
Palladium(0)-catalyzed synthesis of cyclic glucosides†
Xin
Huang
,
Chunling
Fu
and
Shengming
Ma
*
Laboratory of Molecular Recognition and Synthesis, Department of Chemistry, Zhejiang University, Hangzhou 310027, Zhejiang, P. R. China. E-mail: masm@sioc.ac.cn; Fax: (+86)21-6260-9305
First published on 30th October 2014
Abstract
A highly regio- and stereo-selective synthesis of cyclic β-D-glucosides 3via Pd(0)-catalyzed coupling cyclization of allenyl β-D-glucoside 1 and organic iodides in 20–38% yields is reported. After the deacetylation of 3, we obtained cyclic β-D-glucoses 4 in 90–97% yields, which may have SGLT2 inhibition activities.
Nowadays, the number of people suffering from type 2, non-insulin dependent, diabetes mellitus (NIDDMa) is increasing.1 Since the first sodium glucose co-transporter 2 (SGLT2) inhibitor Phlorizin,2 a naturally-occurring O-aryl glycoside was discovered, more and more similar derivatives3 have been developed by chemists. Usually the skeletons of SGLT2 inhibitors are C- or O-glycosides bearing more than two aromatic or heteroaromatic rings (Fig. 1).3 However, there are limited reports on the synthesis of cyclic glycosides with SGLT2 inhibitors.4
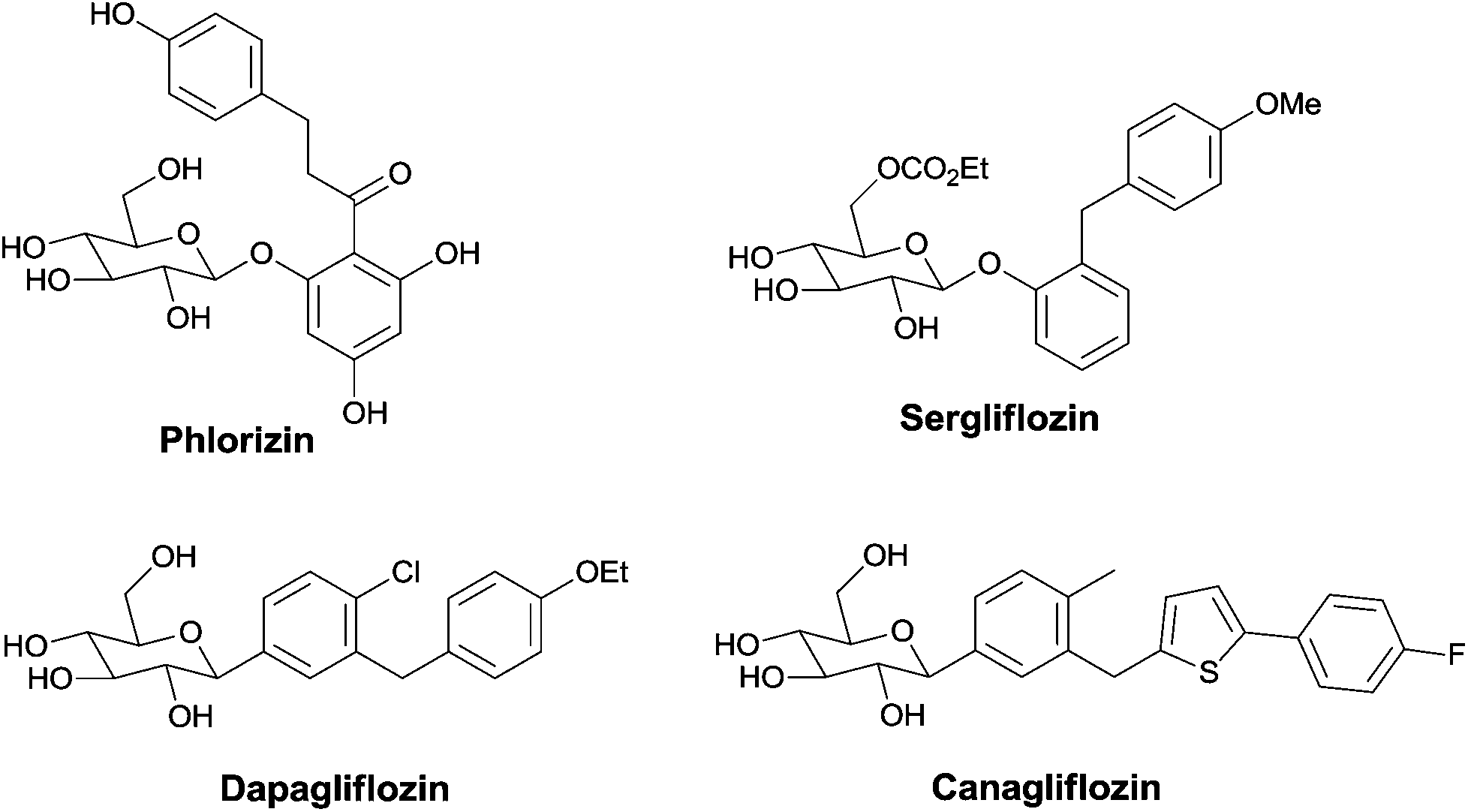 | ||
| Fig. 1 Typical structures of some C-aryl or O-aryl glucoside SGLT2 inhibitors. | ||
Recently, several reports on the Pd-catalyzed synthesis of medium-sized rings or macrocycles via allenes with a nucleophilic functionality and organic halides have been reported.5 We reasoned that this method could be applied to the construction of aryl or heteroaryl substituted cyclic glycosides, which may also have inhibition activities to type 2 diabetes mellitus (Scheme 1). Usually medium sized rings are very hard to prepare due to entropic/enthalpic factors, especially here where an oxygen bridge exists in the designed cyclic glucosides.6
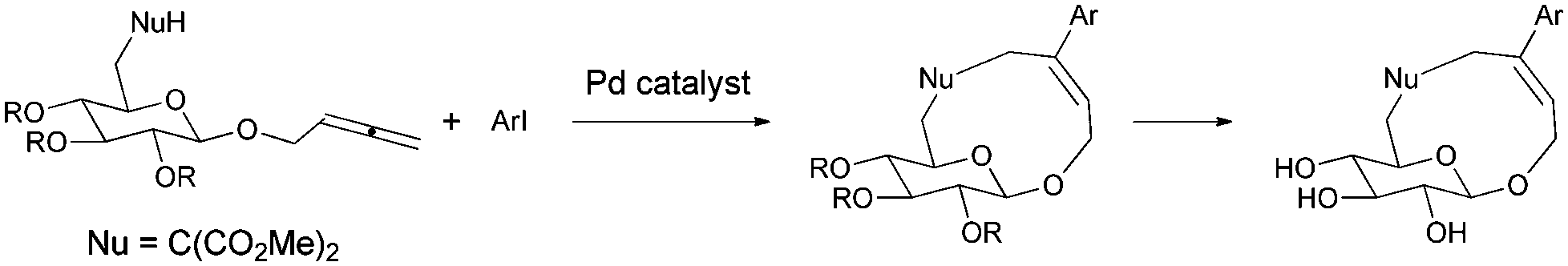 | ||
| Scheme 1 Design of new cyclic glucoside SGLT2 inhibitors. | ||
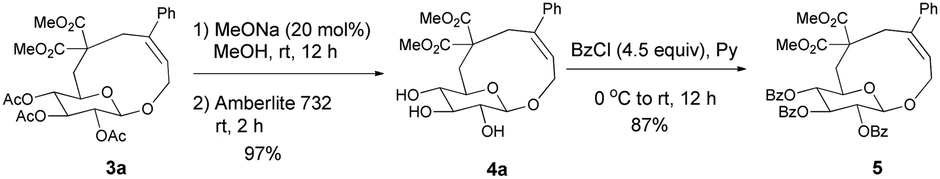
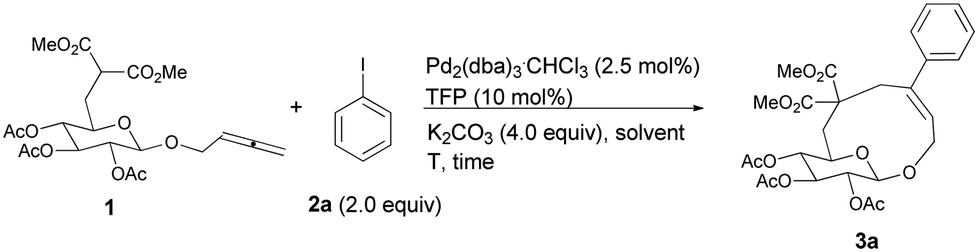
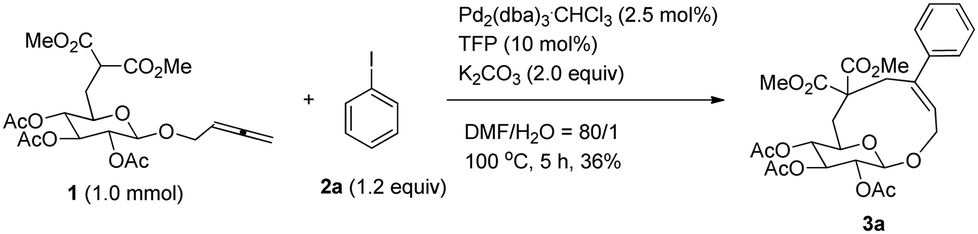
Initially, we conducted the reaction of allenyl β-D-glucoside 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 with PhI (2.0 equiv.) under the catalysis of Pd2(dba)3·CHCl3 (2.5 mol%), TFP (10 mol%, TFP = tris(2-furyl)phosphine) and K2CO3 (4.0 equiv.) at 60 °C. After screening a series of solvents, we found that the reaction using DMF afforded cyclic glucoside 3a in the highest NMR yield of 21% (entries 1–4, Table 1). The skeleton of the cyclic glucosides on the ring was further established by the X-ray diffraction study of compound 5 (Fig. 2), which was synthesized by deacetylation and benzoylation of 3a [eqn (1)].8 Increasing the temperature to 80 °C could shorten the reaction time to 5 h with a similar yield (entry 5, Table 1). The yield could be increased to 29% when the temperature is increased to 100 °C (entry 6, Table 1). Further reaction at 120 °C gave a lower yield (entry 7, Table 1).
7 with PhI (2.0 equiv.) under the catalysis of Pd2(dba)3·CHCl3 (2.5 mol%), TFP (10 mol%, TFP = tris(2-furyl)phosphine) and K2CO3 (4.0 equiv.) at 60 °C. After screening a series of solvents, we found that the reaction using DMF afforded cyclic glucoside 3a in the highest NMR yield of 21% (entries 1–4, Table 1). The skeleton of the cyclic glucosides on the ring was further established by the X-ray diffraction study of compound 5 (Fig. 2), which was synthesized by deacetylation and benzoylation of 3a [eqn (1)].8 Increasing the temperature to 80 °C could shorten the reaction time to 5 h with a similar yield (entry 5, Table 1). The yield could be increased to 29% when the temperature is increased to 100 °C (entry 6, Table 1). Further reaction at 120 °C gave a lower yield (entry 7, Table 1).
 | (1) |
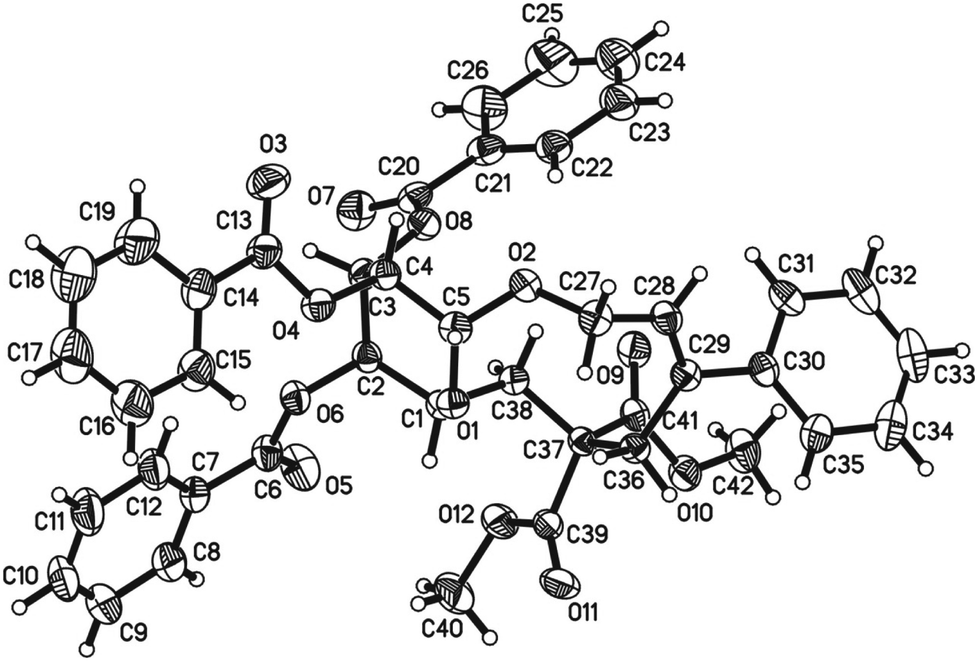 | ||
| Fig. 2 ORTEP representation of compound 5. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | T (°C) | Time (h) | NMR yield of 3ab (%) |
| a The reaction was conducted with a concentration of 1 of 0.067 M. b Determined by 1H NMR analysis of the crude products with mesitylene as the internal standard. c 83% of 1 was recovered. | ||||
| 1 | DMA | 60 | 15 | 17 |
| 2 | Dioxane | 60 | 40 | 0c |
| 3 | DMSO | 60 | 11 | 14 |
| 4 | DMF | 60 | 14 | 21 |
| 5 | DMF | 80 | 5 | 19 |
| 6 | DMF | 100 | 5 | 29 |
| 7 | DMF | 120 | 5 | 19 |
It is worth mentioning that the same yield of cyclic glucoside may be obtained with 2.0 equiv. of K2CO3 (entry 1, Table 2). We then conducted the reaction under diluted conditions and the yield improved to 37% when the concentration of 1 was decreased to 0.025 M (entries 1–5, Table 2). Also the amount of 2a could be lowered to 1.2 equiv. with a 38% yield of 3a (entries 6 and 7, Table 2). It was found that a mixture of DMF–H2O (80/1) is more effective with 39% NMR yield (entry 8, Table 2). Increasing the loading of water led to poorer results (entries 9 and 10). In addition, replacing TFP with PPh3 afforded lower yield of 3a (entry 11, Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | X (equiv.) | Solvent | Conc. of 1 (mol L−1) | Time (h) | NMR yield of 3ab (%) |
| a Reaction conditions: 1 (0.1 mmol), Pd2(dba)3.CHCl3 (2.5 mol%), TFP (10 mol%), K2CO3 (2.0 equiv.), 100 °C. b Determined by 1H NMR analysis of the crude products with mesitylene as the internal standard. c 10 mol% of PPh3 was used instead of TFP. | |||||
| 1 | 2.0 | DMF | 0.067 | 5 | 29 |
| 2 | 2.0 | DMF | 0.050 | 4 | 26 |
| 3 | 2.0 | DMF | 0.033 | 4 | 31 |
| 4 | 2.0 | DMF | 0.025 | 8 | 37 |
| 5 | 2.0 | DMF | 0.014 | 8 | 37 |
| 6 | 1.5 | DMF | 0.025 | 7 | 37 |
| 7 | 1.2 | DMF | 0.025 | 7 | 38 |
| 8 | 1.2 | DMF–H2O (80/1) | 0.025 | 3 | 39 |
| 9 | 1.2 | DMF–H2O (40/1) | 0.025 | 3 | 37 |
| 10 | 1.2 | DMF–H2O (10/1) | 0.025 | 3 | 0 |
| 11c | 1.2 | DMF–H2O (80/1) | 0.025 | 20 | 28 |
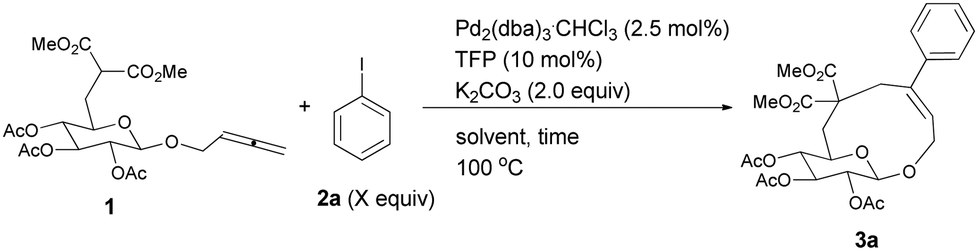
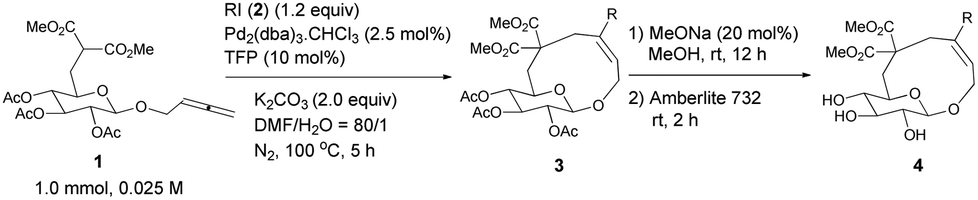
Thus, the following [Pd2(dba)3·CHCl3 (2.5 mol%), TFP (10 mol%), K2CO3 (2.0 equiv.) and 2 (1.2 equiv.) in DMA, 100 °C] were established as the optimized conditions for further study with the typical results summarized in Table 3. The following issues should be noted: (i) yields of cyclic glucosides 3 range from 20% to 38% while the reactions were carried out on a 1.0 mmol scale. This is quite remarkable since the formation of ten-membered cyclic glucosides is not easy. (ii) Not only electron-rich (entries 2 and 3, Table 3) but also electron-deficient (entries 4–10, Table 3) substituents could be introduced into aryl iodides. (iii) Also thienyl iodide (2j) could react with 1, which afforded 3j in 34% yield (entry 11, Table 3). (iv) Vinyl iodide (2k) could also be applied in this reaction affording the corresponding product 3k in 20% isolated yield (entry 12, Table 3). (v) We also investigated the reaction of 1 and 2f on a 4 mmol scale under the optimized conditions and obtained a 34% isolated yield of 3f (entry 7, Table 3). (vi) We prepared cyclic glucosides 4 by deacetylation9 of 3 in 90% to 97% yields (Table 3).
|
|
|||
|---|---|---|---|
| Entry | R | Isolated yields of 3 (%) | Isolated yields of 4 (%) |
| a All reactions were carried out on a 1.0 mmol scale under the standard conditions: 1 (0.025 M), 2 (1.2 equiv.), Pd2(dba)3·CHCl3 (2.5 mol%), TFP (10 mol%), K2CO3 (2.0 equiv.), DMF–H2O (80/1), 100 °C, 5 h. b The reaction was conducted on a 4.0 mmol scale of 1. | |||
| 1 | Ph (2a) | 36 (3a) | 97 (4a) |
| 2 | p-MeC6H4 (2b) | 38 (3b) | 92 (4b) |
| 3 | p-MeOC6H4 (2c) | 23 (3c) | 95 (4c) |
| 4 | p-FC6H4 (2d) | 34 (3d) | 91 (4d) |
| 5 | p-ClC6H4 (2e) | 38 (3e) | 96 (4e) |
| 6 | p-BrC6H4 (2f) | 31 (3f) | 90 (4f) |
| 7b | p-BrC6H4 (2f) | 34 (3f) | — |
| 8 | p-IC6H4 (2g) | 24 (3g) | 92 (4g) |
| 9 | p-MeOOCC6H4 (2h) | 36 (3h) | 94 (4h) |
| 10 | p-MeCOC6H4 (2i) | 34 (3i) | 92 (4i) |
| 11 | 2-thienyl (2j) | 34 (3j) | 92 (4j) |
| 12 |
|
20 (3k) | 91 (4k) |
Conclusions
In conclusion, we have developed a highly regio- and stereo-selective synthesis of cyclic β-D-glucosides via Pd(0)-catalyzed coupling cyclization of functionalized allenyl β-D-glucoside and organic iodides. Studies on the SGLT2 inhibition activity are in progress. Further study in this area is being pursued in our laboratory.Experimental
Pd(0)-catalyzed coupling-cyclization of 1 with different organic iodides 2 under standard conditions. Preparation of 3a.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 5.22–5.08 (m, 2H), 5.03–4.93 (m, 1H), 4.86 (s, 1H), 4.41–4.27 (m, 2H), 3.88 (d, J = 13.8 Hz, 1H), 3.83–3.67 (m, 1H), 3.74 (s, 3H, Me), 3.29 (d, J = 13.8 Hz, 1H), 2.89 (s, 3H, Me), 2.70 (dd, J1 = 14.7 Hz, J2 = 12.3 Hz, 1H), 2.13–1.96 (m, 1H), 2.08 (s, 3H, Me), 2.06 (s, 3H, Me), 2.04 (s, 3H, Me); 13C NMR (75 Hz, CDCl3) δ 170.8, 170.4, 169.7, 169.5, 169.3, 145.0, 141.4, 128.2, 127.8, 127.5, 127.0, 102.0, 73.7, 70.8, 69.7, 64.5, 55.6, 52.5, 51.7, 33.6, 32.9, 20.6, 20.5; IR (KBr) ν (cm−1) 2957, 1754, 1730, 1439, 1370, 1243, 1221, 1117, 1080, 1067, 1041; MS (ESI, m/z) 1119 (2M + Na+), 587 (M + K+), 571 (M + Na+), 566 (M + NH4+), 549 (M + H+); Anal. Calcd for C27H32O12 (%): C 59.12, H 5.88; Found: C 59.13, H 5.92.
), 5.22–5.08 (m, 2H), 5.03–4.93 (m, 1H), 4.86 (s, 1H), 4.41–4.27 (m, 2H), 3.88 (d, J = 13.8 Hz, 1H), 3.83–3.67 (m, 1H), 3.74 (s, 3H, Me), 3.29 (d, J = 13.8 Hz, 1H), 2.89 (s, 3H, Me), 2.70 (dd, J1 = 14.7 Hz, J2 = 12.3 Hz, 1H), 2.13–1.96 (m, 1H), 2.08 (s, 3H, Me), 2.06 (s, 3H, Me), 2.04 (s, 3H, Me); 13C NMR (75 Hz, CDCl3) δ 170.8, 170.4, 169.7, 169.5, 169.3, 145.0, 141.4, 128.2, 127.8, 127.5, 127.0, 102.0, 73.7, 70.8, 69.7, 64.5, 55.6, 52.5, 51.7, 33.6, 32.9, 20.6, 20.5; IR (KBr) ν (cm−1) 2957, 1754, 1730, 1439, 1370, 1243, 1221, 1117, 1080, 1067, 1041; MS (ESI, m/z) 1119 (2M + Na+), 587 (M + K+), 571 (M + Na+), 566 (M + NH4+), 549 (M + H+); Anal. Calcd for C27H32O12 (%): C 59.12, H 5.88; Found: C 59.13, H 5.92.
Acknowledgements
Financial support from the National Natural Basic Research Program of China (2011CB808700) and the National Natural Science Foundation of China (21232006) are greatly appreciated. We thank Mr Wanli Zhang in this group for reproducing the preparation of 3c, 4c, 3h and 4h in Table 3. Shengming Ma is a Qiu Shi Adjunct Professor at the Zhejiang University.Notes and references
- (a) N. J. Wareham and N. G. Forouhi, Diabetologia, 2005, 48, 1454 CrossRef CAS PubMed; (b) S. Wild, G. Roglic, A. Green, R. Sicree and H. King, Diabetes Care, 2004, 27, 1047 CrossRef PubMed; (c) S. H. Golden, K. A. Robinson, I. Saldanha, B. Anton and P. W. Ladenson, J. Clin. Endocrinol. Metab., 2009, 94, 1853 CrossRef CAS PubMed.
- (a) F. W. Mckee and W. B. Hawkins, Physiol. Rev., 1945, 25, 255 CAS; (b) G. Toggenburger, M. Kessler and G. Semenza, Biochim. Biophys. Acta, 1982, 688, 557 CrossRef CAS PubMed.
- (a) H. Kakinuma, T. Oi, Y. Hashimoto-Tsuchiya, M. Arai, Y. Kawakita, Y. Fukasawa, I. Iida, N. Hagima, H. Takeuchi, Y. Chino, J. Asami, L. Okumura-Kitajima, F. Io, D. Yamamoto, N. Miyata, T. Takahashi, S. Uchida and K. Yamamoto, J. Med. Chem., 2010, 53, 3247 CrossRef CAS PubMed; (b) R. P. Robinson, V. Mascitti, C. M. Boustany-Kari, C. L. Carr, P. M. Foley, E. Kimoto, M. T. Leininger, A. Lowe, M. K. Klenotic, J. I. MacDonald, R. J. Maguire, V. M. Masterson, T. S. Maurer, Z. Miao, J. D. Patel, C. Préville, M. R. Reese, L. She, C. M. Steppan, B. A. Thuma and T. Zhu, Bioorg. Med. Chem. Lett., 2010, 20, 1569 CrossRef CAS PubMed; (c) S. Nomura, S. Sakamaki, M. Hongu, E. Kawanishi, Y. Koga, T. Sakamoto, Y. Yamamoto, K. Ueta, H. Kimata, K. Nakayama and M. Tsuda-Tsukimoto, J. Med. Chem., 2010, 53, 6355 CrossRef CAS PubMed; (d) W. Meng, B. A. Ellsworth, A. A. Nirschl, P. J. McCann, M. Patel, R. N. Girotra, G. Wu, P. M. Sher, E. P. Morrison, S. A. Biller, R. Zahler, P. P. Deshpande, A. Pullockaran, D. L. Hagan, N. Morgan, J. R. Taylor, M. T. Obermeier, W. G. Humphreys, A. Khanna, L. Discenza, J. G. Robertson, A. Wang, S. Han, J. R. Wetterau, E. B. Janovitz, O. P. Flint, J. M. Whaley and W. N. Washburn, J. Med. Chem., 2008, 51, 1145 CrossRef CAS PubMed; (e) B. Lv, B. Xu, Y. Feng, K. Peng, G. Xu, J. Du, L. Zhang, W. Zhang, T. Zhang, L. Zhu, H. Ding, Z. Sheng, A. Welihinda, B. Seed and Y. Chen, Bioorg. Med. Chem. Lett., 2009, 19, 6877 CrossRef CAS PubMed; (f) N. C. Goodwin, R. Mabon, B. A. Harrison, M. K. Shadoan, Z. Y. Almstead, Y. Xie, J. Healy, L. M. Buhring, C. M. DaCosta, J. Bardenhagen, F. Mseeh, Q. Liu, A. Nouraldeen, A. G. E. Wilson, S. D. Kimball, D. R. Powell and D. B. Rawlins, J. Med. Chem., 2009, 52, 6201 CrossRef CAS PubMed; (g) B. Xu, Y. Feng, B. Lv, G. Xu, L. Zhang, J. Du, K. Peng, M. Xu, J. Dong, W. Zhang, T. Zhang, L. Zhu, H. Ding, Z. Sheng, A. Welihinda, B. Seed and Y. Chen, Bioorg. Med. Chem., 2010, 18, 4422 CrossRef CAS PubMed; (h) H. Zhou, D. P. Danger, S. T. Dock, L. Hawley, S. G. Roller, C. D. Smith and A. L. Handlon, ACS Med. Chem. Lett., 2010, 1, 19 CrossRef CAS PubMed; (i) S. Y. Kang, K. Song, J. Lee, S. Lee and J. Lee, Bioorg. Med. Chem., 2010, 18, 6069 CrossRef CAS PubMed.
- (a) V. Mascitti and C. Préville, Org. Lett., 2010, 12, 2940 CrossRef CAS PubMed; (b) S. Y. Kang, M. J. Kim, J. S. Lee and J. Lee, Bioorg. Med. Chem., 2011, 21, 3759 CrossRef CAS PubMed.
- (a) X. Jiang, Q. Yang, Y. Yu, C. Fu and S. Ma, Chem. – Eur. J., 2009, 15, 7283 CrossRef CAS PubMed; (b) B. Wan, G. Jia and S. Ma, Adv. Synth. Catal., 2011, 353, 1763 CrossRef CAS; (c) X. Huang, X. Jiang, C. Fu and S. Ma, Adv. Synth. Catal., 2013, 355, 3295 CrossRef CAS.
- (a) G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95 CrossRef CAS; (b) G. A. Molander, Acc. Chem. Res., 1998, 31, 603 CrossRef CAS.
- For the synthesis of allenyl β-D-glucoside 1, see: X. Huang, C. Fu and S. Ma, Synthesis, 2014, 2917 Search PubMed.
- Crystal data for compound 5: C42H38O12, MW = 734.72, monoclinic, space group P21, final R indices [I > 2σ(I)], R1 = 0.0354, wR2 = 0.0945, R indices (all data) R1 = 0.0383, wR2 = 0.0981, a = 7.29191(19) Å, b = 16.5352(4) Å, c = 16.0030(4) Å, α = 90.00°, β = 99.499(3)°, γ = 90.00°, V = 1903.08(8) Å3, T = 293(2) K, Z = 2, reflections collected/unique 7884/3510 (Rint = 0.0336), number of observations [>2σ(I)] 3260, parameters: 490. CCDC-987494 contains the supplementary crystallographic data for this paper.
- N. Pietrzik, D. Schmollinger and T. Ziegler, Beilstein J. Org. Chem., 2008, 4, 30 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of all compounds, and copies of 1H and 13C NMR spectra of selected compounds. CCDC 987494. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob01975j |
| This journal is © The Royal Society of Chemistry 2015 |