
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCell-penetrating poly(disulfide)s: the dependence of activity, depolymerization kinetics and intracellular localization on their length†
Nicolas
Chuard
,
Giulio
Gasparini
,
Aurélien
Roux
,
Naomi
Sakai
and
Stefan
Matile
*
School of Chemistry and Biochemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/ Fax: +41 22 379 5123; Tel: +41 22 379 6523
First published on 6th November 2014
Abstract
We report that the depolymerization kinetics of cell-penetrating poly(disulfide)s depend exclusively on their length and propose a kinetic uptake model to explain why their intracellular destination changes with the increasing length from the endosomes over the cytosol to the nucleoli.
Substrate-initiated cell-penetrating poly(disulfide)s (siCPDs) have been introduced recently as a conceptually innovative approach for the covalent delivery of unmodified substrates into cells.1,2 Inspired by the ease and reliability of ring-opening disulfide-exchange polymerization3 on solid surfaces,4 we found that guanidinium-rich cell-penetrating poly(disulfide)s5 can be grown on substrates of free choice under the mildest conditions (Fig. 1).2 Right after uptake, the siCPD transporter is destroyed by depolymerization with glutathione in the cytosol.1 This liberates the native substrate and minimizes toxicity, overcoming one of the main limitations of classical cell-penetrating peptides (CPPs) and other protein transduction domain (PTD) mimics.6,7 To cross the membranes barrier, siCPDs bind thiols covalently at the surface and then pass through counterion-mediated transient micellar pores (Fig. 2). This promising counterion1,7-thiol-mediated1,8 uptake mechanism drives the concept of covalent delivery of unmodified substrates to the extreme and bypasses endosomal capture via endocytosis, overcoming the second main limitation of CPPs.6
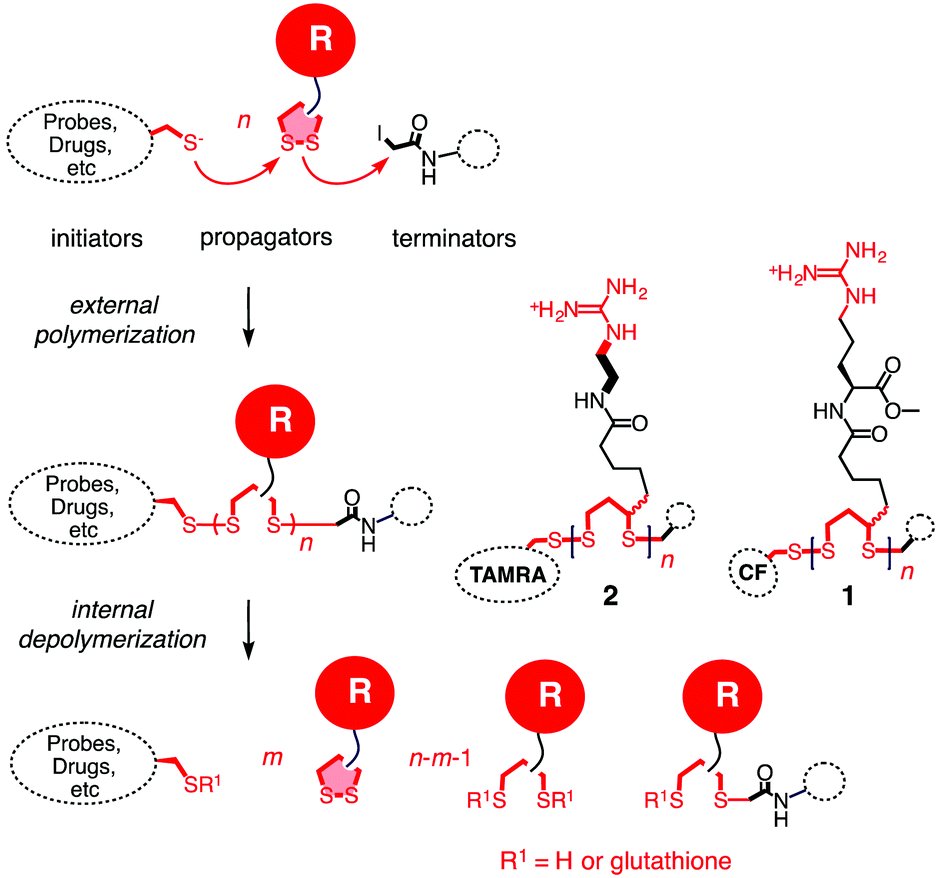 | ||
| Fig. 1 The concept of substrate-initiated CPDs. Fluorescent initiators were used to initiate the polymerization through ring-opening disulfide exchange between guanidinium-containing propagators. The polymerization was quenched using iodoacetamide as the terminator. | ||
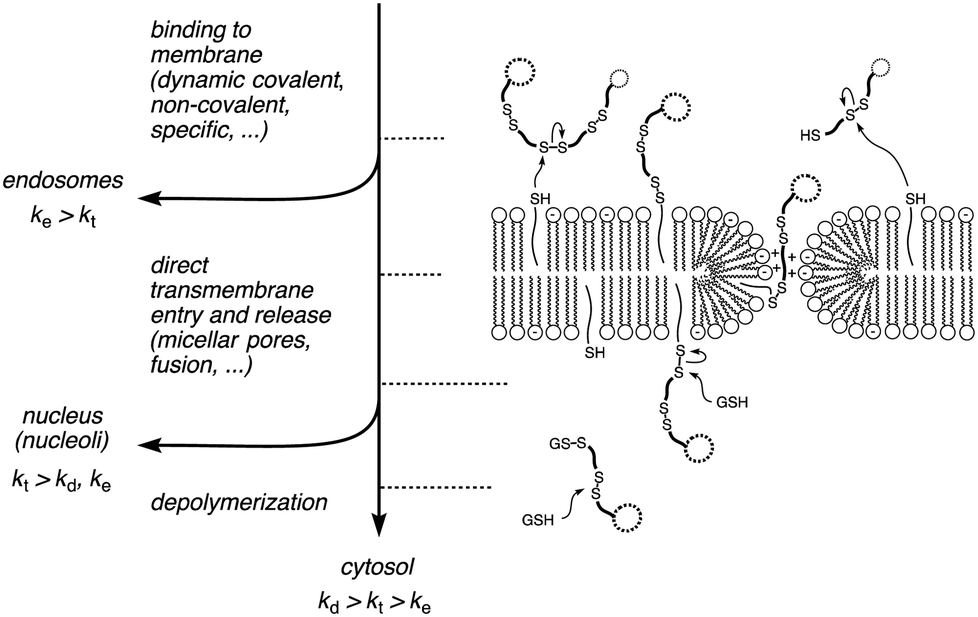 | ||
| Fig. 2 A kinetic model for cellular uptake of siCPDs: if the endocytosis (ke) is faster than the transmembrane penetration (kt), the siCPD stays mainly in the endosomes. If the depolymerization (kd) is faster than kt, the polymer stays in the cytosol, otherwise it can enter into the nucleus. In this model, kt stands for transmembrane translocation into the cytosol as well as entry into the nucleus, a simplifying assumption that remains to be verified. | ||
Interestingly, the intracellular localization of siCPDs with a nearly identical global structure differed significantly, at least within the HeLa cells.1 More hydrophobic siCPDs accumulated in the endosomes. Most remarkably, the original siCPD 113.5, made from lipoic acid and L-arginine with a molecular weight of Mw = 13.5 kDa, was found in the nucleoli, whereas siCPD 25.6 accumulated mainly in the cytosol. These differences suggested that siCPD 25.6 is destroyed immediately after reaching the cytosol, whereas the depolymerization of siCPD 1 is slow enough to enable the diffusion of the polymer into the nucleus and ultimately bind to the oligonucleotides in the nucleoli. Different depolymerization kinetics could originate from differences in the polymer length or structure. Different rates observed for siCPDs 113.5 and 25.6 suggested that either the polymer structure or polymer length would determine depolymerization kinetics. The objective of this study was to clarify this question, and to elaborate on the relationship between depolymerization kinetics and intracellular localization. We report that depolymerization kinetics of siCPDs depend exclusively – and more strongly than expected – on their length. According to a unified kinetic model (Fig. 2), these findings suggest that the increasing length of unbiased siCPDs changes their destination from the endosomes over the cytosol to the nucleoli.
The siCPDs 1 and 2 of different lengths were grown with strained propagators on carboxyfluorescein (CF) or carboxytetramethylrhodamine (TAMRA) initiators as described previously (Scheme S1†).1 The polymerizations were performed in neutral water at room temperature. It was possible to control the polymer size simply by changing the initiator concentration, monomer concentration or polymerization times (Table S1†).
We obtained polymer 1 ranging from 1 to 20 kDa average molecular weight Mw (Table S2†) and polymer 2 with Mw = 1–5 kDa (Table S3†), with a polydispersity index lower than PDI = 1.4 in most of the cases, according to GPC analysis.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1,2 So far we were unable to obtain polymer 2 with a Mw higher than 5 kDa, but comparisons between different functional groups were still possible because of the similarity in Mw to at least the shorter versions of polymer 1.
1,2 So far we were unable to obtain polymer 2 with a Mw higher than 5 kDa, but comparisons between different functional groups were still possible because of the similarity in Mw to at least the shorter versions of polymer 1.
The transport activity of the synthesized polymers was determined as fluorescent signals increased upon the addition of siCPDs to fluorescent vesicles (LUVs) composed of egg yolk phosphatidylcholine (EYPC) and loaded with 5(6)-carboxyfluorescein (CF). This well-established method allowed us to correlate the transmembrane transport activity (EC50) of siCPDs carrying the two different functional groups with their chain length in a rapid and straightforward way. Interference from the CF initiator in the siCPD transporter was negligible because the concentration of CF used in the assay was much higher (50 mM intravesicular CF against <1 μM transporter). The activity of both classes of polymers decreased with decreasing length, resulting in an increase of the EC50 (Fig. 3A). Interestingly, the activity in vesicles also depended on the type of functional group carried by the polymers. The simple guanidinium cation in propagator 2 gave more effective polymers than the arginine derivative 1. This was evident by comparing polymers 11.9 and 21.1. Although being formed by the same number of monomer units, the activity was more than 10 times higher when the short linker of 2 was present between the guanidinium cation and the lipoic acid.
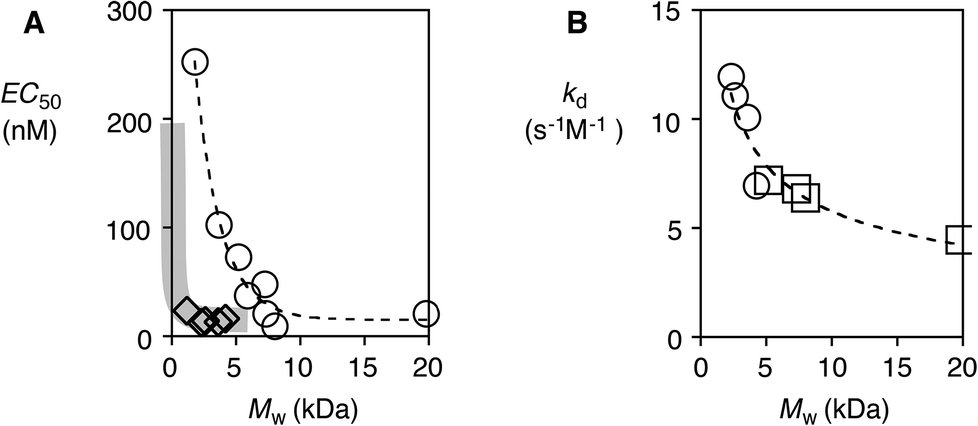 | ||
| Fig. 3 (A) Dependence of EC50 on the molecular weight of polymers 1 (○) and 2 (◊). (B) The rate constant of depolymerization as a function of the molecular weight of polymers 1 (□) and 2 (○). | ||
To determine whether differences in the transmembrane activity or differences in the length are responsible for the different localizations within cells,1 we investigated the depolymerization kinetics of polymers of different lengths under conditions that mimic the cytoplasm of HeLa cells. Polymers were incubated at 37 °C in buffered solutions (pH = 7.4) containing different amounts of glutathione (GSH, 0.1–5.0 mM). The depolymerization kinetics were then followed monitoring the loss in activity in LUVs with depolymerization.9 GPC and product analysis by HPLC2b confirmed that the observed decrease in activity in LUVs indeed originates from depolymerization. For each polymer, the half-life time t50 was determined as a function of the concentration of GSH (Table S4, Fig. S4†) by comparing the maximal transport activity Y of the polymer in LUVs with incubation times, using the Hill equation (eqn (S3)†). Once the t50 for each polymer at different concentrations of GSH was obtained, linear regression was applied. Using the following equation (eqn (1)), the depolymerization rate constants kd were determined for each polymer (Table S5†).
| logt50 = (1 − n) log[GSH] + log((2n−1 − 1)/kd(n − 1)) | (1) |
Depolymerization was found to follow second-order kinetics (1: n = 1.84–2.07, 2: n = 1.77–1.89). The kd decreased with the polymer length as expected (Fig. 3B). Most importantly, depolymerization kinetics were independent of the nature of the polymer (Fig. 3B, dashed line). For example, polymers 15.7 and 25.0, containing the same number of monomers, had a kd of 7.19 and 6.96 s−1 M−1, respectively, although the latter showed five times higher transport activity. This result demonstrated that the depolymerization of siCPDs by GSH depends only on their length, whereas their transport activity can depend on both the polymer length as well as the structure of their side chains.
To better understand the influence of functional groups on the intracellular localization, HeLa cells were incubated with polymers 15.7 and 25.0. Because these two polymers are composed of the same number of monomers, eventual differences in intracellular localization would have to originate from their different structures. In the images recorded by confocal laser scanning microscopy (CSLM) after 15 minutes of incubation, the localization of the fluorophores was reported independent of the degree of depolymerization of the siCPD transporters. As shown in Fig. 4A and B, both polymers 15.7 and 25.0 localized mainly in the cytosol, although they could also be seen in the nucleoli and endosomes. This distribution for polymer 25.0 was in agreement with previously obtained results with siCPD 25.6 of similar length (Fig. 4D).1 However, localization of 15.7 in the cytosol and also endosomes differed clearly from the previously found accumulation of the clearly longer siCPD 113.5 in the nucleoli (Fig. 4C).1 The fluorescence patterns observed for the endosomes (particularly Fig. 4A) and nucleoli (particularly Fig. 4C) were characteristic to an extent that co-localization experiments did not appear necessary.
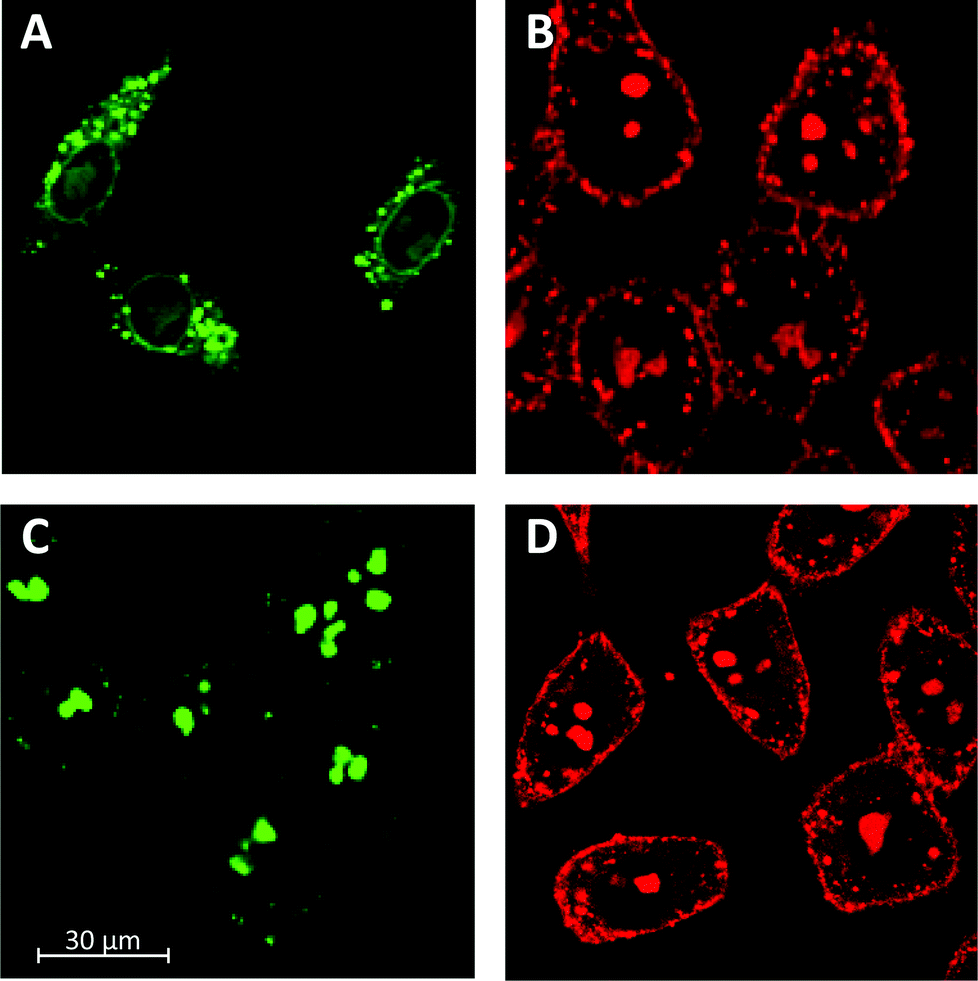 | ||
| Fig. 4 CLSM images of HeLa cells after 15 min of incubation with 500 nM polymer 15.7 (A) and polymer 25.0 (B) compared to polymer 113.5 (C) and 25.6 (D) at 37 °C in Leibovitz medium. | ||
Taken together, depolymerization kinetics of siCPDs depend on their length and not on their structure, their activity in vesicles and their intracellular localization can depend on their length and on their structure. Slow transmembrane translocation increases endosomal capture (e.g.15.7, Fig. 3A○ and 4A). Fast transmembrane translocation and slow depolymerization lead to selective delivery to the nucleoli (e.g., 113.5, Fig. 3A○, B□ and 4C). Fast transmembrane translocation and fast depolymerization increase accumulation in the cytosol (e.g.25.0, 25.6, Fig. 3A◊, B○, 4B and D). These insights support a simple kinetic model for cellular uptake of unbiased transporters, i.e. without specifically added recognition motifs for intracellular targeting (Fig. 2).7 If endocytosis is faster than direct transmembrane translocation (ke > kt), transporters go to the endosomes. If entry into cytosol and nucleus are faster than endocytosis and depolymerization (kt > ke, kd), transporters go to the nucleus. If entry into cytosol and nucleus are faster than endocytosis but slower than depolymerization (kd > kt > ke), transporters go to the cytosol. Examples of endosomal delivery with (ke > kt) include too hydrophobic or too short and inactive siCPDs (Fig. 4A, or CPPs, etc.).1,7 Redirection of endosomal delivery (i.e., endosomal bypass rather than endosomal escape) has been achieved by siCPD elongation (Fig. 4C) or an accelerated direct transmembrane entry (pyrenebutyrate trick).7 siCPDs of intermediate length go to the cytosol because of kd > kt > ke, longer siCPDs are found in the nucleoli because of kt > kd, ke. We feel that this unified kinetic model for unbiased siCPD delivery, although certainly oversimplifying in many situations, provides a useful general basis to guide future studies on this important topic.
The kinetic model assumes that the velocities of direct transmembrane translocation into the cytosol and entry into the nucleus are in the same range. This assumption is very speculative. The apparent labeling of the nuclear envelope, a double lipid bilayer membrane, with the shorter, less active polymer 15.7 is an intriguing observation from this point of view (Fig. 4A). More detailed studies on the nuclear entry of siCPDs are ongoing and will be reported in due course.
Acknowledgements
We thank Gevorg Sargsyan, Eun-Kyoung Bang and Sandra Ward for their assistance with synthesis and experiments, the NMR and the Sciences Mass Spectrometry (SMS) platforms for services, and the University of Geneva, the European Research Council (ERC Advanced Investigator), the National Centre of Competence in Research (NCCR) Chemical Biology, the NCCR Molecular Systems Engineering and the Swiss NSF for financial support.Notes and references
- G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- (a) E.-K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed; (b) E.-K. Bang, S. Ward, G. Gasparini, N. Sakai and S. Matile, Polym. Chem., 2014, 5, 2433–2441 RSC.
- E.-K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC.
- (a) H. Hayashi, A. Sobczuk, A. Bolag, N. Sakai and S. Matile, Chem. Sci., 2014, 5, 4610–4614 RSC; (b) E. Orentas, M. Lista, N.-T. Lin, N. Sakai and S. Matile, Nat. Chem., 2012, 4, 746–750 CrossRef CAS PubMed; (c) N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 18542–18545 CrossRef CAS PubMed; (d) M. Lista, J. Areephong, N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 15228–15231 CrossRef CAS PubMed; (e) N. Sakai, M. Lista, O. Kel, S. Sakurai, D. Emery, J. Mareda, E. Vauthey and S. Matile, J. Am. Chem. Soc., 2011, 133, 15224–15227 CrossRef CAS PubMed.
- (a) D. Oupicky and J. Li, Macromol. Biosci., 2014, 14, 908–922 CrossRef CAS PubMed; (b) T.-I. Kim and S. W. Kim, React. Funct. Polym., 2011, 71, 344–349 CrossRef CAS PubMed; (c) M. Piest and J. F. J. Engbersen, J. Controlled Release, 2011, 155, 331–340 CrossRef CAS PubMed; (d) C. R. Drake, A. Aissaoui, O. Argyros, M. Thanou, J. H. G. Steinke and A. D. Miller, J. Controlled Release, 2013, 171, 81–90 CrossRef CAS PubMed; (e) H. Zeng, H. C. Little, T. N. Tiambeng, G. A. Williams and Z. Guan, J. Am. Chem. Soc., 2013, 135, 4962–4965 CrossRef CAS PubMed; (f) S. Son, R. Namgung, J. Kim, K. Singha and J. W. Kim, Acc. Chem. Res., 2012, 45, 1100–1112 CrossRef CAS PubMed; (g) Y. Lee, H. Mo, H. Koo, J.-Y. Park, M. Y. Cho, G.-W. Jin and J.-S. Park, Bioconjugate Chem., 2007, 18, 13–18 CrossRef CAS PubMed; (h) K. Miyata, Y. Kakizawa, N. Nishiyama, A. Harada, Y. Yamasaki, H. Koyama and K. Kataoka, J. Am. Chem. Soc., 2004, 126, 2355–2361 CrossRef CAS PubMed.
- (a) E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef CAS PubMed; (b) F. Sgolastra, B. M. Deronde, J. M. Sarapas, A. Som and G. N. Tew, Acc. Chem. Res., 2013, 135, 2977–2987 CrossRef PubMed; (c) S. Futaki, H. Hirose and I. Nakase, Curr. Pharm. Des., 2013, 19, 2863–2868 CrossRef CAS; (d) E. Lei, M. P. Pereira and S. O. Kelley, Angew. Chem., Int. Ed., 2013, 52, 9660–9663 CrossRef CAS PubMed; (e) R. Weinstain, E. N. Savariar, C. N. Felsen and R. Y. Tsien, J. Am. Chem. Soc., 2014, 136, 874–877 CrossRef CAS PubMed; (f) M. Lnoue, W. Tong, J. D. Esko and Y. Tor, ACS Chem. Biol., 2013, 8, 1383–1388 CrossRef PubMed; (g) J. S. Appelbaum, J. R. LaRochelle, B. A. Smith, D. M. Balkin, J. M. Holub and A. Schepartz, Chem. Biol., 2012, 19, 819–830 CrossRef CAS PubMed; (h) J. Fernandez-Carneado, M. Van Gool, V. Martos, S. Castel, P. Prados, J. De Mendoza and E. Giralt, J. Am. Chem. Soc., 2005, 127, 869–874 CrossRef CAS PubMed.
- (a) T. Takeuchi, M. Kosuge, A. Tadokoro, Y. Sugiura, M. Nishi, M. Kawata, N. Sakai, S. Matile and S. Futaki, ACS Chem. Biol., 2006, 1, 299–303 CrossRef CAS PubMed; (b) N. Sakai, S. Futaki and S. Matile, Soft Matter, 2006, 2, 636–641 RSC.
- (a) A. G. Torres and M. J. Gait, Trends Biotechnol., 2012, 30, 185–190 CrossRef CAS PubMed; (b) T. Li and S. Takeoka, Int. J. Nanomedicine, 2014, 9, 2849–2861 Search PubMed; (c) I. D. Alves, A. Walran, C. Bechara and S. Sagan, Curr. Protein Pept. Sci., 2012, 13, 658–671 CrossRef CAS.
- (a) T. Miyatake, M. Nishihara and S. Matile, J. Am. Chem. Soc., 2006, 128, 12420–12421 CrossRef CAS PubMed; (b) G. Das and S. Matile, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5183–5188 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02060j |
| This journal is © The Royal Society of Chemistry 2015 |