EC. Concentration of 2H-chromene-3-carbaldehyde 5a (■), 3,4-dihydro 2H benzopyran-3-carboxylic acid 5d (●), 2H-chromene-3-carboxylic acid 5e (□), chromane-3-carbaldehyde 5b (◆) and enantiomeric excess of 3,4-Dihydro 2H-benzopyran-3-carboxylic acid (S)-5d (○). Experimental conditions: reaction volume = 1 mL, 50 mM KPi pH 7.0, 30 °C, [OYE2] & [GluOx] = 10 μM, [Ald-DH-EC] = 3 μM, [NADH] = 100 μM, [5a] = 5 mM; extraction under acidic conditions with EtOAc (2 × 400 μL) and derivatization with (trimethylsilyl)diazomethane to methylester. Conversion measured by achiral GC (DB-Wax) and ee value by chiral GC (Restek Rt-ßDEXsm). | ||
The best results for the one-pot two-enzyme cascade reaction for substrates 1a–5a are summarized in Table 5. Although the substrates investigated are structurally diverse, a suitable adjustment of the reaction conditions allowed for obtaining quantitative conversion in four cases out of five. Chemoselectivity varied from good to excellent. Moreover, the chemoselectivity might be improved if other Ald-DHs will be discovered or engineered to accept only the saturated aldehyde intermediate. The stereoselectivity was also in general elevated, although there is an urgent demand for stereocomplementary ERs that would lead to the opposite enantiomer with equally high ees.
| Fig. | Substrate | ER | Ald-DH | NADH [μM] | Conv. [%] | Reac. time [h] | Chemoselectivity [%] | ee [%] | Config. |
|---|---|---|---|---|---|---|---|---|---|
| 1 |
|
OYE2 10 μM | EC 5 μM | 25 | >99 | 1.33 | 95 | 99 | (S) |
| 2 |
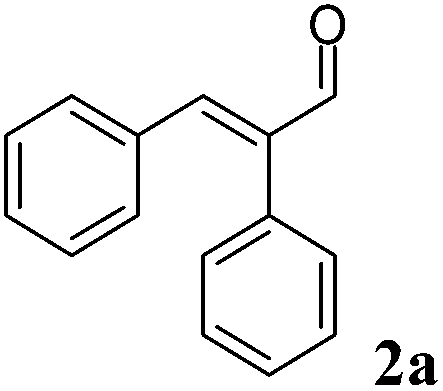
|
XenB 25 μM | BOV 4 μM | 50 | 91 | 5 | 95 | >98 | (R) |
| 3 |
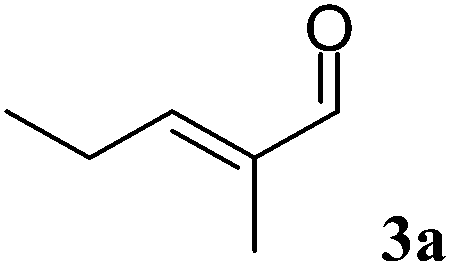
|
OYE2 10 μM | BOV 3 μM | 25 | >99 | 12 | 92 | >98 | (S) |
| 4 |
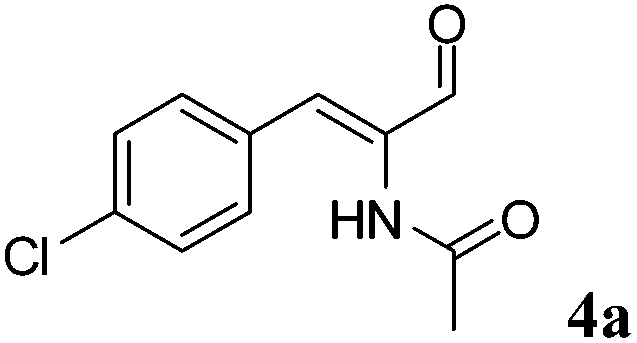
|
OYE2 10 μM | BOV 10 μM | 50 | 99 | 6 | 81 | 95 | (S) |
| 5 |
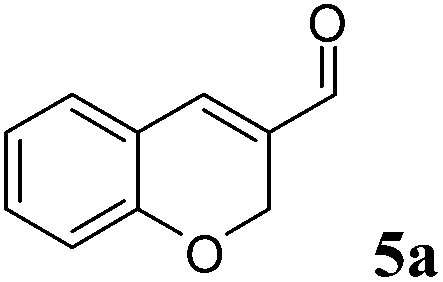
|
OYE2 10 μM | EC 3 μM | 100 | >99 | 2.5 | 81 | 95 | (S) |
| GluOx 10 μM | EC 3 μM | 100 | >99 | 2.5 | 82 | 95 | (S) |
As previously mentioned, substrates 1a and 3a have been tested in a concomitant work by another group.12 It is important to point out that the overall strategy depicted in Chart 1 was not developed and the enzymes were simply mixed together without any additional accurate study. As a consequence, in this study, substrate 1a and 3a were indeed quantitatively converted but at the expense of the ees that were only 64% and 78%, respectively. In contrast, our study demonstrates that it is possible to reach up to 99% stereoselectivity after selecting the best suitable ER and systematically tuning the reaction parameters (Table 5, entry 1 and 3). Additionally, our procedure generally allowed for reduction of the reaction time from 24 h to few hours. The substrate concentration was increased as well. Both factors lead to a dramatic increase of the productivity. Substrate 5a was instead the object of a two-step cascade combining ERs with alcohol dehydrogenases to give enantioenriched β-substituted alcohols.45 The alcohol was recovered with 91% ee, whereas in our study the related carboxylic acid was obtained with 95% ee (Table 5, entry 5). Therefore, our proposed methodology allows one to obtain the highest chemoselectivity and ee for a given hydrogen-borrowing cascade.
Finally, 1d was produced on a preparative scale (100 mg of starting material 1a) resulting in quantitative conversion after 2 h reaction time with 96% chemoselectivity and >98% ee (S). After work-up, the isolated yield was 88%.
Materials and methods
Synthesis of substrates and reference compounds, sources and cloning/expression/purification conditions for ERs and Ald-DHs used in this study, analytical methods as well as chiral GC and HPLC chromatograms can be found in the ESI.†Materials
α-Methyl-trans-cinnamaldehyde (1a), α-methylhydrocinnamic acid (1d), α-methylcinnamic acid (1e), α-phenylhydrocinnamic acid (2d), α-phenylcinnamic acid (2e), trans-2-methyl-2-pentenal (3a), 2-methylpentanal (3b), 2-methylpentanoic acid (3d), trans-2-methyl-2-pentenoic acid (3e), 2-acetyl-4-chloro-DL-phenylalanine (4d), 3,4-dihydro 2H-benzopyran-3-carboxylic acid (5d) and (trimethylsilyl)diazomethane (2 M in hexanes) were purchased from Sigma Aldrich (Gillingham, Dorset, UK). 2H-Chromene-3-carbaldehyde (5a) was purchased from Thermo Fisher Scientific (Loughborough, UK). Glucose dehydrogenase (GDH, CDX-901) was bought from Codexis (Redwood City, CA, USA). Synthetic genes for the Ald-DHs were bought from GenScript (Piscataway, NJ, USA).α-methylhydrocinnamaldehyde (1b), α-methylhydrocinnamic alcohol (1c), α-phenylcinnamaldehyde (2a), α-phenyl-hydrocinnamaldehyde (2b), (Z)-N-(1-(4-chloro-phenyl)-3-oxoprop-1-en-2-yl)acetamide (4a) and chromane-3-carbaldehyde (5b) were chemically synthesized as described in the ESI.†
N-(1-(4-chlorophenyl)-3-oxopropan-2-yl)acetamide (4b) was obtained through a biotransformation of the related unsaturated substrate (4a) using ER and glucose as the final reducing reagent. (Z)-2-acetamido-3-(4-chloro-phenyl)-acrylic acid (4e) and 2H-chromene-3-carboxylic acid (5e) were obtained through a biotransformation of the related substrates (4a and 5a, respectively) using Ald-DH and stoichiometric amounts of NAD+ (see ESI†).
General procedure for biotransformations
The substrates were dissolved as stock solutions in ethanol or DMSO, and then diluted to 5 mM in the reaction (2% final co-solvent concentration). Standard reactions (1.0 mL) were performed in phosphate buffer (50 mM KH2PO4/K2HPO4, pH 7.0) and reactions were shaken at 30 °C at 160 rpm in an orbital shaker. The reaction was terminated by the extraction with EtOAc or MTBE (2 × 500 μL) under acidic conditions, the extracts were dried using anhydrous MgSO4 and analyzed by GC or HPLC as described in the ESI† to determine the conversion and enantiomeric excess. (I) Asymmetric bioreduction using ERs: 1 mL reaction volume consisted of aldehydes 1a to 5a (5 mM), ER (2 to 4 μM), NAD(P)H (11 mM). If the GDH recycling system was applied, the reaction mixture consisted of NAD(P)H (10 μM), glucose (15 mM), and GDH (10 U). (II) Oxidation of aldehydes using Ald-DHs: The reaction mixture consisted of substrate 1b to 5b (5 mM), Ald-DH (2 to 5 μM) and NAD(P)+ (6 to 7 mM). After extraction with MTBE or EtOAc (2 × 400 μL) under acidic conditions and drying with anhydrous MgSO4, the carboxylic acids were derivatized to the corresponding methylester and analyzed by GC or HPLC. (III) One-pot two enzyme cascade reaction: 1 mL reaction mixture consisted of substrate (1a to 5a, 5 mM), ER (2–25 μM), Ald-DH (2–10 μM) and cofactor NADH (10–500 μM). A two-step extraction protocol was performed, extracting the aldehydes under basic conditions with MTBE or EtOAc (2 × 500 μL) and in a second step the carboxylic acids under acidic conditions (2 × 400 μL MTBE or EtOAc). The acids were derivatized using the protocol described below.Derivatization of carboxylic acids to methylester
To the extracts (800 μL) MeOH (200 μL) and (trimethylsilyl)diazomethane (10 μL) were added and the reaction was shaken at 30 °C, 160 rpm for 60 min. The excess of the derivatization reagent was destroyed by the addition of acetic acid (2 μL) and the reaction was again shaken at 30 °C for another 25 min prior to analysis of the compounds by GC or HPLC.Conclusions
In summary, we have developed a systematic strategy to set up an efficient two-step hydrogen-borrowing cascade in terms of conversion, chemoselectivity, stereoselectivity, reaction time and substrate concentration. The biocatalytic cascade possesses the highest atom efficiency since the hydride consumed in the first reductive step is produced in the following oxidative step. The only additional reagent is a water molecule and no waste (e.g. gluconolactone, carbon dioxide, etc.) is produced. This approach was applied to the conversion of α-substituted α,β-unsaturated aldehydes into the related optically active saturated carboxylic acids. Our methodology proved to be successful towards a panel of diverse substrates and can be applied for the production of chiral substituted cinnamic acids, aliphatic acids, heterocycles and even acetylated amino acids. Future work should aim at further improving the chemoselectivity by selecting or engineering other aldehyde dehydrogenases or reversing the stereoselective outcome of the reaction by engineering stereocomplementary ene-reductases.Abbreviations
| ER | Ene-reductase |
| Ald-DH | Aldehyde dehydrogenase |
Acknowledgements
The work was funded by the UK Biotechnology and Biological Sciences Research Council (BBSRC; BB/K0017802/1) and GlaxoSmithKline. N.S.S. received funding as an Engineering and Physical Sciences Research Council (EPSRC) Established Career Fellow (EP/J020192/1) and a Royal Society Wolfson Merit Award. F.G.M. has received funding from the European Union's Seventh Framework Programme FP7/2007–2013 under grant agreement no. 266025 (BIONEXGEN) and no. 115360 (CHEM21). Prof. Peter Macheroux (University of Technology, Graz) is gratefully acknowledged for providing the plasmid DNA for XenA, XenB and NerA, LeOPR1 and YqjM.Notes and references
- R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, in Green Chemistry and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, pp. 1–47 Search PubMed.
- P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452–1461 RSC.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- P. A. Santacoloma, G. Sin, K. V. Gernaey and J. M. Woodley, Org. Process Res. Dev., 2010, 15, 203–212 CrossRef.
- J. H. Schrittwieser, J. Sattler, V. Resch, F. G. Mutti and W. Kroutil, Curr. Opin. Chem. Biol., 2011, 15, 249–256 CrossRef CAS PubMed.
- C. V. Voss, C. C. Gruber, K. Faber, T. Knaus, P. Macheroux and W. Kroutil, J. Am. Chem. Soc., 2008, 130, 13969–13972 CrossRef CAS PubMed.
- V. Köhler, Y. M. Wilson, M. Dürrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knörr, D. Häussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2013, 5, 93–99 CrossRef PubMed.
- F. G. Mutti, A. Orthaber, J. H. Schrittwieser, J. G. de Vries, R. Pietschnig and W. Kroutil, Chem. Commun., 2010, 46, 8046–8048 RSC.
- J. H. Sattler, M. Fuchs, K. Tauber, F. G. Mutti, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 9156–9159 CrossRef CAS PubMed.
- S. Gargiulo, D. J. Opperman, U. Hanefeld, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2012, 48, 6630–6632 RSC.
- T. Winkler, H. Gröger and W. Hummel, ChemCatChem, 2014, 6, 961–964 CrossRef CAS.
- H. S. Toogood and N. S. Scrutton, Curr. Opin. Chem. Biol., 2014, 19, 107–115 CrossRef CAS PubMed.
- C. K. Winkler, G. Tasnadi, D. Clay, M. Hall and K. Faber, J. Biotechnol., 2012, 162, 381–389 CrossRef CAS PubMed.
- H. Toogood, D. Mansell, J. M. Gardiner and N. S. Scrutton, in Comprehensive Chirality, ed. N. J. Turner, 2012, vol. 1, pp. 216–255 Search PubMed.
- H. S. Toogood, J. M. Gardiner and N. S. Scrutton, ChemCatChem, 2010, 2, 892–914 CrossRef CAS.
- Y. Fu, K. Castiglione and D. Weuster-Botz, Biotechnol. Bioeng., 2013, 110, 1293–1301 CrossRef CAS PubMed.
- D. J. Mansell, H. S. Toogood, J. Waller, J. M. X. Hughes, C. W. Levy, J. M. Gardiner and N. S. Scrutton, Adv. Synth. Catal., 2013, 3, 370–379 CAS.
- E. Burda, T. Ress, T. Winkler, C. Giese, X. Kostrov, T. Huber, W. Hummel and H. Groger, Angew. Chem., Int. Ed., 2013, 52, 9323–9326 CrossRef CAS PubMed.
- H. S. Toogood, A. Fryszkowska, V. Hare, K. Fisher, A. Roujeinikova, D. Leys, J. M. Gardiner, G. M. Stephens and N. S. Scrutton, Adv. Synth. Catal., 2008, 350, 2789–2803 CrossRef CAS PubMed.
- K. Durchschein, S. Wallner, P. Macheroux, W. Schwab, T. Winkler, W. Kreis and K. Faber, Eur. J. Org. Chem., 2012, 4963–4968 CrossRef CAS.
- Y. Yanto, H. H. Yu, M. Hall and A. S. Bommarius, Chem. Commun., 2010, 46, 8809–8811 RSC.
- P. Ferraboschi, P. Grisenti, R. Casati, A. Fiecchi and E. Santaniello, J. Chem. Soc., Perkin Trans. 1, 1987, 1743–1748 RSC.
- S. Koul, D. H. G. Crout, W. Errington and J. Tax, J. Chem. Soc., Perkin Trans. 1, 1995, 2969–2988 RSC.
- M. Utaka, S. Konishi, A. Mizuoka, T. Ohkubo, T. Sakai, S. Tsuboi and A. Takeda, J. Org. Chem., 1989, 54, 4989–4992 CrossRef CAS.
- C. Stueckler, M. Hall, H. Ehammer, E. Pointner, W. Kroutil, P. Macheroux and K. Faber, Org. Lett., 2007, 9, 5409–5411 CrossRef CAS PubMed.
- E. Brenna, F. G. Gatti, A. Manfredi, D. Monti and F. Parmeggiani, Catal. Sci. Technol., 2013, 3, 1136–1146 CAS.
- C. Stueckler, C. K. Winkler, M. Bonnekessel and K. Faber, Adv. Synth. Catal., 2010, 352, 2663–2666 CrossRef CAS.
- C. K. Winkler, D. Clay, N. G. Turrini, H. Lechner, W. Kroutil, S. Davies, S. Debarge, P. O'Neill, J. Steflik, M. Karmilowicz, J. W. Wong and K. Faber, Adv. Synth. Catal., 2014, 356, 1878–1882 CrossRef CAS.
- C. Stueckler, C. K. Winkler, M. Hall, B. Hauer, M. Bonnekessel, K. Zangger and K. Faber, Adv. Synth. Catal., 2011, 353, 1169–1173 CrossRef CAS.
- E. Brenna, G. Fronza, C. Fuganti, D. Monti and F. Parmeggiani, J. Mol. Catal. B: Enzym., 2011, 73, 17–21 CAS.
- E. Brenna, F. G. Gatti, A. Manfredi, D. Monti and F. Parmeggiani, Org. Process Res. Dev., 2012, 16, 262–268 CrossRef CAS.
- G. Tasnadi, C. K. Winkler, D. Clay, N. Sultana, W. M. F. Fabian, M. Hall, K. Ditrich and K. Faber, Chem. – Eur. J., 2012, 18, 10362–10367 CrossRef CAS PubMed.
- C. E. French and N. C. Bruce, Biochem. J., 1994, 301, 97–103 CAS.
- B. V. Adalbjornsson, H. S. Toogood, A. Fryszkowska, C. R. Pudney, T. A. Jowitt, D. Leys and N. S. Scrutton, ChemBioChem, 2010, 11, 197–207 CrossRef PubMed.
- P. A. Karplus, K. M. Fox and V. Massey, FASEB J., 1995, 9, 1518–1526 CAS.
- D. S. Blehert, B. G. Fox and G. H. Chambliss, J. Bacteriol., 1999, 181, 6254–6263 CAS.
- J. Straßner, A. Fürholz, P. Macheroux, N. Amrhein and A. Schaller, J. Biol. Chem., 1999, 274, 35067–35073 CrossRef PubMed.
- J. R. Snape, N. A. Walkley, A. P. Morby, S. Nicklin and G. F. White, J. Bacteriol., 1997, 179, 7796–7802 CAS.
- N. Richter, H. Groger and W. Hummel, Appl. Microbiol. Biotechnol., 2011, 89, 79–89 CrossRef CAS PubMed.
- T. B. Fitzpatrick, N. Amrhein and P. Macheroux, J. Biol. Chem., 2003, 278, 19891–19897 CrossRef CAS PubMed.
- C. E. French, S. Nicklin and N. C. Bruce, J. Bacteriol., 1996, 178, 6623–6627 CAS.
- A. Fryszkowska, H. Toogood, M. Sakuma, J. M. Gardiner, G. M. Stephens and N. S. Scrutton, Adv. Synth. Catal., 2009, 351, 2976–2990 CrossRef CAS.
- M. Hall, C. Stueckler, H. Ehammer, E. Pointner, G. Oberdorfer, K. Gruber, B. Hauer, R. Stuermer, W. Kroutil, P. Macheroux and K. Faber, Adv. Synth. Catal., 2008, 350, 411–418 CrossRef CAS.
- E. Brenna, F. G. Gatti, L. Malpezzi, D. Monti, F. Parmeggiani and A. Sacchetti, J. Org. Chem., 2013, 78, 4811–4822 CrossRef CAS PubMed.
- H. H. Ting and M. J. Crabbe, Biochem. J., 1983, 215, 351–359 CAS.
- H. H. Ting and M. J. Crabbe, Biochem. J., 1983, 215, 361–368 CAS.
- J. Eckfeldt, L. Mope, K. Takio and T. Yonetani, J. Biol. Chem., 1976, 251, 236–240 CAS.
- J. H. Eckfeldt and T. Yonetani, Arch. Biochem. Biophys., 1976, 173, 273–281 CrossRef CAS.
- J. E. Jo, S. Mohan Raj, C. Rathnasingh, E. Selvakumar, W. C. Jung and S. Park, Appl. Microbiol. Biotechnol., 2008, 81, 51–60 CrossRef CAS PubMed.
- E. Brenna, F. G. Gatti, D. Monti, F. Parmeggiani and A. Sacchetti, Chem. Commun., 2012, 48, 79–81 RSC.
- D. J. Bougioukou, A. Z. Walton and J. D. Stewart, Chem. Commun., 2010, 46, 8558–8560 RSC.
- E. Brenna, F. G. Gatti, D. Monti, F. Parmeggiani and A. Sacchetti, ChemCatChem, 2012, 4, 653–659 CrossRef CAS.
- G. Maksay, E. Palosi, Z. Tegyey and L. Otvos, J. Med. Chem., 1981, 24, 499–502 CrossRef CAS.
- G. Maksay, Z. Tegyey, V. Kemeny, I. Lukovits, L. Otvos and E. Palosi, J. Med. Chem., 1979, 22, 1436–1443 CrossRef CAS.
- M. S. Poslusney, C. Sevel, T. J. Utley, T. M. Bridges, R. D. Morrison, N. R. Kett, D. J. Sheffler, C. M. Niswender, J. S. Daniels, P. J. Conn, C. W. Lindsley and M. R. Wood, Bioorg. Med. Chem. Lett., 2012, 22, 6923–6928 CrossRef CAS PubMed.
- N. B. Carter, L. M. D. Turnbull, P. J. Crowley and M. R. Cordingley, Syngenta Limited, WO2008/9908, 2008.
- Z. Feng, J. H. Kim and S. F. Brady, J. Am. Chem. Soc., 2010, 132, 11902–11903 CrossRef CAS PubMed.
- B. Kongkathip, S. Akkarasamiyo, K. Hasitapan, P. Sittikul, N. Boonyalai and N. Kongkathip, Eur. J. Med. Chem., 2013, 60, 271–284 CrossRef CAS PubMed.
- H.-Y. Hung, E. Ohkoshi, M. Goto, K. F. Bastow, K. Nakagawa-Goto and K.-H. Lee, J. Med. Chem., 2012, 55, 5413–5424 CrossRef CAS PubMed.
- M. Reis, R. J. Ferreira, M. M. M. Santos, D. J. V. A. dos Santos, J. Molnár and M.-J. U. Ferreira, J. Med. Chem., 2013, 56, 748–760 CrossRef CAS PubMed.
- J. L. Evenden, E. M. Hammarberg, H. S. Hansson, S. E. Hellberg, L. G. Johansson, J. R. M. Lundkvist, S. B. Ross, D. D. Sohn and S. O. Thorberg, U.S. Patent5616610, 1997 Search PubMed.
- H.-J. Federsel, Org. Process Res. Dev., 2000, 4, 362–369 CrossRef CAS.
- A. Beliaev, D. A. Learmonth and P. Soares-da-Silva, J. Med. Chem., 2006, 49, 1191–1197 CrossRef CAS PubMed.
- S. Kousteni, The Trustees of Columbia University in the city of New York, WO2013/74889, 2013.
- M. D. Gershon and K. Margolis, Lexicon Pharmaceuticals, Inc., WO2013/148978, 2013.
- G. Karsenty, P. F. Duciy, V. K. Yadav, D. Landry and S. X. Deng, The Trustees of Columbia University in the city of New York, WO2010/56992, 2010 Search PubMed.
- Q. Liu, Q. Yang, W. Sun, P. Vogel, W. Heydorn, X.-Q. Yu, Z. Hu, W. Yu, B. Jonas, R. Pineda, V. Calderon-Gay, M. Germann, E. O'Neill, R. Brommage, E. Cullinan, K. Platt, A. Wilson, D. Powell, A. Sands, B. Zambrowicz and Z.-c. Shi, J. Pharmacol. Exp. Ther., 2008, 325, 47–55 CrossRef CAS PubMed.
- E. C. Dykhuizen, J. F. May, A. Tongpenyai and L. L. Kiessling, J. Am. Chem. Soc., 2008, 130, 6706–6707 CrossRef CAS PubMed.
- K. Takiguchi, K. Yamada, M. Suzuki, K.-i. Nunami, K. Hayashi and K. Matsumoto, Agric. Biol. Chem., 1989, 53, 77–82 CrossRef CAS.
- T. Murata, M. Umeda, S. Yoshikawa, K. Urbahns, J. Gupta and O. Sakurai, Bayer Healthcare AG, WO2004/43926, 2004.
- M. B. Aspinall, W. R. Mound, J. S. Wailes, W. G. Whittingham, J. Williams, C. L. Winn and P. A. Worthington, Syngenta Limited, WO2009/81112, 2009.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of substrates and reference compounds, sources and cloning/expression/purification conditions for ERs and Ald-DHs used in this study, analytical methods as well as chiral GC and HPLC chromatograms. See DOI: 10.1039/c4ob02282c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |