
Solid phase synthesis of 1,3,4-oxadiazin-5 (6R)-one and 1,3,4-oxadiazol-2-one scaffolds from acyl hydrazides†
Bani Kanta
Sarma
a,
Xiaodan
Liu
b,
Hao
Wu
b,
Yu
Gao
b and
Thomas
Kodadek
*b
aDepartment of Chemistry, School of Natural Sciences, Shiv Nadar University, Dadri, Uttar Pradesh-201314, India
bDepartments of Chemistry and Cancer Biology, Scripps Research Institute, Scripps Florida, 130 Scripps Way, #3A2, Jupiter, Fl 33458, USA. E-mail: kodadek@scripps.edu
First published on 30th October 2014
Abstract
Solid phase synthesis of 1,3,4-oxadiazin-5(6R)-one and 1,3,4-oxadiazol-2-one scaffolds from resin-bound acyl hydrazides is described. We demonstrate here that the reactions of resin-bound aryl or hetero-aromatic acyl hydrazides with 2-substituted-2-bromoacetic acids and 4-nitrophenyl chloroformate and subsequent treatment with DIEA lead to intramolecular cyclization reactions to produce six-membered 1,3,4-oxadiazin-5(6R)-ones and five-membered 1,3,4-oxadiazol-2-ones, respectively. We also show that acyl hydrazide-derived 1,3,4-oxadiazol-2-ones may be useful serine hydrolase inhibitors.
To facilitate the rapid and inexpensive discovery of bioactive compounds, several laboratories have focused on the development of large libraries of oligomeric compounds displayed on hydrophilic TentaGel beads.1–6 These can be screened for ligands to a protein of interest at a fraction of the cost of a functional, automated high-throughput screen. However, the chemical diversity of these libraries is limited by the availability of reactions efficient enough to produce the extremely high yields needed for the production of high quality libraries by split and pool solid-phase synthesis. In particular, heterocyclic compounds, which are so commonly found in bioactive small molecules, are underrepresented in such libraries.7,8 Here we report facile methods for the solid-phase synthesis of six-membered 1,3,4-oxadiazin-5-one and five-membered 1,3,4-oxadiazol-2-one rings (Fig. 1A) using exceedingly efficient reactions suitable for the construction of combinatorial libraries by split and pool synthesis. These units are found in several bioactive molecules9–17 and the latter is a source of serine hydrolase (SH) inhibitors.18–22 For example, compound 7600 and CAY10499 (Fig. 1B) are potent inhibitors of hormone-sensitive lipases (HSLs) that function via a covalent, but reversible, mechanism.19,20,23,24
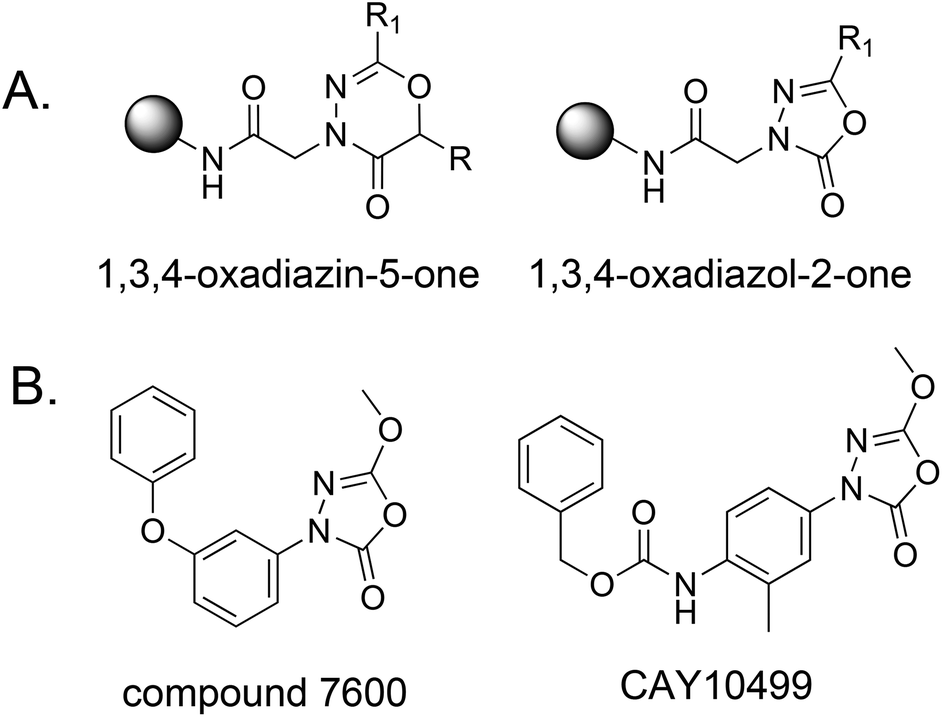 | ||
| Fig. 1 Structures and function of the target molecules of this study. (A) General structures of the two types of heterocycles created here. (B) Structures of two 1,3,4-oxadiazol-2-ones reported to be inhibitors of serine hydrolases. | ||
This work proceeded from our recent observation that 2-substituted-1,3,4-oxadiazin-5(6H)-ones (R = H in Fig. 1A) were often formed as a side product when resin-bound N-bromoacylated aryl or hetero-aromatic acyl hydrazides were treated with a primary amine.25,26
While this was originally encountered as an unwanted side reaction during the synthesis of N-azapeptoid chains, a useful synthesis of 2-substituted-1,3,4-oxadiazin-5(6H)-one-capped oligomers was effected by simply replacing the primary amine with diisopropylethylamine (DIEA), which provided exclusively the cyclic product (Fig. 1A; R = H).26 Since enantiomerically pure 2-bromo carboxylic acids are readily available from amino acids,27 this chemistry should provide access to a variety of chiral, 2,6-disubstituted 1,3,4-oxadiazin-5-ones. To test this idea, the peptoid sequence (Nbsa-Nmea-Nmea) [Nbsa = 4-(2-aminoethyl)benzenesulfonamide; Nmea = 2-methoxyethylamine] was synthesized on the solid support using the standard peptoid sub-monomer synthesis28 (Fig. 2A). After acylation with bromoacetic acid, the terminal bromide was displaced with different aryl and hetero-aromatic acyl hydrazides using our previously reported method.25 The resin-bound acyl hydrazides were then treated with different chiral 2-substituted-2-bromoacetic acids (1–5) (10 equiv.) in the presence of diisopropylcarbodiimide (DIC) (10 equiv.) in DMF at 37 °C for 1 h. The beads were washed and then treated with a 1 M DIEA solution in N-methylpyrrolidinone (NMP) for ∼15 hours at 60 °C, which led to efficient formation of the 6-membered oxadiazinone ring-containing compounds 6–13 (Fig. 2A), as evidenced by their HPLC traces and mass spectra (Fig. 2B and ESI, Fig. S7†).
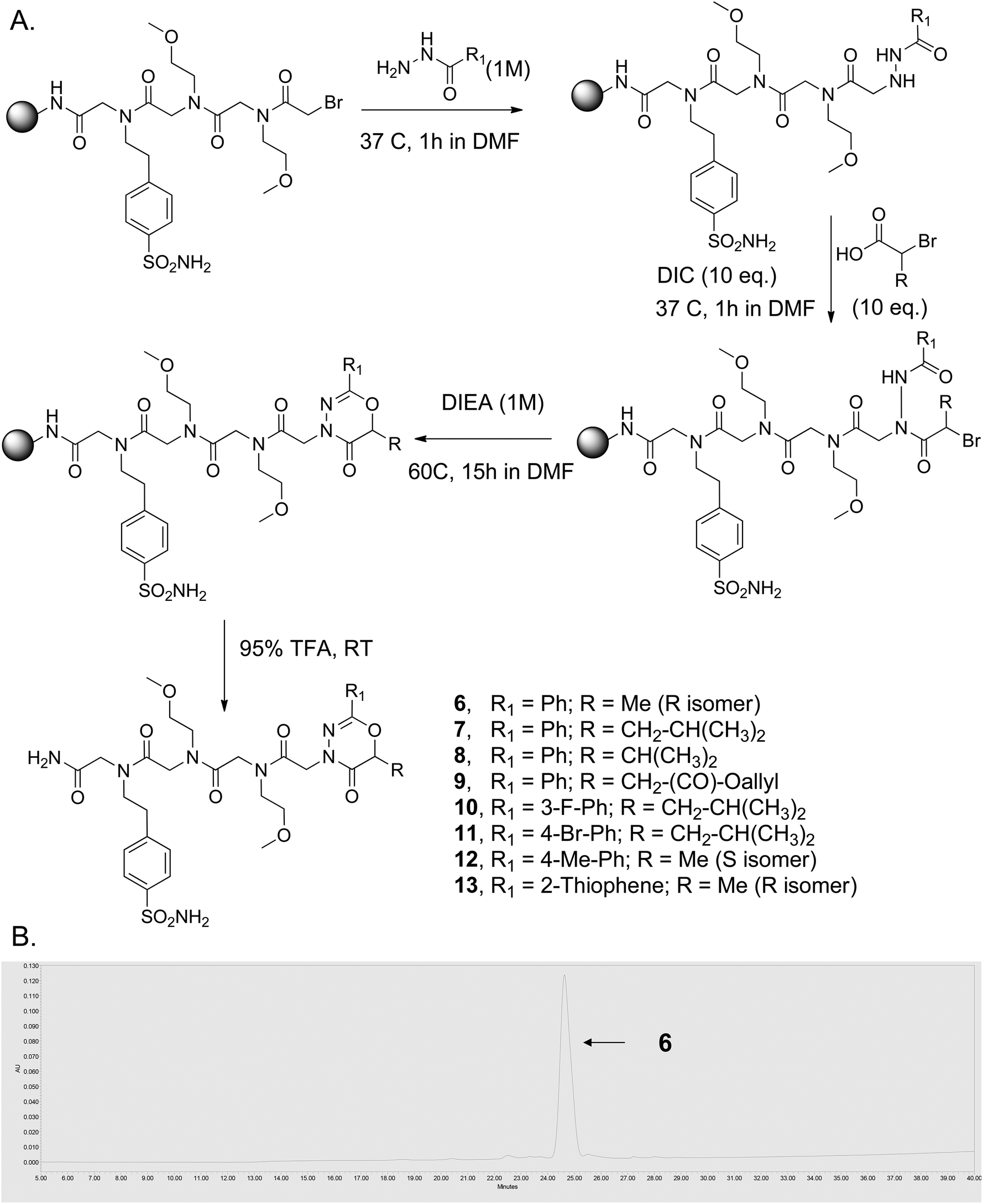 | ||
| Fig. 2 Solid-phase synthesis of chiral, 6-substituted 2-aryl-1,3,4-oxadiazin-5-ones. (A) Synthetic scheme. (B) HPLC trace of crude compound 6 after release from the solid support. | ||
Several efficient synthetic routes for the solution phase synthesis of 1,3,4-oxadiazol-2-one moieties are known in the literature.29–31 To incorporate 1,3,4-oxadiazol-2-one moieties on the solid phase, we followed a similar sequence of reactions as discussed above for compounds 6–13. After incorporating a peptoid linker (Nbsa-Nmea-Nmea) and different acyl hydrazides, the acyl hydrazide-bound resin was treated with 4-nitrophenyl chloroformate (5 equiv.) in the presence of DIEA (7 equiv.) in dichloromethane (DCM) at room temperature for 3 hours (Fig. 3A). The beads were washed with DMF and DCM and then treated with 1 M DIEA for ∼15 hours at 60 °C, which resulted in the highly efficient formation of the 5-membered oxadiazolone ring-containing compounds (14–18) (purity >95%; see Fig. 3B and the ESI, Fig. S8†).
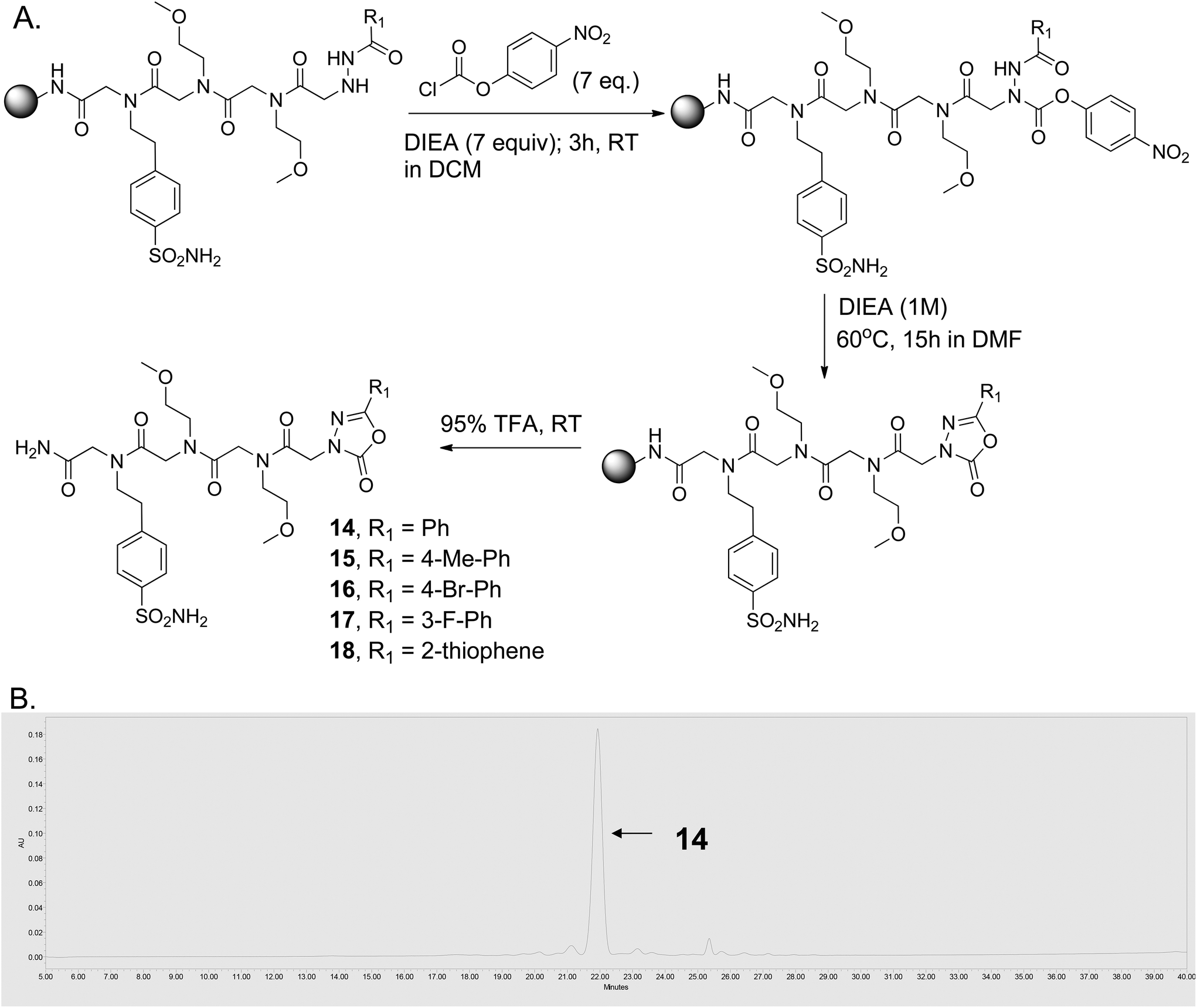 | ||
| Fig. 3 Solid-phase synthesis of 1,3,4-oxadiazol-2-ones. (A) Synthetic scheme. (B) HPLC trace of crude compound 14 after release from the solid support. | ||
As mentioned above, the 1,3,4-oxadiazol-2-one ring can function as the core unit of a reversible covalent inhibitor of SHs. A pan-selective, moderate activity covalent modifier of SHs is of interest to us for use as a “warhead” with which to cap libraries of oligomers. These libraries could then be screened for reversible covalent inhibitors that derive high selectivity for a particular SH through a combination of the 1,3,4-oxadiazol-2-one ring coupling with the active site and the oligomer binding to a surface nearby that might be unique to the particular SH of interest. However, the compounds that have been well-characterized in this regard, such as compound 7600, contain the heterocycle fused directly to a phenyl ring, whereas our chemistry most naturally places an acetamide group on the C-terminal side of the ring. Therefore, we set out to evaluate whether this acetamide-containing unit can serve as a general unit for the inhibition of SHs. For this purpose, we set out to synthesize compounds 19–36 (Fig. 4) using the method discussed above. All were obtained in excellent yield and purity with the exception of 36, which places a methoxy group at the 5-position as in compound 7600 and CAY 10499. In this case, unfortunately, the only product obtained was 37, indicating that an oxygen substituent at the 5-position strongly inhibits the ring-closing reaction.
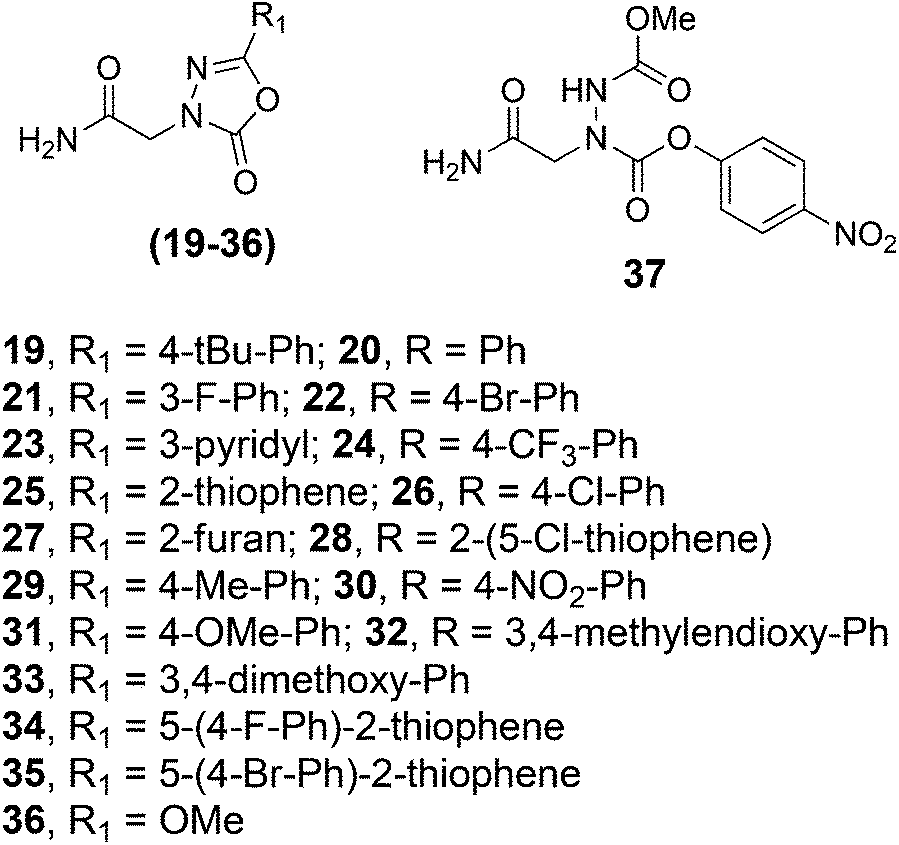 | ||
| Fig. 4 Structures of various 1,3,4-oxadiazol-2-ones prepared. Compound 36 was not obtained from the Fig. 3 protocol. Instead, only compound 37 was obtained. | ||
To evaluate the activity of these molecules, they were analysed using an activity-based protein-profiling (ABPP) gel-based assay32 in which the compound being tested was asked to inhibit coupling of SH enzymes in a HeLa cell lysate to a fluorescently-labelled fluorophosphonate.33 Thus, a diminution of labelling represents engagement of the SH active site with the compound. Since these species may be reversible covalent inhibitors, they were pre-incubated with the enzymes for 30 minutes prior to addition of the labelled fluorophosphonate, after which the reactions were allowed to proceed for 10 minutes before being quenched. Control experiments demonstrated that this was well within the linear range of the labelling reaction (not shown). As shown in Fig. 5A, none of the compounds except 35 had a noticeable effect on the SHs that can be visualized by ABPP in this assay at a concentration of 100 μM. 35 reduced the activity of most of the visible SHs by about 50% at this concentration. To complement these data, we also analyzed the effect of compounds 19–35 (200 μM) on five purified SHs using the same assay. Again, compound 35 inhibited all the five of the SHs studied (Fig. 5B). Many of the compounds had no effect on any of the enzymes, but several inhibited FAM108b selectively, particularly compound 28.
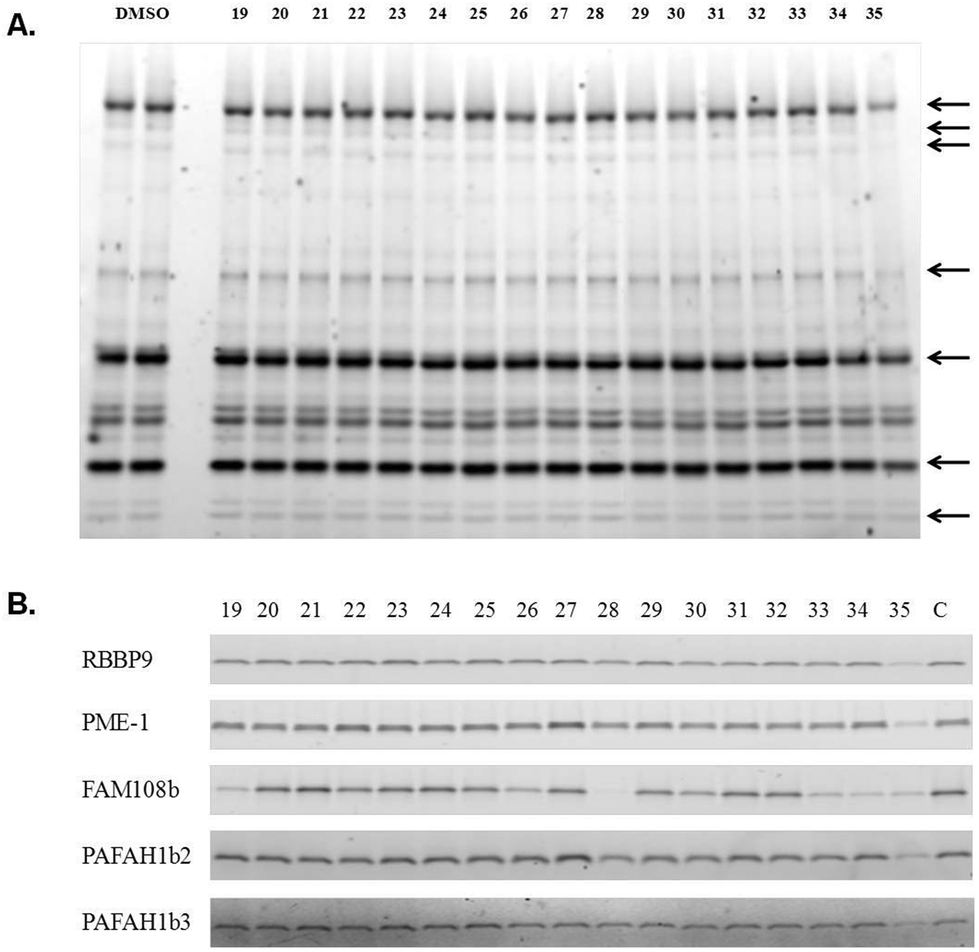 | ||
| Fig. 5 Inhibition of SHs by 1,3,4-oxadiazol-2-ones. (A) ABPP Gel-based inhibition assay of compounds 19–35 using a HeLa cell lysate. The lysate (1 mg mL−1 total protein) was incubated with 100 μM of each compound (19–35) for 30 minutes. Rhodamine fluorophosphonate (FP-Rh) (100 nM) was then added and the reaction mixture was incubated for 10 minutes before being quenched with a denaturing buffer. The samples were subjected to SDS-PAGE and the rhodamine-labeled protein bands were visualized using a Typhoon gel scanner. (B) A similar experiment as in part A was conducted except that five purified, recombinant SH enzymes (2 μM each) were employed in place of the cell lysate. In this case the compounds were employed at a concentration of 200 μM. | ||
In summary, we have developed efficient protocols for the solid-phase synthesis of oligomers terminated with diverse 2-R1-1,3,4-oxadiazin-5(6R)-one and 2-R1-1,3,4-oxadiazol-2-one scaffolds. Several of the latter class of molecules were tested for SH inhibitor activity using an ABPP assay, revealing compound 35 as a weak, pan-selective SH inhibitor (Fig. 5) All of the other compounds studied failed to show significant activity at 200 μM with the SHs analysed, except for some that inhibited FAM108b (Fig. 5B). Of course, there are a great many SHs that have yet to be tested against this suite of compounds, so lead compounds could yet arise from this collection or similar species. Future work will focus on the creation of complex combinatorial libraries capped with these species, which may provide a source of highly selective SH inhibitors.
Acknowledgements
This work was supported by a grant from the NIH (GM090294).Notes and references
- K. S. Lam and M. Renil, Curr. Opin. Chem. Biol., 2002, 6, 353–358 CrossRef CAS.
- R. Liu, J. Marik and K. S. Lam, J. Am. Chem. Soc., 2002, 124, 7678–7680 CrossRef CAS PubMed.
- W. Lian, P. Upadhyaya, C. A. Rhodes, Y. Liu and D. Pei, J. Am. Chem. Soc., 2013, 135, 11990–11995 CrossRef CAS PubMed.
- C. Aquino, M. Sarkar, M. J. CHalmers, K. Mendes, T. Kodadek and G. Micalizio, Nat. Chem., 2011, 4, 99–104 CrossRef PubMed.
- M. Hintersteiner, T. Kimmerlin, F. Kalthoff, M. Stoeckli, G. Garavel, J. M. Seifert, N. C. Meisner, V. Uhl, C. Buehler, T. Weidemann and M. Auer, Chem. Biol., 2009, 16, 724–735 CrossRef CAS PubMed.
- Y. Gao and T. Kodadek, Chem. Biol., 2013, 20, 360–369 CrossRef CAS PubMed.
- A. Aditya and T. Kodadek, ACS Comb. Sci., 2012, 14, 164–169 CrossRef CAS PubMed.
- S. Suwal and T. Kodadek, Org. Biomol. Chem., 2013, 11, 2088–2092 CAS.
- S. Ke, X. Cao, Y. Liang, K. Wang and Z. Yang, Mini-Rev. Med. Chem., 2011, 11, 642–657 CrossRef CAS.
- F. Mazouz, S. Gueddari, C. Burstein, D. Mansuy and R. Milcent, J. Med. Chem., 1993, 36, 1157–1167 CrossRef CAS.
- F. Mazouz, L. Lebreton, R. Milcent and C. Burstein, Eur. J. Med. Chem., 1988, 23, 441–451 CrossRef CAS.
- S. M. Ismail, R. A. Baines, R. G. Downer and M. A. Dekeyser, Pestic. Sci., 1996, 46, 163–170 CrossRef CAS.
- M. A. Dekeyser, D. M. Borth, R. C. Moore and A. Mishra, J. Agric. Food Chem., 1991, 39, 374–379 CrossRef CAS.
- M. A. Dekeyser, W. A. Harrison, P. T. McDonald and R. G. Downer, Pestic. Sci., 1993, 38, 309–314 CrossRef CAS.
- M. A. Dekeyser, P. T. McDonald, G. W. Angle and R. G. Downer, J. Agric. Food Chem., 1993, 41, 1329–1331 CrossRef CAS.
- M. A. Dekeyser, A. Mishra and R. C. Moore, Google Patents, 1987 Search PubMed.
- M. A. Dekeyser, D. S. Mitchell and R. G. Downer, J. Agric. Food Chem., 1994, 42, 1783–1785 CrossRef CAS.
- J. Z. Patel, T. Parkkari, T. Laitinen, A. A. Kaczor, S. M. Saario, J. R. Savinainen, D. Navia-Paldanius, M. Cipriano, J. Leppanen and I. O. Koshevoy, J. Med. Chem., 2013, 56, 8484–8496 CrossRef CAS PubMed.
- Y. Ben Ali, H. Chahinian, S. Petry, G. Muller, R. Lebrun, R. Verger, F. Carrière, L. Mandrich, M. Rossi and G. Manco, Biochemistry, 2006, 45, 14183–14191 CrossRef CAS PubMed.
- V. Delorme, S. V. Diomandé, L. Dedieu, J.-F. Cavalier, F. Carrière, L. Kremer, J. Leclaire, F. Fotiadu and S. Canaan, PLoS One, 2012, 7, e46493 CAS.
- V. Point, K. Pavan Kumar, S. Marc, V. Delorme, G. Parsiegla, S. Amara, F. Carrière, G. Buono, F. Fotiadu and S. Canaan, Eur. J. Med. Chem., 2012, 58, 452–463 CrossRef CAS PubMed.
- A. Minkkilä, J. R. Savinainen, H. Käsnänen, H. Xhaard, T. Nevalainen, J. T. Laitinen, A. Poso, J. Leppänen and S. M. Saario, ChemMedChem, 2009, 4, 1253–1259 CrossRef PubMed.
- Y. Ben Ali, R. Verger, F. Carrière, S. Petry, G. Muller and A. Abousalham, Biochimie, 2012, 94, 137–145 CrossRef CAS PubMed.
- G. G. Muccioli, G. Labar and D. M. Lambert, ChemBioChem, 2008, 9, 2704–2710 CrossRef CAS PubMed.
- B. K. Sarma and T. Kodadek, Chem. Commun., 2011, 47, 10590–10592 RSC.
- B. K. Sarma and T. Kodadek, ACS Comb. Sci., 2012, 14, 558–564 CrossRef CAS PubMed.
- N. Izumiya and A. Nagamatsu, Bull. Chem. Soc. Jpn., 1952, 25, 265–267 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- S. Osamu, T. Arakaki, H. Kamio and K. Tanji, Chem. Commun., 2014, 50, 7314–7317 RSC.
- V. Chudasama, J. M. Ahern, D. V. Dhokia, R. J. Fitzmaurice and S. Caddick, Chem. Commun., 2011, 47, 3269–3271 RSC.
- M. J. Mulvihill, D. V. Nguyen, B. S. MacDougall, D. G. Weaver, W. D. MathisRohm and H. Company, Synthesis, 2001, 1965–1970 CrossRef CAS.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- X. Liu, M. Dix, A. E. Speers, D. A. Bachovchin, A. M. Zuhl, B. F. Cravatt and T. J. Kodadek, ChemBioChem, 2012, 13, 2082–2093 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, HPLC chromatograms, MALDI TOF MS spectra and NMR spectra of compounds. See DOI: 10.1039/c4ob01883d |
| This journal is © The Royal Society of Chemistry 2015 |