
Development of a novel fluorescence probe capable of assessing the cytoplasmic entry of siderophore-based conjugates†
Hyeon Seok
Kim
,
Woon Young
Song
and
Hak Joong
Kim
*
Department of Chemistry, College of Science, Korea University, Seoul 136-701, Republic of Korea. E-mail: hakkim@korea.ac.kr; Tel: +82-2-3290-3148
First published on 22nd October 2014
Abstract
A novel fluorescence probe capable of assessing the cytoplasmic entry of siderophore-based conjugates was synthesized and evaluated by photochemical characterization and cell-based assays. The specific responsiveness to the cytoplasmic entry of the probe was implemented by adopting a disulfide linker, whose cleavage under the reducing conditions of the cytoplasm induced the display of a distinctive fluorescence signal.
Siderophores are high affinity iron chelators produced and utilized by most bacteria as the primary means of assimilating the iron from the aerobic environment, in which the solubility of Fe(III) is extremely low.1,2 Because of the essentiality of the iron for many biological functions, including oxygen storage, energy transfer, and catalysis, the mechanisms related to the siderophore utilization, such as biosynthesis, secretion, and active uptake, have been considered as promising novel antibiotic targets.3 One of the actively pursued approaches is to utilize the siderophore as the drug delivery vector by constructing siderophore-drug conjugates (SDCs).4–7 This design, inspired by natural product antibiotics such as salmycins and albomycins, has been anticipated to be effective in treating Gram-negative pathogens because their drug resistance is often associated with the restricted membrane permeability.8,9
Indeed, many artificial SDCs have been prepared and tested for their antibiotic activity against various Gram-negative bacteria. Interestingly, increased or similar activity, compared to the parent antibiotic drug, has been observed mostly when the SDCs feature drug functions on periplasmic targets, such as penicillin-binding proteins (PBPs).10–13 The conjugation with quinolone antibiotics targeting DNA gyrase in the cytoplasm has been found ineffective for Gram-negative bacteria, suggesting that the attachment of a siderophore might attenuate the activity of the parent drug,14 or the corresponding SDCs might have limited access to the cytoplasm.12,15 Most siderophores, except for pyoverdines, are known to deliver the iron intracellularly by penetration into the cytoplasm through designated uptake systems composed of TonB-dependent outer membrane receptors, periplasmic binding proteins, and ATP-dependent inner membrane permease complexes.16 Thus, if the conjugation of drug molecules to the siderophore has detrimental effects on the interactions of siderophores with uptake machineries located at the periplasm and/or the inner membrane, the cytoplasmic entry of SDCs can be inhibited.
The restricted access of SDCs only to the periplasm can be problematic in finding the potential of SDC antibiotics because the repertoire of drugs to be conjugated becomes significantly limited. Nonetheless, there has been no systematic study to address this issue until very recently, when Nolan and co-workers have found that the size of conjugated molecules is an important parameter that affects the transport of enterobactin-based conjugates into the cytoplasm of Escherichia coli and Pseudomonas aeruginosa.14 In this regard, we herein report the development of a novel fluorescence siderophore probe responsive to the cytoplasmic entry, thus allowing the detailed analysis of the trafficking of siderophore-based conjugates. In particular, the design concept, preparation of a model probe 6, photochemical characterization, as well as cell-based evaluation are described.
The concept of our new probe design is depicted in Fig. 1. Essentially, its structural organization composed of a fluorophore, a siderophore, and a linker, is similar to the previously known siderophore–fluorophore conjugates,17–21 but distinctive because of the presence of a disulfide bond on the linker. In bacteria, such as E. coli, the cytoplasm is maintained under reducing conditions by free thiols, such as the thioredoxins and glutathione, whereas the periplasm is maintained in an oxidative state primarily by a disulfide-forming enzyme DsbA.22 Accordingly, our probe can be specifically responsive upon entry into the cytoplasm, where cellular thiols cleave the disulfide bond, triggering the liberation of a free amine form of the fluorophore with altered photochemical property (State I or II to State III, Fig. 1). This disulfide cleavage-based strategy has been exploited in the development of a number of intracellular thiol probes and drug delivery systems, particularly in mammalian cells.23–25 In addition, the new probe is also capable of detecting the reversible Fe(III) binding, similar to other siderophore–fluorophore conjugates because this event often accompanies the fluorescence quenching (State I to State II, Fig. 1). Thus, our novel probe design allows the simultaneous monitoring of two separate behaviours of a siderophore-based conjugate, chelation to Fe(III) as well as cytoplasmic entry.
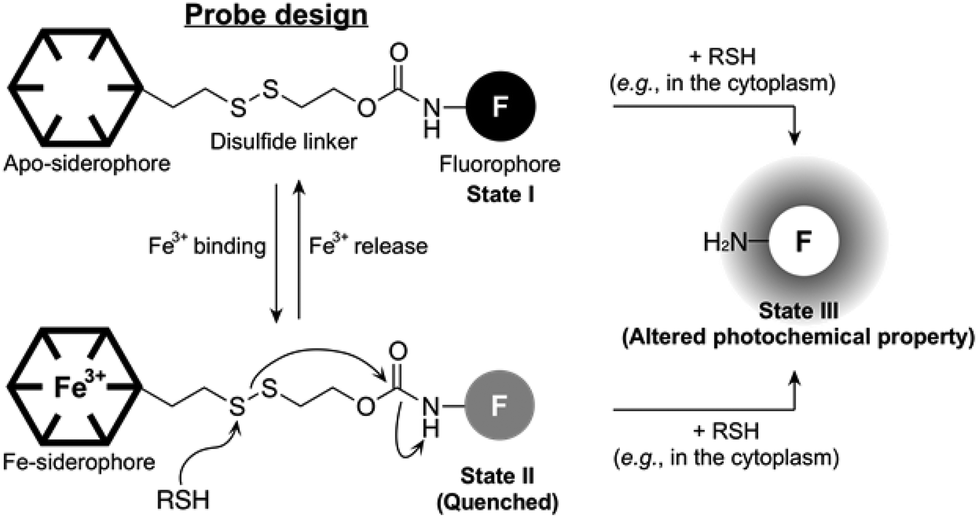 | ||
| Fig. 1 The concept of the new probe design. | ||
As a model probe, we decided to synthesize compound 6, featuring 4-amino-1,8-naphthalimide (1) and desferrioxamine B (DFOB, 5) as the fluorophore and the siderophore, respectively, which are connected via a disulfide linker 2. The synthesis of 6 relied on successive carbamate formations on both free hydroxy ends of 2 with fluorophore 1 and DFOB (5), as described in Scheme 1. Briefly, the connection between 1 and 2 was effected by treatment with triphosgene to afford 3. The second carbamate linkage was elaborated by priming the free hydroxyl group as the NHS anhydride. The subsequent application of DFOB (5) to the activated intermediate 4 in a buffered solution successfully yielded probe 6 after purification by reverse-phase HPLC.
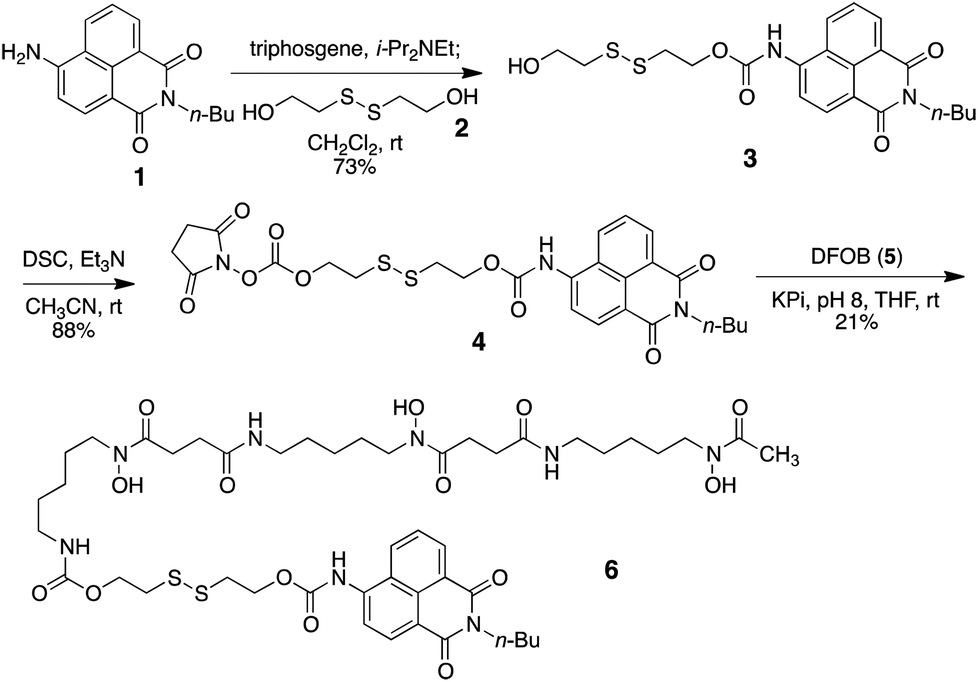 | ||
| Scheme 1 Synthesis of a model probe 6. | ||
After the synthesis of probe 6, spectroscopic characterization was conducted to validate the probe design in vitro. In particular, the changes in the absorption and fluorescence profiles upon exposure to glutathione (GSH), a representative cellular free thiol, or Fe(III) or both were monitored. As shown in Fig. 2A, the treatment of 6 with GSH induced a redshift (from 370 nm to 440 nm) in absorption, consistent with the liberation of 1 (Fig. S2†). The fluorescence also underwent changes, in which probe 6 alone displayed a strong emission (max at ca. 470 nm) upon excitation at 370 nm, whereas only marginal emission was observed from the sample treated with GSH (Fig. 2B). In contrast, the excitation at 440 nm gave rise to a strong emission (max at ca. 540 nm) from the GSH-treated sample, in contrast to 6 alone that showed no response (Fig. 2C). Then, the effect of the iron binding was tested. While no apparent change in the position of the absorption maximum was observed upon exposure to FeCl3 (Fig. 2A and S1†), the Fe(III) chelation to 6 was found to effectively quench the fluorescence emission at 370 nm excitation, as anticipated (Fig. 2B). The cleavage of the disulfide bond by GSH appeared to be not affected by association with Fe(III) (Fig. 2C). Collectively, these spectral data support that probe 6 is differentially, though not fully independently, responsive to the Fe(III)-binding and the cytoplasmic entry as designed, where the events can be readily assessed by monitoring two separate fluorescence channels: excitation near 370 nm and 440 nm, respectively.
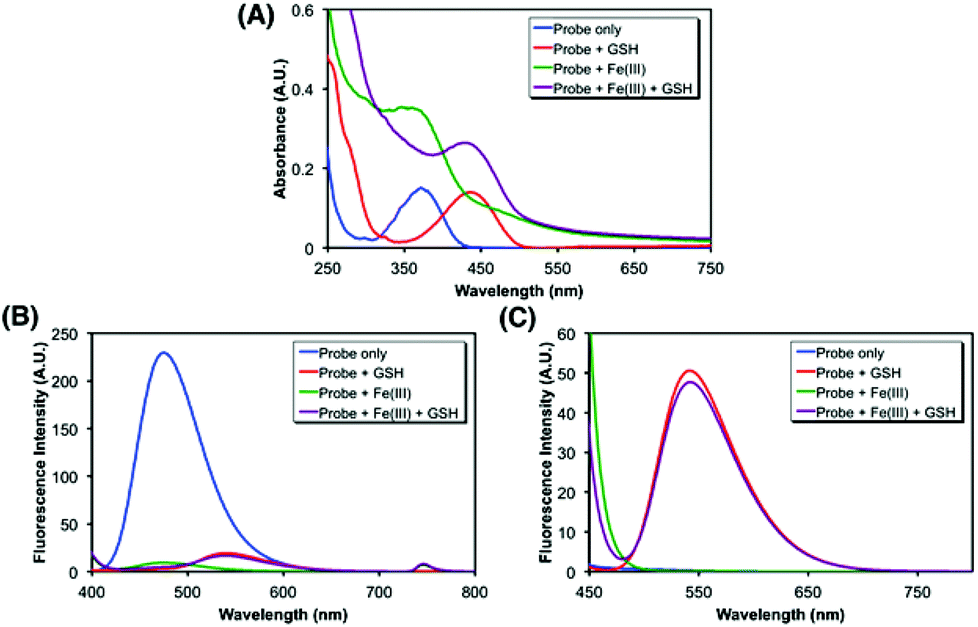 | ||
| Fig. 2 Spectroscopic characterization of 6 (10 μM, blue) under different conditions selectively involving glutathione (5 mM, red) or FeCl3 (100 μM, green) or both (violet) at 37 °C in 100 mM Tris·HCl (pH 8, 50% methanol). (A) UV/Vis absorption spectra; fluorescence spectra upon the excitation (B) at 370 nm, (C) at 440 nm. | ||
To validate the function of probe 6 in the cell-based settings, two Gram-negative strains, Pseudomonas putida ATCC 12633 and E. coli BW25113, were selected. Although they do not endogenously produce DFOB, they are capable of utilizing it as a xenosiderophore.26,27E. coli BW25113, a K-12 derivative, depends on FhuCDB for the inner membrane transport of DFOB, but this strain lacks a high affinity outer membrane receptor for DFOB. Thus, we transformed E. coli with pFU2 containing the foxA gene, an outer membrane ferrioxamine receptor from Yersinia enterolitica, to increase the DFOB uptake capacity.28
Each strain was cultured in an iron-deficient succinate medium up to the mid-exponential phase, and Fe(III)-6 (5 μM) was added after the adjustment of OD600 to 1. After incubating for 1.5 h at 37 °C, the fluorescence emissions were monitored in a 96-well microplate reader. As shown in Fig. 3A and B, the incubation of Fe(III)-6 with P. putida exhibited discernible fluorescence increases upon excitation at both 355 nm and 460 nm, which correspond to the Fe(III) release from Fe(III)-6 and the liberation of fluorophore 1via the cleavage of the disulfide bond in the probe, respectively. Treatment with NaN3, a known inhibitor for energy-dependent processes, significantly decreased the emissions by 39% and 31% at 355 nm and 460 nm excitations, respectively. In addition, co-treatment with ferrioxamine B (FOB) also led to slight fluorescence decreases at 355 nm and 460 nm excitations, both by 13%. These antagonizing effects by NaN3 and FOB collectively support that the observed fluorescence changes are driven by the actions of DFOB-specific uptake machineries. Fluorescence increase at 460 nm excitation, requiring the presence of free thiols, is particularly noteworthy because it indicates that Fe(III)-6 is capable of penetrating inside the cytoplasm of P. putida. The fluorescence increase at 355 nm excitation suggests that the dissociation of Fe(III) from the ferrioxamine scaffold in Fe(III)-6 would precede the disulfide cleavage upon its delivery to the cytoplasm, and some portions of iron-free 6 would remain intact. In contrast, the incubation of Fe(III)-6 with E. coli did not elicit any fluorescence change either upon excitation at 355 nm or at 460 nm (Fig. 3C and D). The possibility of insufficient cellular thiol contents in the E. coli cytoplasm or the slow kinetics of the thiol-promoted disulfide cleavage could be ruled out because on treatment with Fe(III)-6, the E. coli lysates displayed strong fluorescence emission at 460 nm excitation (Fig. S5†). These results suggest that the E. coli strain used herein is incapable of accepting Fe(III)-6, which is indicative of the adverse effects of the fluorophore appendage in Fe(III)-6 on interactions with DFOB-uptake machineries in E. coli. The difference in the transport capacity for Fe(III)-6 between P. putida and E. coli is intriguing, and it suggests that the DFOB-specific uptake machineries in these two organisms have different ligand specificity. While the sequence alignments could provide some insight into this observation (see the ESI, Fig. S9†), a more rigorous answer would require extensive structural analysis and binding studies.
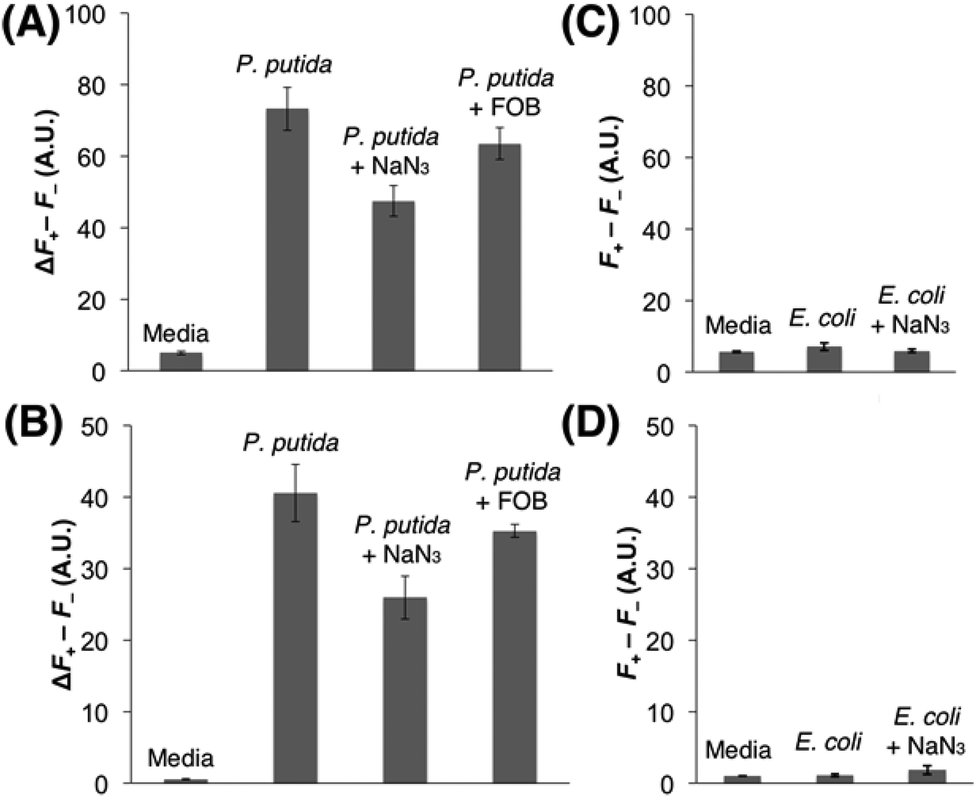 | ||
| Fig. 3 Evaluation of the cellular uptake of Fe(III)-6 in Pseudomonas putida [(A), (B)] and Escherichia coli [(C), (D)]. The differences in the fluorescence signals between the presence and the absence of the probe, Fe(III)-6, (F+–F−) are presented. (A), (C) emission at 460 nm upon excitation at 355 nm. (B), (D) emission at 535 nm upon excitation at 460 nm. The test condition is indicated on the top of each column. All the experiments were performed at least in triplicates and the error bars indicate the standard deviations. | ||
Conclusions
Herein, we presented a novel fluorescence probe design capable of assessing the cytoplasmic entry of siderophore-based conjugates. The model probe based on DFOB, Fe(III)-6, was observed to be accessible into the cytoplasm of P. putida, but not of E. coli. These contrasting results clearly demonstrate that our probe design can be readily utilized to compare the cytoplasmic entry of siderophore-based conjugates among different bacteria. Our probe design is distinguished from other commonly used methods, to investigate the cellular uptake of siderophores, based on the use of radioactive Fe(III)29 or conventional siderophore–fluorophore conjugates,17–21,30 in which its response is directly related to the cellular location of the probe itself, while the other methods correspond to the trajectory of Fe(III), and not that of the siderophore. Thus, our new probe design would allow a more detailed tracing of the cellular uptake pathway of siderophore-based conjugates, particularly the cytoplasmic entry. This disulfide linker-based design is readily adaptable to other siderophores, and thus can be used for the direct comparison of the cytoplasmic uptake efficiency of various different siderophore-based conjugates, providing information that should make significant contributions to the discovery of new potent SDC antibiotics.Acknowledgements
This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF-2012R1A1A1042665, NRF20100020209) and TJ Science Fellowship of POSCO TJ Park Foundation. We would also like to thank Prof. Klaus Hantke for providing us pFU2 plasmid.Notes and references
- M. Miethke and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2007, 71, 413 CrossRef CAS PubMed.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637 RSC.
- T. L. Foley and A. Simeonov, Expert Opin. Drug Discovery, 2012, 7, 831 CrossRef CAS PubMed.
- M. G. P. Page, Ann. N. Y. Acad. Sci., 2013, 1277, 115–126 CrossRef CAS PubMed.
- C. Ji, R. E. Juárez-Hernández and M. J. Miller, Future Med. Chem., 2012, 4, 297 CrossRef CAS PubMed.
- J. M. Roosenberg, Y. M. Lin, Y. Lu and M. J. Miller, Curr. Med. Chem., 2000, 7, 159 CrossRef CAS.
- A. Górska, A. Sloderbach and M. P. Marszałł, Trends Pharmacol. Sci., 2014, 1 Search PubMed.
- A. H. Delcour, Biochim. Biophys. Acta, 2009, 1794, 808 CrossRef CAS PubMed.
- M. N. Alekshun and S. B. Levy, Cell, 2007, 128, 1037 CrossRef CAS PubMed.
- P. G. Higgins, D. Stefanik, M. G. P. Page, M. Hackel and H. Seifert, J. Antimicrob. Chemother., 2012, 67, 1167 CrossRef CAS PubMed.
- C. Ji, P. A. Miller and M. J. Miller, J. Am. Chem. Soc., 2012, 134, 9898 CrossRef CAS PubMed.
- T. A. Wencewicz and M. J. Miller, J. Med. Chem., 2013, 56, 4044 CrossRef CAS PubMed.
- T. Zheng and E. M. Nolan, J. Am. Chem. Soc., 2014, 136, 9677 CrossRef CAS PubMed.
- T. Zheng, J. L. Bullock and E. M. Nolan, J. Am. Chem. Soc., 2012, 134, 18388 CrossRef CAS PubMed.
- R. E. Juárez-Hernández, P. A. Miller and M. J. Miller, ACS Med. Chem. Lett., 2012, 3, 799 CrossRef PubMed.
- I. J. Schalk and L. Guillon, Amino Acids, 2013, 44, 1267 CrossRef CAS PubMed.
- H. Weizman, O. Ardon, B. Mester, J. Libman, O. Dwir, Y. Hadar, Y. Chen and A. Shanzer, J. Am. Chem. Soc., 1996, 118, 12368 CrossRef CAS.
- R. Nudelman, O. Ardon, Y. Hadar, Y. Chen, J. Libman and A. Shanzer, J. Med. Chem., 1998, 41, 1671 CrossRef CAS PubMed.
- H. Kornreich-Leshem, C. Ziv, E. Gumienna-Kontecka, R. Arad-Yellin, Y. Chen, M. Elhabiri, A.-M. Albrecht-Gary, Y. Hadar and A. Shanzer, J. Am. Chem. Soc., 2005, 127, 1137 CrossRef CAS PubMed.
- H. Ouchetto, M. Dias, R. Mornet, E. Lesuisse and J.-M. Camadro, Bioorg. Med. Chem., 2005, 13, 1799 CrossRef CAS PubMed.
- M. Hannauer, Y. Barda, G. L. A. Mislin, A. Shanzer and I. J. Schalk, J. Bacteriol., 2010, 192, 1212 CrossRef CAS PubMed.
- J. C. Bardwell, K. McGovern and J. Beckwith, Cell, 1991, 67, 581 CrossRef CAS.
- M. H. Lee, J. H. Han, P.-S. Kwon, S. Bhuniya, J. Y. Kim, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 1316 CrossRef CAS PubMed.
- M. H. Lee, J. Y. Kim, J. H. Han, S. Bhuniya, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 12668 CrossRef CAS PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071 CrossRef CAS PubMed.
- E. Jurkevitch, Y. Hadar, Y. Chen, P. Yakirevitch, J. Libman and A. Shanzer, Microbiology, 1994, 140, 1697 CrossRef CAS PubMed.
- M. Sauer, K. Hantke and V. Braun, J. Bacteriol., 1987, 169, 2044 CAS.
- A. J. Bäumler and K. Hantke, Mol. Microbiol., 1992, 6, 1309 CrossRef PubMed.
- L. A. de Weger, J. J. van Arendonk, K. Recourt, G. A. van der Hofstad, P. J. Weisbeek and B. Lugtenberg, J. Bacteriol., 1988, 170, 4693 CAS.
- S. Noël, L. Guillon, I. J. Schalk and G. L. A. Mislin, Org. Lett., 2011, 13, 844 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data for all new compounds, copies of spectra, and results of photochemical characterization and cell-based assays. See DOI: 10.1039/c4ob01810a |
| This journal is © The Royal Society of Chemistry 2015 |