
Design and synthesis of fluorescent 7-deazaadenosine nucleosides containing π-extended diarylacetylene motifs†
Sara
De Ornellas
 ab,
John M.
Slattery
a,
Robert M.
Edkins
c,
Andrew
Beeby
c,
Christoph G.
Baumann
*bd and
Ian J. S.
Fairlamb
*ad
ab,
John M.
Slattery
a,
Robert M.
Edkins
c,
Andrew
Beeby
c,
Christoph G.
Baumann
*bd and
Ian J. S.
Fairlamb
*ad
aDepartment of Chemistry, University of York, York, YO10 5DD, UK. E-mail: ian.fairlamb@york.ac.uk; Tel: +44 (0)1904 324091
bDepartment of Biology, University of York, York, YO10 5DD, UK. E-mail: christoph.baumann@york.ac.uk; Tel: +44 (0)1904 328828
cDepartment of Chemistry, University of Durham, South Road, Durham, DH1 3LE, UK. E-mail: andrew.beeby@durham.ac.uk
dBiological Physical Sciences Institute (BPSI), University of York, York, YO10 5DD, UK
First published on 10th November 2014
Abstract
C-modified 7-deazaadenosines containing a diphenylacetylene moiety have been synthesised using cross-coupling approaches. The C-modified nucleosides exhibit remarkable fluorescence properties, including high quantum yields. Solvatochromic studies show a near linear correlation between the Stokes shift and solvent polarity which is indicative of intramolecular charge transfer. DFT calculations have allowed us to correlate the experimentally observed photophysical properties with the calculated HOMO–LUMO energy gaps within a series of real and model compounds.
Introduction
Fluorescently-labelled nucleosides and nucleotides are invaluable tools in the study of biological systems.1 The addition of compact substituents at C8 in adenosine, e.g. phenyl-(1) or phenylethynyl-(2), induces significant fluorescence.2 This property cannot be exploited fully however, as C8-modified analogues are incompatible with base-pair formation (due to an unfavourable conformational preference).3On the other hand, 7-deazaadenosine nucleosides adopt conformations commensurate with adenosine, allowing the C7-position to be utilised for functionalisation.4 C7-groups orientate into the major groove of double helices, resulting in minimal duplex destabilisation.5 There are a limited number of 7-deaza-2′-deoxyadenosine analogues incorporating compact fluorophores (e.g.3), but they typically exhibit weak fluorescence.6 It is therefore necessary to develop highly fluorescent C7-modified analogues, which is the principal aim of the current study.
In this paper, we report the synthesis and photophysical properties of novel 7-deazaadenosine nucleosides containing compact π-extended fluorophores. An extrinsic fluorophore, i.e. a diarylacetylene group (tolan), has been introduced at the C7-position (as illustrated for generic compound 4 in Fig. 1). The design of the series of compounds 4a–e was guided by computational studies, which predict that such molecules should function as classical ‘push–pull’ diarylacetylene systems, which are known to exhibit strong fluorescence.7 The theoretical predictions go some way to explain the experimentally observed photophysical properties of compounds within the series 4a–e. The fluorescence lifetimes and quantum yields make these artificial C-modified nucleosides potentially useful as probes in biological systems.
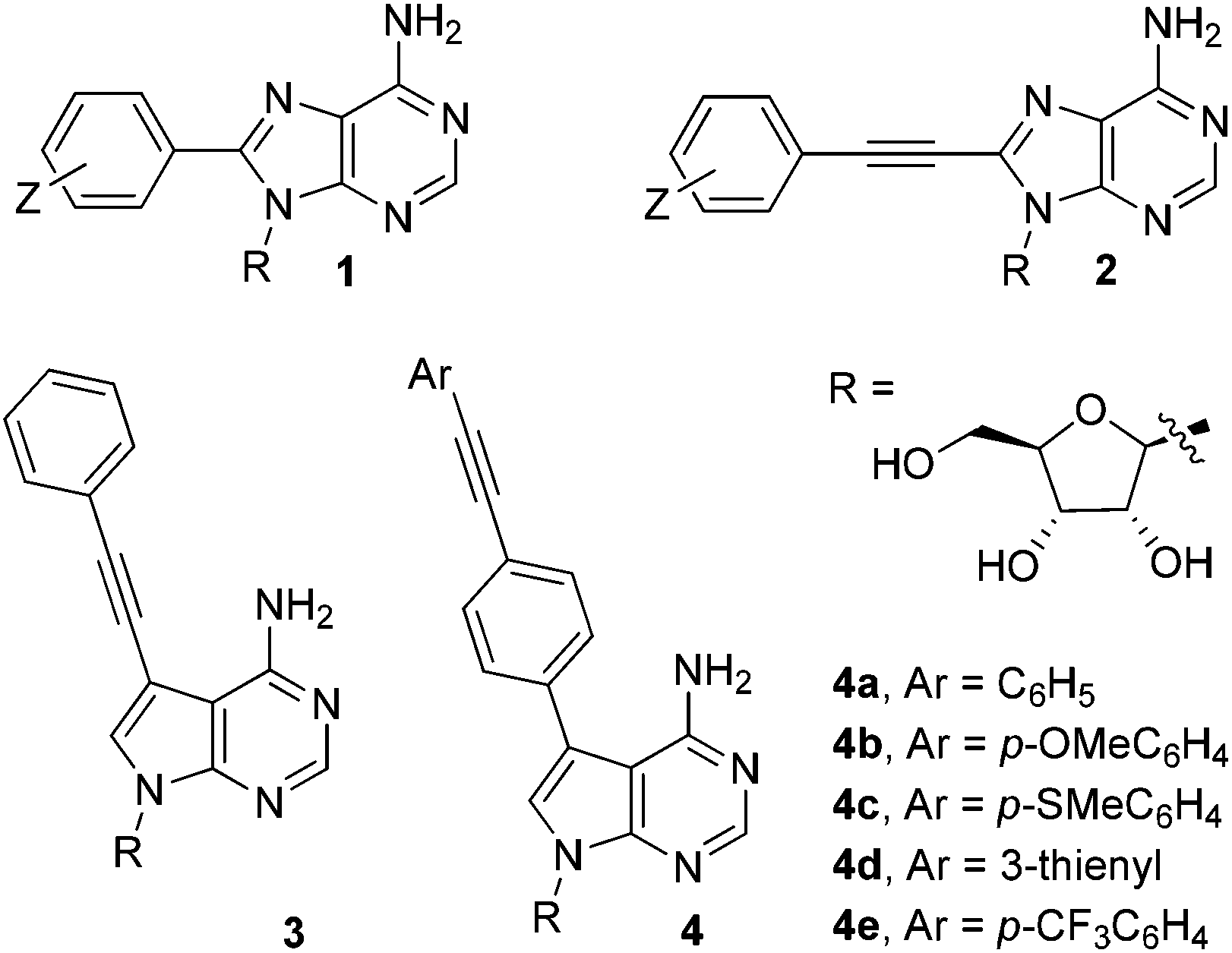 | ||
| Fig. 1 Fluorescent nucleosides, including C7 and C8 modifications. The series of compounds 4a–e are the target molecules in this paper. | ||
Results and discussion
The following section is divided into theoretical (DFT calculations) and experimental (synthesis and photophysical properties) parts. The theoretical work establishes the credibility of 4 as a potentially useful new class of fluorescent nucleosides.Density functional theory (DFT) calculations
We studied structures containing both N-ribose and N-methyl groups, i.e.3 and 4a, and 5 and 6a, respectively. Several conformations within the ribose sugar and the π-system within each molecule were considered in the calculations. In these derivatives, the ribose with a C3′-endo conformation was selected. The methyl substituent was viewed as an electronically similar group to the more conformationally-flexible ribose. Decoupling the potential complexity of the latter was necessary, especially when studying a larger series of compounds (4a–e), vide infra. All of the calculated structures were optimised at the (RI-)PBE0/def2-TZVPP level and their frontier molecular orbitals (MOs) and vertical excitation energies (from TDDFT studies at the same level of theory) were used to probe the effect of chemical structure on their photophysical properties.Interestingly, a qualitative analysis of the MOs of 3 and 5 (Fig. 2) goes some way to explaining a general preference for non-radiative decay and the weak fluorescence observed6 experimentally. The HOMO/LUMO in both 3 and 5 are characteristic of a weaker push–pull system and suggests limited intramolecular charge transfer upon excitation.
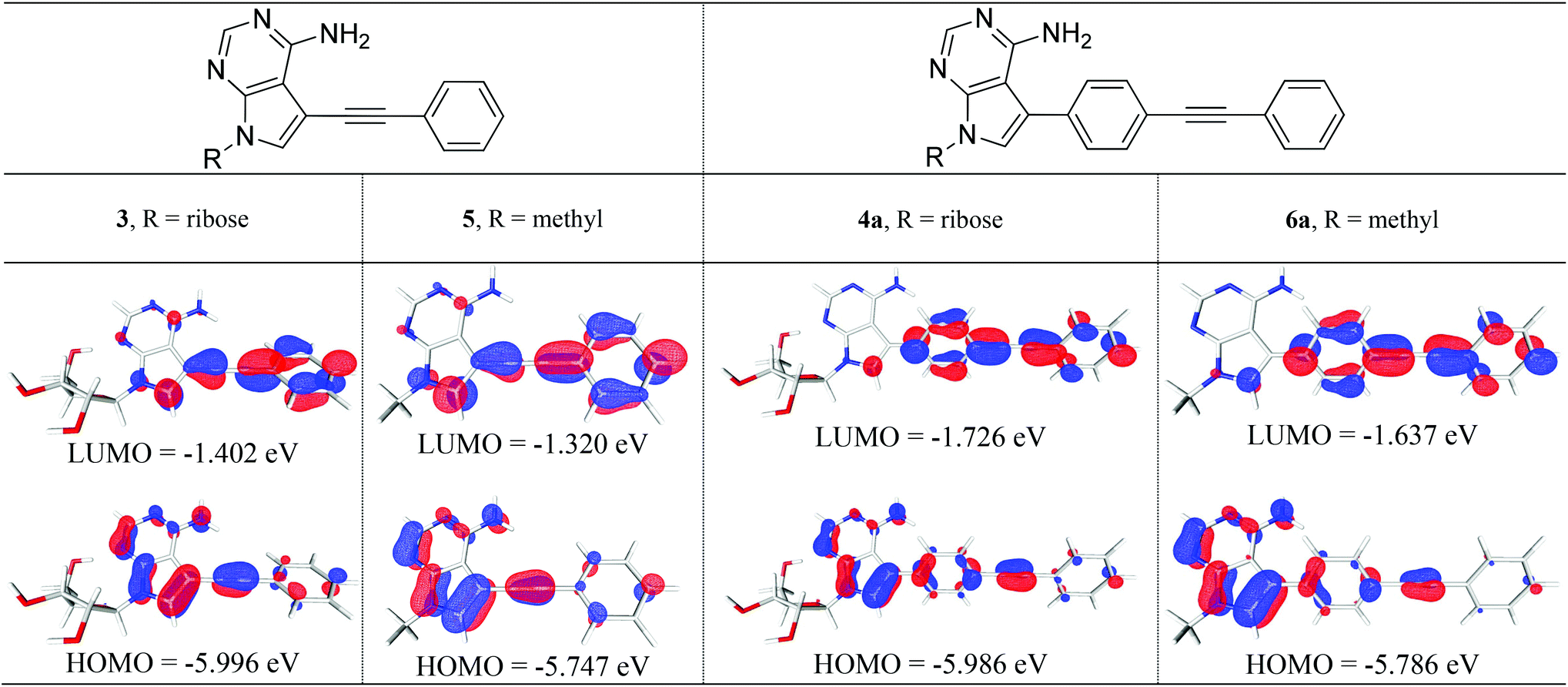 | ||
| Fig. 2 (RI-)PBE0/def2-TZVPP optimised structures for N-substituted-7-phenylethynyl-7-deazaadenosines 3 and 5 (left) and N-substituted-7-[4-(phenylethynyl)-phenyl]-7-deazaadenosines 4a and 6a (right). In all cases, the frontier MOs (HOMO/LUMO) are illustrated (isosurface at 0.05 a.u.). | ||
By contrast, the MOs for ribose derivative 4a and N-methyl derivative 6a (shown in Fig. 2) show localisation of the HOMO primarily on the 7-deazaadenine unit and the LUMO on the diarylacetylene. This observation suggests that there is potential for intramolecular charge transfer upon excitation in this type of structure, i.e. as expected in a ‘push–pull’-type system, and that the ribose and methyl groups do not appreciably affect the distribution of electron density within the π-system.
Synthesis of target compounds (4a–4e)
We set about synthesising the core structure of 4a {and derivatives (4b–e)} using 7-iodo-7-deazaadenosine 11 as a convenient starting material (Scheme 1). The synthesis of 11 involves reaction of 6-chloro-7-deazapurine 7 with iodine in DMF to give 8. An alternative iodination method using N-iodosuccinimide required recrystallisation of 8 from MeOH, which leads to the undesired formation of 7-iodo-6-methoxy-7-deazapurine. Compound 8 was coupled to protected ribose sugar 9 using Vorbrüggen glycosylation conditions to give 10.8 Optimal yields of 10 were achieved using sub-stoichiometric quantities of trimethylsilyl triflate and N,O-bis(trimethylsilyl)acetamide (BSA). Sugar deprotection of 10 and installation of the exocyclic amine was carried out with aqueous NH3 in dioxane (in a sealed tube at 60 °C), which avoids methanolic ammonia and an autoclave, affording 11 in good overall yield.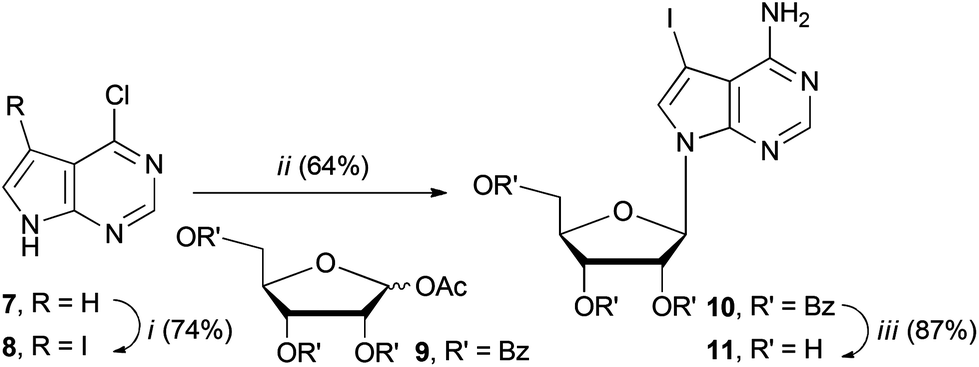 | ||
Scheme 1 Synthesis of key starting material 11. Reaction conditions: i. I2 (1.1 eq.), KOH (2.5 eq.), DMF, rt, 30 min. ii. 9 (1.1 eq.), BSA (0.5 eq.), TMSOTf (0.5 eq.), MeCN, 80 °C, 1.5 h. iii. NH3 (aq, 25%)/dioxane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 60 °C, 3 days. :1), 60 °C, 3 days. | ||
For the diarylacetylene arm, an organoboron species was required for coupling to 11. Diarylacetylene boronate esters 12a–e were synthesised by chemoselective Pd-catalysed cross-coupling of 4-bromophenyl boronic acid neopentyl glycol ester 13.9 Two approaches were used. Firstly, 13 was cross-coupled with three terminal arylacetylenes to give 12a, 12b and 12d, respectively (Scheme 2).
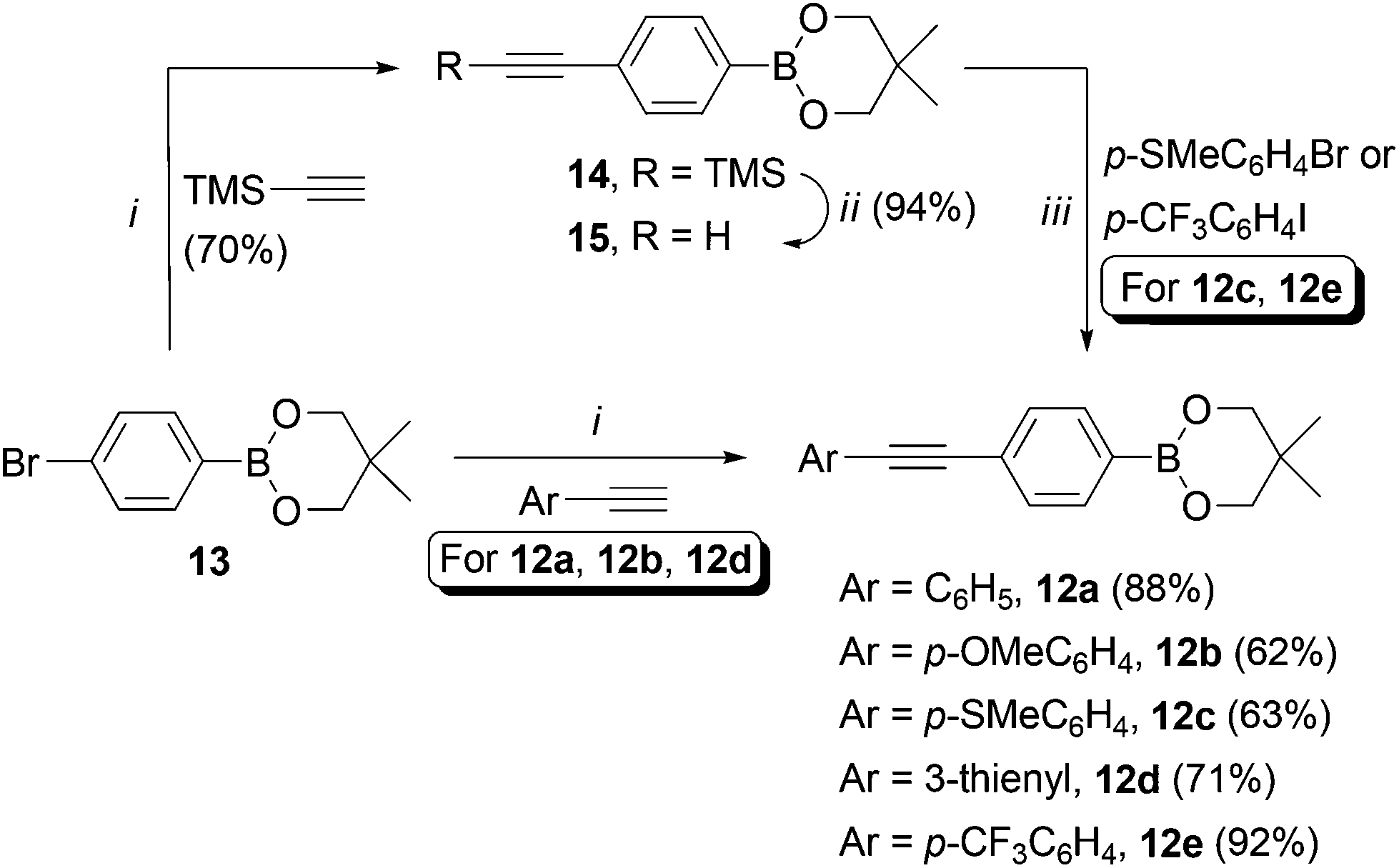 | ||
| Scheme 2 Synthesis of compounds 12a–e. Reaction conditions: i. PdCl2(PPh3)2 (5 mol%), CuI (5 mol%), PPh3 (20 mol%), Et2NH, DMF, 120 °C (MW), 20 min. ii. TBAF, THF. iii. As for i, without DMF, 25 min. | ||
For access to 12c and 12e, 13 was cross-coupled with trimethylsilylacetylene to give 14, which was then deprotected quantitatively with TBAF to reveal terminal acetylene 15. The latter compound was then cross-coupled with the respective aryl halide to give boronate esters 12c and 12e (Scheme 2).
With the diarylacetylene boronate esters 12a–e in hand, they were cross-coupled with 7-iodo-7-deazaadenosine 11 under reported aqueous conditions,10,4a with the water-soluble phosphine ligand TPPTS (Scheme 3). Pure C7-diarylacetylene functionalised nucleosides 4a–4e were all isolated by column chromatography on silica gel or by preparative thin-layer chromatography in good yields (see ESI† for characterisation details).
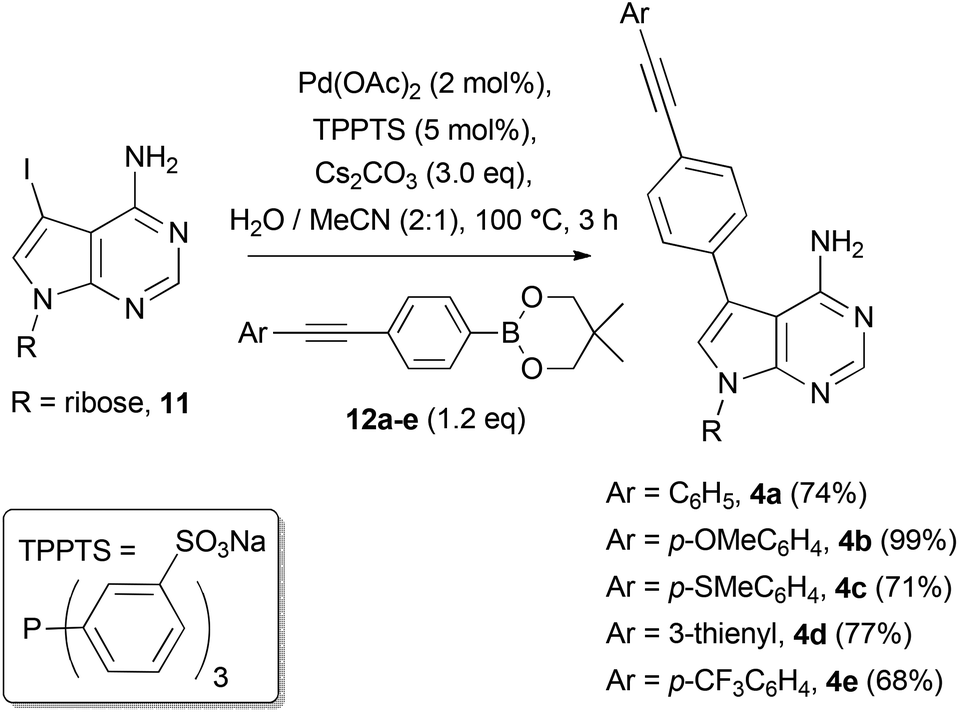 | ||
| Scheme 3 Synthesis of target compounds 4a–e. | ||
Photophysical studies
We turned our attention to studying the photophysical properties of 7-modified-7-deazaadenosines 4a–e. UV-visible absorption spectra of the modified nucleosides exhibit maxima at wavelengths >310 nm, with the lowest λmax observed for the thiophene-containing compound 4d (Table 1). The absorbance maxima of 4a–c (at ca. 320 nm) are shifted away from the intrinsic absorbance of nucleic acids and proteins (ca. >280 nm), which is important when considering the potential applications of these novel nucleosides as fluorescent probes in biological systems. The 7-modified-7-deazaadenosines 4a–e possess high molar absorbance coefficients (ca. 2 × 104 M−1 cm−1), suggesting the diphenylacetylene motif connected to the 7-deaza-adenine unit is an effective chromophore.| Cpd | λ max/nm (exp.) | λ max/nm (calc.)a | ε/104 M−1 cm−1 | λ em/nm | Stokes shift/ cm−1 | Φ | τ/ns |
|---|---|---|---|---|---|---|---|
| a Calculated λmax are for model complexes 6a–6e. | |||||||
| 4a | 321 | 345 | 2.2 | 428 | 7790 | 0.74 | 2.2 (92%), 0.5 (8%) |
| 4b | 321 | 341 | 2.3 | 405 | 6460 | 0.78 | 1.6 (90%), 0.5 (10%) |
| 4c | 331 | 353 | 2.5 | 431 | 7010 | 0.76 | 2.0 (92%), 0.5 (8%) |
| 4d | 313 | 349 | 2.1 | 404 | 7200 | 0.32 | 1.5 (88%), 0.5 (12%) |
| 4e | 328 | 363 | 1.5 | 491 | 10000 |
0.41 | 2.1 (87%), 0.8 (12%) |
The 7-modified-7-deazaadenosines 4a–e exhibit promising fluorescence properties (Table 1). High emission quantum yields were recorded for all compounds in DMSO, but of particular note are compounds 4a, 4b and 4c, which have quantum yields >0.70. The thiophene-containing analogue 4d has a somewhat lower quantum yield (Φ = 0.32), as does the trifluoromethyl-compound 4e (Φ = 0.41). The variation in wavelength of the emission maxima for these compounds is much larger than the variation in their absorption maxima. This gives rise to large differences in their Stokes shifts, e.g. trifluoromethyl compound 4e, has a remarkably large Stokes shift of 1 × 104 cm−1.
In order to further explore the substituent effects seen in 4a–e, the model structures (6a–e, R = Me rather than ribose) were optimised computationally at the (RI-)PBE0/def2-TZVPP level, allowing their UV-visible absorption spectra to be calculated in the gas phase (TDDFT at the same level of theory, 50 singlet excitations considered). Vertical excitation energies were used to probe the effects of various substituents on the photophysical properties of these molecules. The frontier MOs and their energies are shown in Fig. 3. The calculated λmax values for 6a–e are given in Table 1, where there are differences with the experimental data. Crucially, the trend within the series can be considered. For example, when all calculated (which includes 4a, 5 and 6a–e) and experimental λmax data are plotted against each other, there is a good correlation between the experimentally and theoretically derived data (Fig. 4). Even in the case of the 3-thienyl-system (4d and 6d), a correlation is seen within the compound series. Interestingly, the 3-thienyl group in 6d contributes to the LUMO, but not appreciably to the HOMO (see Fig. 3), which is perhaps surprising given the electron-rich nature of the thienyl group.
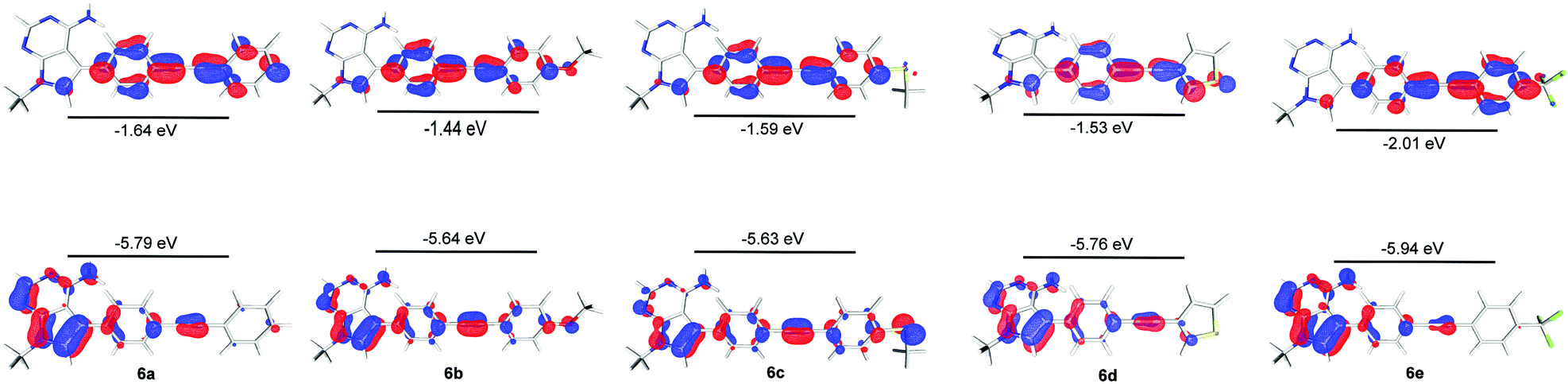 | ||
| Fig. 3 HOMO and LUMO surfaces (isosurface at 0.05 a.u.) and energies for compounds 6a–6e, calculated by DFT at the (RI-)PBE0/def2-TZVPP) level. | ||
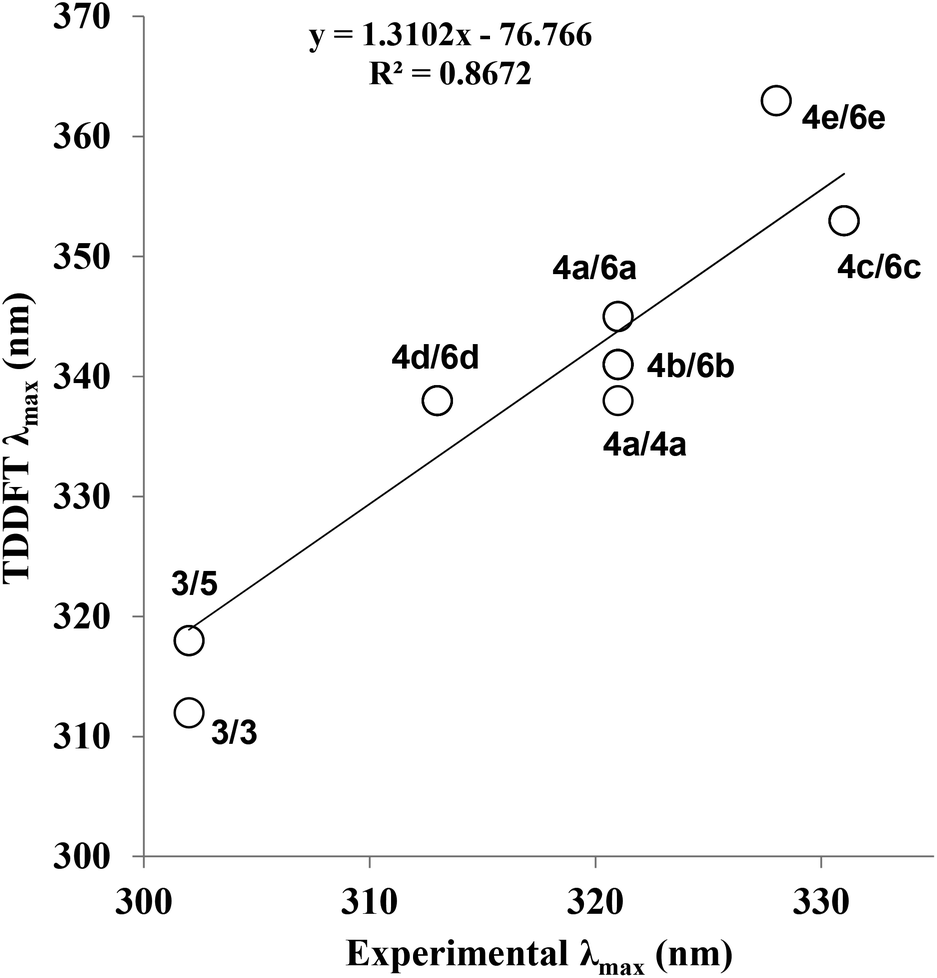 | ||
| Fig. 4 Correlation between calculated and experimental λmax (nm) for all optimised structures and conformational isomers (see ESI† for details). The structures that are plotted against each other are indicated as their compound numbers (e.g.4d/6d). TDDFT refers to Time-Dependent Density Functional Theory; calculated λmax values for structures 5, 6a–e are shown on the y-axis. The experimental λmax values for compounds 3 and 4a–e are shown on the x-axis. | ||
Fluorescence lifetimes were measured using time-correlated single photon counting (TCSPC), see Table 1.11 All of the 7-modified-7-deazaadenosines 4a–e were found to exhibit bi-exponential decays, with a major component that varied according to the substituent at the 7-position, and a minor component (ca. 10%) that was consistently ca. 0.5 ns. The fluorescence lifetime values recorded are in keeping with measurements on related tolan derivatives made by Marder and co-workers.12
Solvatochromism of fluorophores can be informative about the nature of the excited state and the electronic transitions giving rise to the absorption and emission spectra. For example, solvatochromic fluorescent probes can be used to gain insight into the polarity of the local environment.11 The variation in the absorption and emission spectra of 7-[4-(phenylethynyl)-phenyl]-7-deazaadenosine 4a was, therefore, investigated in a variety of solvents (Table 2).
| Solvent | λ max/nm | ε/104 M−1 cm−1 | λ em/nm | Stokes shift/cm−1 | Φ | Solvent dielectric constantb |
|---|---|---|---|---|---|---|
| a Solutions made from stocks of 4a in DMSO (2.5 × 10−3 M), due to poor compound solubility in non-polar solvents, which were diluted to 1–100 μM, so that A = 0.1 for each sample. The % (v/v) DMSO in the final solutions was typically <1%. b Measured at 20 °C. | ||||||
| H2Oa | 302 | 2.0 | 429 | 9800 | 0.15 | 80.1 |
| DMSO | 321 | 2.2 | 428 | 7790 | 0.74 | 47.2 |
| MeCN | 308 | 2.4 | 409 | 8020 | 0.62 | 36.6 |
| MeOH | 306 | 2.5 | 407 | 8110 | 0.46 | 33.0 |
| EtOH | 304 | 2.6 | 393 | 7450 | 0.31 | 25.3 |
| iPrOH | 304 | 3.1 | 396 | 7640 | 0.29 | 20.2 |
| CH2Cl2a | 312 | 2.5 | 394 | 6670 | 0.61 | 9.0 |
| EtOAca | 311 | 2.4 | 389 | 6450 | 0.33 | 6.1 |
| DMF | 318 | 2.7 | 420 | 7640 | 0.78 | 38.3 |
Both the absorption and emission maxima vary slightly when the solvent is changed. Whilst no distinct correlation was observed between wavelength and solvent polarity, the data shows a reasonable correlation between (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) A–F) and the orientation polarizability (Δf) in a Lippert–Mataga plot (Fig. 5), indicating the sensitivity of the fluorophore to its local chemical environment. The linear correlation is improved if one omits water (R2 = 0.89), which could suggest that 4a is behaving quite differently in this solvent. It is of particular note that the fluorescence quantum yield of 4a in water was found to be 0.15, which is significantly higher than 7-phenylethynyl-7-deaza-2′-deoxyadenosine (Φ = 0.02)6 and, indeed, the majority of other bespoke fluorescent C-modified nucleosides reported to date.
A–F) and the orientation polarizability (Δf) in a Lippert–Mataga plot (Fig. 5), indicating the sensitivity of the fluorophore to its local chemical environment. The linear correlation is improved if one omits water (R2 = 0.89), which could suggest that 4a is behaving quite differently in this solvent. It is of particular note that the fluorescence quantum yield of 4a in water was found to be 0.15, which is significantly higher than 7-phenylethynyl-7-deaza-2′-deoxyadenosine (Φ = 0.02)6 and, indeed, the majority of other bespoke fluorescent C-modified nucleosides reported to date.
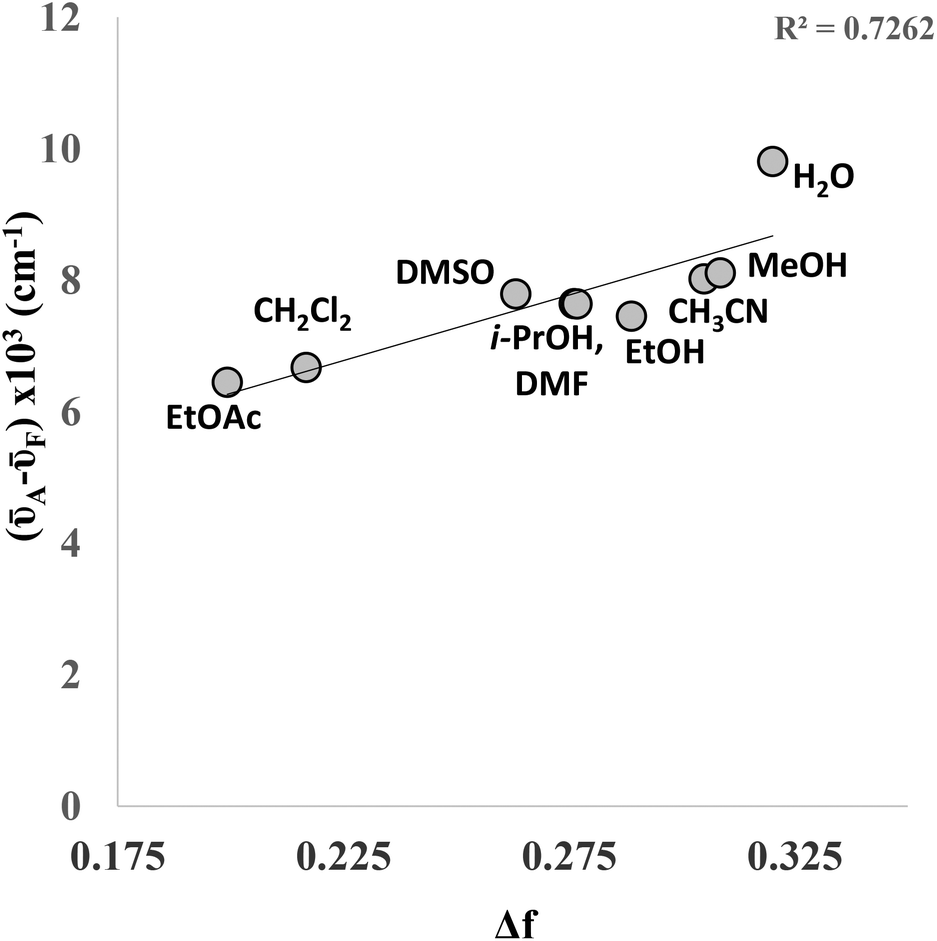 | ||
| Fig. 5 Lippert–Mataga plot for compound 4a. | ||
In conclusion, a novel class of 7-modified-7-deazaadenosine nucleosides 4a–e have been designed and synthesised from the corresponding 7-iodo nucleoside 11, using multi-step Pd-catalysed cross-coupling approaches. The nucleosides exhibit promising UV-visible absorption and fluorescence properties, with large quantum yields and absorption maxima shifted away from the intrinsic absorption associated with amino acid and nucleic acid residues. The changes in the absorption and emission wavelengths in different solvents indicate that the fluorescence properties of 4a are quite complex, whereas the Stokes shift exhibits a reasonable linear correlation with the orientation polarizability of the solvent. DFT calculations provided valuable insight into the fluorescent properties observed experimentally for reported compound 3, and the herein described novel compounds 4a–e.
We were particularly pleased with the quantum yield for 4a in water (Φ = 0.15). The incorporation of a compact fluorophore means that the chemical modification should not have a pronounced effect on either: (i) the biological compatibility of the nucleoside, or (ii) the stability of nucleic acids containing these C7-modified nucleotides. Applications for this class of fluorescent biomolecules will be reported in due course.
Acknowledgements
We thank BBSRC (S. De O./ref. BB/D527034/1), University of York and the Royal Society (I. J. S. F.) for funding this work. R. M. E. thanks Durham University for a Durham Doctoral Fellowship. We are grateful to EPSRC for funding the computational equipment used in this study (ref. EP/H011455/1). This work was supported in-part by EPSRC ‘ENERGY’ grant, ref. no. EP/K031589/1.Notes and references
- (a) C. R. Cremo, in Methods in Enzymology, ed. I. P. Gerard Marriott, Academic Press, 2003, vol. 360, pp. 128–177 Search PubMed; (b) D. M. Jameson and J. F. Eccleston, in Methods in Enzymology, ed. M. L. J. Ludwig Brand, Academic Press, 1997, vol. 278, pp. 363–390 Search PubMed; (c) C. R. Bagshaw, J. Cell Sci., 2001, 114, 459–460 CAS.
- (a) A. G. Firth, I. J. S. Fairlamb, K. Darley and C. G. Baumann, Tetrahedron Lett., 2006, 47, 3529–3533 CrossRef CAS PubMed; (b) T. E. Storr, J. A. Strohmeier, C. G. Baumann and I. J. S. Fairlamb, Chem. Commun., 2010, 46, 6470–6472 RSC.
- (a) H. Cahova, R. Pohl, L. Bednarova, K. Novakova, J. Cvacka and M. Hocek, Org. Biomol. Chem., 2008, 6, 3657–3660 RSC; (b) A. G. Firth, Synthesis and characterisation of fluorescent ribonucleotide substrates for DNA-dependent RNA polymerases, University of York, 2008 Search PubMed.
- (a) M. Vrabel, R. Pohl, I. Votruba, M. Sajadi, S. A. Kovalenko, N. P. Ernsting and M. Hocek, Org. Biomol. Chem., 2008, 6, 2852–2860 RSC; (b) S. Jäger, G. Rasched, H. Kornreich-Leshem, M. Engeser, O. Thum and M. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef PubMed; (c) G. F. Kaufmann, M. M. Meijler, C. Sun, D.-W. Chen, D. P. Kujawa, J. M. Mee, T. Z. Hoffman, P. Wirsching, R. A. Lerner and K. D. Janda, Angew. Chem., Int. Ed., 2005, 44, 2144–2148 CrossRef CAS PubMed. For a recent discussion of photoswitchable 7-substituted 7-deazaadenosine systems, see: (d) M. Singer and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8372–8377 CrossRef CAS PubMed.
- F. Seela and M. Zulauf, Helv. Chim. Acta, 1999, 82, 1878–1898 CrossRef CAS.
- F. Seela, M. Zulauf, M. Sauer and M. Deimel, Helv. Chim. Acta, 2000, 83, 910–927 CrossRef CAS.
- (a) A. E. Stiegman, V. M. Miskowski, J. W. Perry and D. R. Coulter, J. Am. Chem. Soc., 1987, 109, 5884–5886 CrossRef CAS; (b) A. E. Stiegman, E. Graham, K. J. Perry, L. R. Khundkar, L. T. Cheng and J. W. Perry, J. Am. Chem. Soc., 1991, 113, 7658–7666 CrossRef CAS.
- F. Seela and X. Ming, Tetrahedron, 2007, 63, 9850–9861 CrossRef CAS PubMed.
- S.-L. Zheng, S. Reid, N. Lin and B. Wang, Tetrahedron Lett., 2006, 47, 2331–2335 CrossRef CAS PubMed.
- E. C. Western, J. R. Daft, E. M. Johnson, P. M. Gannett and K. H. Shaughnessy, J. Org. Chem., 2003, 68, 6767–6774 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- C. D. Entwistle, J. C. Collings, A. Steffen, L.-O. Palsson, A. Beeby, D. Albesa-Jove, J. M. Burke, A. S. Batsanov, J. A. K. Howard, J. A. Mosely, S.-Y. Poon, W.-Y. Wong, F. Ibersiene, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, J. Mater. Chem., 2009, 19, 7532–7544 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation data of new compounds and detailed flurorescence measurements and spectra. See DOI: 10.1039/c4ob02081b |
| This journal is © The Royal Society of Chemistry 2015 |