
Gold and gold–palladium alloy nanoparticles on heterostructured TiO2 nanobelts as plasmonic photocatalysts for benzyl alcohol oxidation†
Tongtong
Jiang
a,
Chuancheng
Jia
b,
Lanchun
Zhang
a,
Shuren
He
a,
Yuanhua
Sang
c,
Haidong
Li
c,
Yanqing
Li
c,
Xiaohong
Xu
*a and
Hong
Liu
*c
aKey Laboratory of Colloid and Interface Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, 27 Shandanan Road, Jinan 250100, China. E-mail: xhxu@sdu.edu.cn
bCenter for Nanochemistry, Beijing National Laboratory for Molecular Sciences, State Key Laboratory for Structural Chemistry of Unstable and Stable Species, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
cState Key Laboratory of Crystal Materials, Shandong University, 27 Shandanan Road, Jinan, 250100, China. E-mail: hongliu@sdu.edu.cn
First published on 3rd November 2014
Abstract
Plasmonic photocatalysts composed of Au and bimetallic Au–Pd alloy nanoparticles (NPs) on one-dimensional TiO2 nanobelts (TiO2-NBs) were used for the aerobic oxidation of benzyl alcohol under visible light irradiation. Remarkable light-promoted activity was observed for the as-synthesized M/TiO2-NB (M = Au, Au–Pd) nanostructures based on the TiO2(B)/anatase heterostructured nanobelt. The difference in band structure and the well matched interface between the TiO2(B) and anatase phases, coupled with the one-dimensional nanostructure, enable an enhanced charge transfer within the heterostructured nanobelt. This inter-phase charge transfer greatly facilitates the flow of hot electrons from the metal NPs to TiO2 and promotes benzyl alcohol oxidation. This efficient electron transfer was identified by the much higher photocurrent response measured for the Au/TiO2-NB nanostructure with the TiO2(B)/anatase heterojunction than those with either of the single phases under visible light irradiation. Alloying Au with Pd in Au–Pd/TiO2-NB results in a significant improvement in the visible light-promoted activity compared to the monometallic Au/TiO2-NB sample. It is supposed that the plasmon-mediated charge distribution within the alloy NPs is mainly responsible for the enhanced photocatalytic activity of the bimetallic nanostructures.
1. Introduction
It is always a challenge to develop an environmentally benign catalytic process with high product selectivity. In the past few decades, the rapid expansion of gold (Au) catalysis has developed many new approaches to achieve chemical transformations under milder reaction conditions, such as low temperature CO oxidation,1–3 propene epoxidation,4–6 aerobic oxidation of alcohols and other organic matters.7–10 However, some of these reactions are energy intensive and the product selectivity is relatively low. Fortunately, the conduction electrons of Au resonating with visible light (localized surface plasmon resonance, LSPR) can generate hot electrons and holes which can drive chemical reactions at room temperature with high product selectivity.11Plasmonic-metal/semiconductor heterojunction structures, such as the integration of Au NPs with titanium dioxide (TiO2), have been proven to be promising visible light-driven photocatalysts.12–17 It has been shown that ideal Au/TiO2 plasmonic photocatalysts must allow an efficient separation and transfer of the photoinduced charge carriers, which is advantageous to achieve high efficiency of the plasmon-mediated catalysis process.18 When irradiated with visible light, the hot electrons excited in Au NPs can be injected into the conduction band of TiO2 over the Schottky barrier, thus separation of the photoinduced charge carriers can be achieved.19–21 However, the hot electron transfer would lead to an electron accumulation in the conduction band of TiO2, which would prohibit the consecutive electron transfer from the Au NPs to TiO2.18,22 It is essential to promote charge transfer within TiO2 to smooth the injection of hot electrons. As reported in the literature, mixed TiO2 phases, such as the well-known P25 composed of anatase and rutile phases, exhibited superior photocatalytic activity compared to either of the component phases.23 It is believed that the promoted charge transfer between different TiO2 phases resulted from the difference between their band structure and accounts for the excellent photocatalytic performance.24–26 In fact, the structure of TiO2 has also shown a strong impact on the photocatalytic activity of plasmonic Au/TiO2 catalysts by mediating the transfer of hot electrons.18,27,28 For example, Hirai et al. found that Au NPs located at the interface of anatase and rutile in P25 are much more efficient for the aerobic oxidation of benzyl alcohol than those located on pure anatase and rutile TiO2 owing to the consecutive electron transfer in this heterojunction.27 One-dimensional TiO2 nanobelts, which are synthesized by the controllable hydrothermal method, can readily form a heterostructure composed of anatase and the metastable phase of TiO2(B) by controlling the calcination temperature of the H2Ti3O7 nanobelts.29 Just like the case of P25, the TiO2(B)/anatase heterostructure with a coherent boundary bicrystalline structure has shown enhanced photocatalytic activity due to efficient inter-phase charge transfer.29–34 Moreover, compared with nanoparticles, the special one-dimensional nanostructure of TiO2 nanobelts and nanorods would lead to distinctive electronic properties, like the free transport of electrons with high speed along the axial direction.35–37 As a result, efficient electron transfer within TiO2 nanobelts can be expected. Furthermore, TiO2 nanobelts obtained by the hydrothermal method normally have clean and smooth surfaces, which allow high dispersal of the catalytically active metal NPs or clusters.38,39
As is well known, for conventional heterogeneous catalysis, bimetallic catalysts often show tunable and synergistic effects compared to their monometallic counterparts.40–42 Moreover, it has been shown that the incorporation of a second metal can also effectively improve the photocatalytic performance of Au-based plasmonic photocatalysts.43–48 Some bimetallic nanostructures, such as Au–Pt,43 Au–Cu,44 Au–Ag45,46 and Au–Pd47,48 NPs supported on TiO2, exhibited much higher light-promoted activities for various chemical reactions than their monometallic counterparts.
Herein, Au and Au–Pd alloy NPs which are highly dispersed on TiO2 nanobelts were synthesized. As expected, the visible light-promoted activity of Au/TiO2-NB photocatalysts strongly depends on the structure of the TiO2 nanobelt, and the highest activity enhancement for benzyl alcohol oxidation was obtained with the TiO2(B)/anatase heterostructured nanobelt under visible light irradiation. Compared to the monometallic Au/TiO2-NB sample, alloying Au with Pd in bimetallic Au–Pd/TiO2-NB nanostructures resulted in improved catalytic activity. Photoelectrochemical measurements were used to discern the role of the plasmon-mediated electron transfer process in the light-promoted activity of M/TiO2-NB photocatalysts.
2. Experimental section
2.1 Materials
H2Ti3O7 nanobelts were synthesized via a typical hydrothermal process in concentrated NaOH aqueous solution with commercial P25 (Evonik, 50 m2 g−1, 80% anatase, 20% rutile) as the starting material.29 Calcining the H2Ti3O7 nanobelts at 673, 873 and 1173 K, TiO2 nanobelts with different structures were obtained, which were labeled as T-1, T-2 and T-3, respectively. HAuCl4 and PdCl2 (Acros Chemicals) were used as the gold and palladium precursors. Urea of analytical reagent grade was used as the precipitation agent. All of the reagents were directly used without further treatment. Ultrapure water was used throughout our experiments.2.2 Synthesis of M/TiO2-NB nanostructures
For all nanostructures, subscript numbers denote the molar fractions of Au or Pd which were calculated on the basis of the metal atom amount in the precursor suspension.
2.3 Characterization
The metal loading was detected with an inductively coupled plasma spectrometer (ICP-AES) on an IRIS Intrepid II XSP instrument (Thermo Electron Corporation). X-ray diffraction (XRD) analysis was conducted on a German Bruker D8 Advance powder X-ray diffractometer with Cu-Kα (λ = 0.15406 nm). Transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HR-TEM) images were obtained with a JOEL JEM 2100 microscope. The UV-visible (UV-vis) absorption spectra were recorded on a Shimadzu U-2400PC spectrophotometer with barium sulfate as the standard for the background correction. X-ray photoelectron spectroscopy (XPS) data were acquired on a Thermo ESCALAB 250 X-ray photoelectron spectrometer and the binding energies were determined utilizing the C1s spectrum as a reference at 284.6 eV.2.4 Catalytic reaction test
The photocatalytic activities of the as-synthesized M/TiO2-NB nanostructures were estimated via the aerobic oxidation of benzyl alcohol under visible light irradiation. The catalyst (20 mg) was dispersed well by ultrasonication in 5 ml toluene containing 40 μmol benzyl alcohol within a Pyrex glass tube (15 mm in diameter with a capacity of 20 ml). After purging the suspension with O2 for 5 min, the tube was sealed with a rubber septum cap. The irradiation was carried out under vigorous magnetic stirring for 5 h using a 500 W Xenon lamp with a 450 nm long pass filter (160 mW cm−2) as the light source. The temperature of the system was controlled by a water bath (303 ± 0.5 K) running through the outer casing of the Pyrex glass tube to avoid light induced heating. The reaction in the dark was tested under the same conditions just in the absence of light. After the reaction, the suspension was separated by centrifugation, and the solution was analyzed with a Shimadzu Type GC-14C equipped with a flame ionization detector, using a SGE-30QC2/AC5 capillary column and N2 as the carrier gas. GC-MS (Thermo Trace GC Ultra DSQ) was also employed to determine the reaction products.2.5 Photoelectrochemical performance measurements
The photoelectrochemical measurements were performed using a classical three-electrode cell consisting of M/TiO2-NB nanostructures as the photoanode (working electrode), Pt counter electrode and a saturated calomel reference electrode (SCE). The photocurrent response was recorded on a CHI 660C electrochemical workstation (the current density was normalized by the geometric surface area of the electrode) using 0.2 M Na2SO4 as the electrolyte solution. The working electrode was irradiated by a 300 W Xenon lamp with a 450 nm long pass filter.3. Results and discussion
One-dimensional TiO2 nanobelts with several tens of micrometers in the axial direction were synthesized via the hydrothermal process (Fig. 1a). Their structure was tuned by controlling the calcination temperature of the H2Ti3O7 nanobelts. As shown in Fig. S1,† TiO2(B) (T-1) and anatase (T-3) nanobelts were formed at 673 and 1173 K respectively, and TiO2(B)/anatase heterostructured nanobelts (T-2) with a composition of 40% TiO2(B) and 60% anatase were obtained at 873 K. The HR-TEM image (Fig. 1e) displays two clear lattice fringes continuously spaced across the surface of the T-2 nanobelts. The interplanar distances of 3.60 and 3.70 Å can be respectively indexed to the (101) plane of anatase and the (110) plane of TiO2(B), and the interface of the two phases is well matched. This reveals the coherent boundary bicrystalline TiO2(B)/anatase heterostructure of T-2 nanobelts.29,32 TEM observation (Fig. 1b–d) shows that a large amount of small-sized Au NPs were uniformly dispersed over the surface of the TiO2 nanobelts in all of the as-synthesized Au/TiO2-NB nanostructures. The morphologies of the Au/TiO2-NB nanostructures were scarcely affected by the nanobelt structure, and comparable dAu (average diameter of Au NPs) values were measured for Au1/T-1 (3.3 nm), Au1/T-2 (2.8 nm) and Au3.5/T-3 (3.0 nm) (Fig. 1f–h).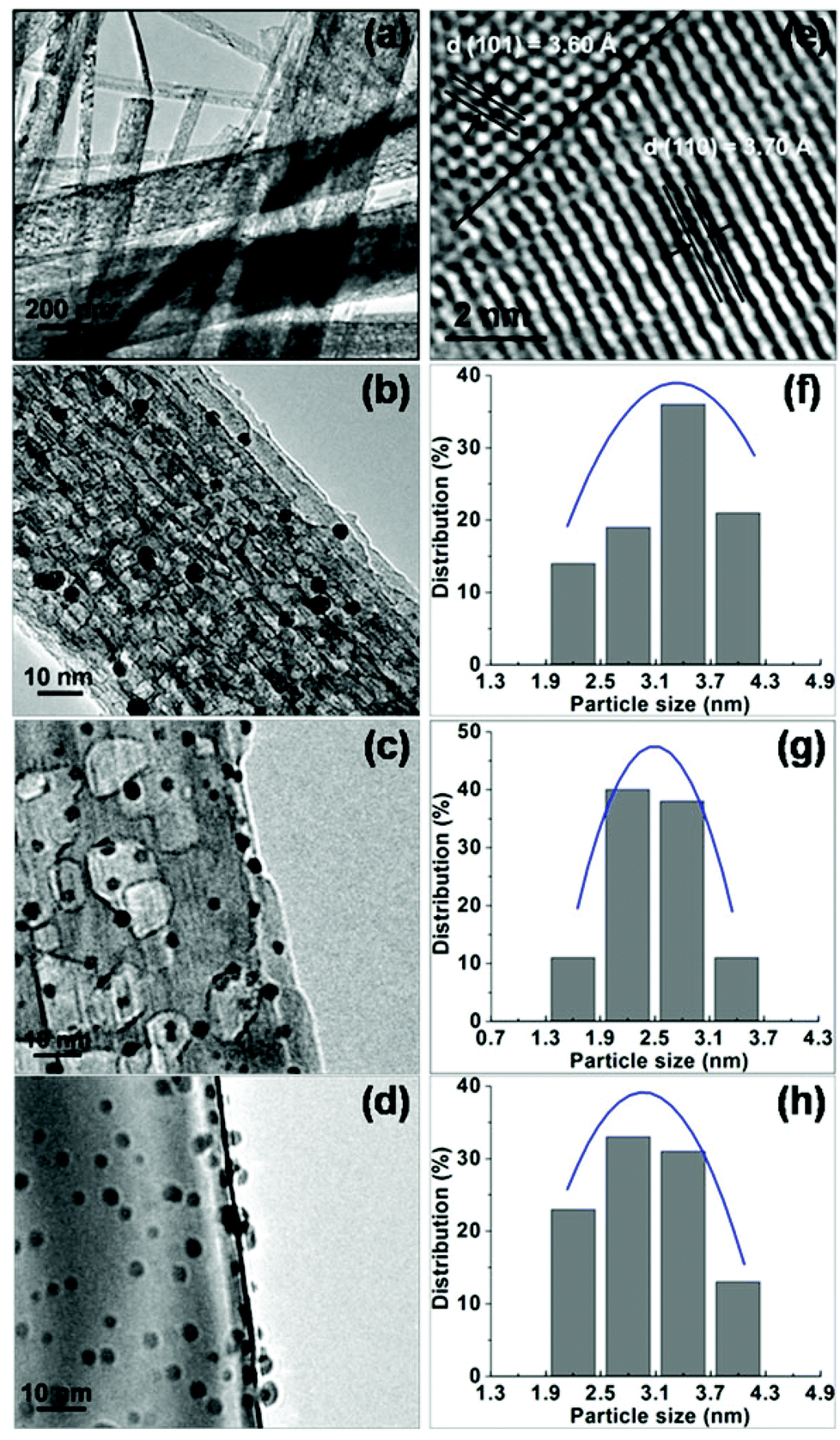 | ||
| Fig. 1 Representative TEM images of (a) TiO2 nanobelts, (b) Au1/T-1, (c) Au1/T-2 and (d) Au1/T-3, and HR-TEM image of (e) T-2 nanobelt and the corresponding particle size distributions of (f) Au1/T-1, (g) Au1/T-2 and (h) Au1/T-3. | ||
Fig. 2a presents the diffuse reflectance UV-vis spectra of the TiO2 nanobelts and Au/TiO2-NB nanostructures. In contrast to only absorption at wavelengths below 450 nm for bare TiO2 nanobelts, Au/TiO2-NB nanostructures showed a wide absorption peak at around 570 nm assigned to the LSPR of the Au NPs.7,49 XPS analysis (Fig. 2b) reveals that the binding energies of Au 4f7/2 and 4f5/2 in the Au/TiO2-NB nanostructures shifted to lower values (such as 83.2 and 86.9 eV in Au1/T-2) compared with the XPS data of bulk gold (84.0 and 87.7 eV). This implies that a Schottky barrier exists at the Au–TiO2 interface, which will lead to electron transfer from TiO2 nanobelts to the Au NPs and thus gold is negatively charged.27,50,51
 | ||
| Fig. 2 (a) Diffuse reflectance UV-vis spectra of TiO2 nanobelts and Au/TiO2-NB nanostructures, and (b) XPS spectra of Au 4f in Au1/T-1, Au1/T-2 and Au1/T-3. | ||
The efficacy of the as-synthesized Au/TiO2-NB nanostructures for plasmonic photocatalysis was assessed by the aerobic oxidation of benzyl alcohol under visible light irradiation (Xe lamp, λ > 450 nm). Remarkably, benzyl alcohol was converted to benzaldehyde with high selectivity (>99% over all samples), while no other products were detected. For comparison, a control experiment in the absence of photocatalysts was also performed, and no conversion of benzyl alcohol was observed. Fig. 3a shows that all bare TiO2 nanobelts exhibited no activity in the dark, while the oxidation of benzyl alcohol to benzaldehyde can be observed under visible light irradiation. This visible light driven activity can be attributed to the complex formed by the adsorption of benzyl alcohol onto the TiO2 surface.52–54 The ligand-to-metal charge transfer of the surface complex rather than the direct excitation of TiO2 should account for the visible light-promoted activity of the TiO2 nanobelts.55,56
 | ||
| Fig. 3 Amount of benzaldehyde formed during the benzyl alcohol oxidation in the dark (grey) and under visible light irradiation (shadow) over Au/TiO2-NB photocatalysts with (a) different TiO2 nanobelts and (b) different Au loadings. | ||
It is noted that all Au/TiO2-NB nanostructures showed enhanced catalytic activity for benzyl alcohol aerobic oxidation both in the dark and under visible light irradiation. As expected, the amounts of Au loaded markedly affected the catalytic activity of Au/TiO2-NB with or without light irradiation. Take the Au/T-2 catalysts for example, the amount of benzaldehyde increased monotonically with the increment of Au loading ≤3.5 mol% but decreased at higher loading in both cases (Fig. 3b). The dAu values for Au1/T-2, Au3.5/T-2 and Au4.5/T-2 are 2.8, 4.0 and 5.5 nm, respectively (Fig. S2, ESI†), implying that the large Au NPs with dAu > 5 nm are less active in the reaction. Moreover, the light-promoted activity enhancement showed the same dependence on Au loading. The highest activity enhancement obtained over Au3.5/T-2, produced 30.1 μmol of benzaldehyde under visible light irradiation, which is 3.7 times that obtained in the dark (8.2 μmol).
However, remarkable visible light-promoted activity was obtained only for Au/T-2 catalysts based on the TiO2(B)/anatase heterostructured nanobelt. As depicted in Fig. 3a, the light-promoted activity of Au/T-2 is much higher than those of the Au/T-1 and Au/T-3 catalysts both with low and high Au loadings. Under visible light irradiation, Au1/T-2 produced 12.5 μmol of benzaldehyde, while even the high-loaded Au3.5/T-1 and Au3.5/T-3 samples produced <9 μmol of benzaldehyde. It should be noted that the Au/TiO2-NB nanostructures based on different TiO2 nanobelts are composed of comparably sized Au NPs when they have the same Au loading (Fig. 1b–d, f–h). Obviously, the structure of the TiO2 nanobelts has a strong impact on the visible light-promoted activity of the Au/TiO2-NB nanostructures. For the Au/TiO2 plasmonic photocatalysts, it has been reported that oxygen molecules adsorbed on TiO2 can capture the electrons injected from Au NPs and form O–O− species, which can promote the oxidation of benzyl alcohol.27 We infer that the excellent visible light-promoted activity of Au/T-2 photocatalysts should result from the efficient transfer of hot electrons from the Au NPs to the heterostructured nanobelt.
In order to elucidate the role of the plasmon-mediated electron transfer process in the visible light-promoted activity of Au/TiO2-NB nanostructures, a series of photoelectrochemical tests were performed. Au/TiO2-NB nanostructures and bare TiO2 nanobelts were used as the working photoanodes. The I–t curves responding to light turning on and off measured at zero bias voltage are presented in Fig. 4a. As can be seen, bare TiO2 nanobelts (T-2) did not produce any photocurrent signal, while all Au/TiO2-NB nanostructures exhibited a significant anodic photocurrent response under visible light irradiation. For example, the measured photocurrent density is about 1.50 μA cm−2 for Au1/T-2, which quickly decreased as soon as the light irradiation was stopped. This result demonstrates that bare TiO2 nanobelts cannot absorb visible light with wavelengths above 450 nm and produce photoinduced charge carriers. Therefore, the photocurrent responses of the Au/TiO2-NB nanostructures can be attributed to the injection of hot electrons mediated via plasmonic excitation of the Au NPs.18–21 It is noted that the transient photocurrent response of Au1/T-2 is much stronger than those of Au1/T-1 and Au1/T-3, indicating an efficient interfacial electron transfer between these Au NPs and the heterostructured nanobelt. This photocurrent response of the respective catalysts is consistent with the catalytic activity under visible light irradiation, suggesting that electron transfer from the Au NPs to the TiO2 nanobelt mediated the photocatalytic activity of the Au/TiO2-NB nanostructures for the benzyl alcohol aerobic oxidation.
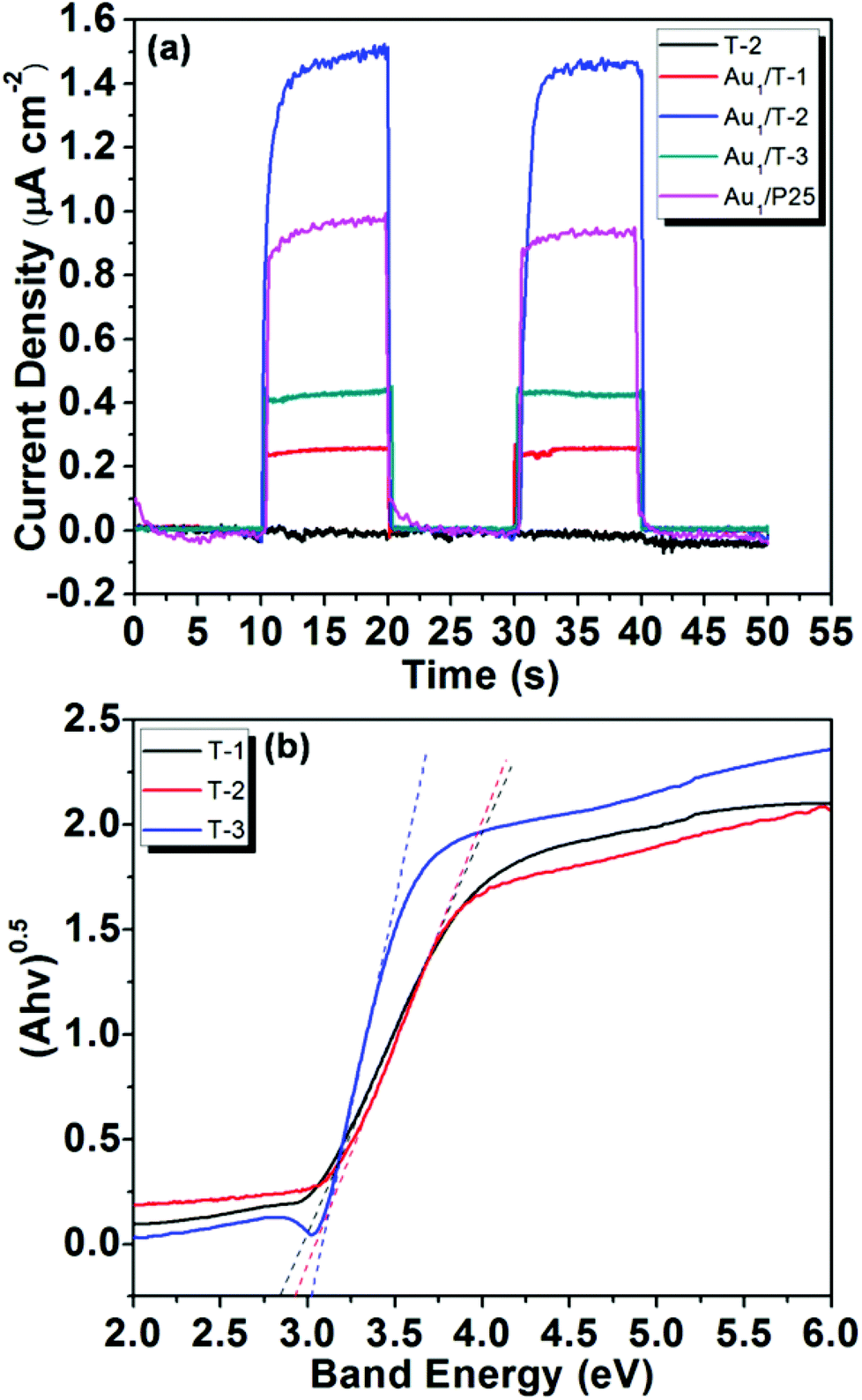 | ||
| Fig. 4 (a) The photocurrent responses of the Au/TiO2-NB and Au/P25 nanostructures under visible light irradiation, and (b) plots of (Ahv)0.5vs. hv of the TiO2 nanobelts. | ||
According to the UV-vis spectra of the TiO2 nanobelts obtained at different calcination temperatures, the band gaps of the T-1, T-2 and T-3 nanobelts were calculated to be 2.83, 2.92 and 3.02 eV, respectively (Fig. 4b). This means that there should be a difference between the band structure of the TiO2(B) and anatase phases, which would result in an inter-phase charge transfer when they are in contact. Up to now, efficient charge separation and promoted photocatalytic activity have been observed for various TiO2(B)/anatase heterostructures, and several models regarding charge transfer in these cases have been proposed.29–34 Among them, Ke et al. have demonstrated that the potentials of conduction and the valence band in TiO2(B) are higher than the corresponding ones of anatase.34 The higher conduction band edge of TiO2(B) provides a higher Schottky barrier,57 which is unfavourable for electron migration from the Au NPs to the TiO2, corresponding to the much lower photocurrent density of Au1/T-1 than that of Au1/T-3. The electron transfer from excited Au NPs to TiO2 (<240 fs) is much faster than the reverse process, indicating electron accumulation in the conduction band of TiO2.18,22 This will inhibit the consecutive electron transfer from the Au NPs to TiO2.
In our previous study, the heterostructured nanobelts show an anatase-TiO2(B)-anatase sandwich structure.29 Accordingly, for Au/T-2 samples, Au/TiO2-NB nanostructures with the heterostructured nanobelt, the HR-TEM images in Fig. S3† show that the Au NPs were mainly deposited on the anatase phase. When the Au/T-2 catalysts were irradiated by visible light, most of the hot electrons were injected into the conduction band of anatase. The electron accumulation in the conduction band of anatase can realign energy levels in the TiO2(B)/anatase heterostructure, enabling electron transfer from anatase to TiO2(B). Accordingly, the TiO2(B)/anatase heterojunction enables efficient electron transport within the heterostructured nanobelt, which will in turn facilitate the injection of hot electrons from the Au NPs into TiO2. This enhanced consecutive transfer of electrons between the dual heterojunction contributes to the much higher photocurrent response and visible light-promoted activity of Au1/T-2 than those of Au1/T-1 and Au1/T-3 based on a single-phase nanobelt.
For comparison, Au/P25 with the same Au loading was prepared and tested. The amounts of benzaldehyde formed over Au1/P25 in the dark and under visible light irradiation were 13.5 and 21.6 μmol respectively. However, the light enhanced benzaldehyde formation obtained over Au1/P25 (8.1 μmol) is lower than that over Au1/T-2 (11.3 μmol, see Fig. 3a). Correspondingly, a weaker transient photocurrent response of Au1/P25 (0.95 μA cm−2) than that of Au1/T-2 (1.50 μA cm−2) was observed (Fig. 4a). This demonstrates that a more efficient electron transfer occurred in Au1/T-2, which can be ascribed to the anisotropic electron flow in the heterostructured nanobelts. Moreover, due to the one-dimensional structure of the TiO2 nanobelts, Au/TiO2-NB nanostructures can be easily separated from the resulting solution after reaction by sedimentation in less than 5 min, while the suspension with the Au/P25 is still turbid (Fig. S4, ESI†). This additionally suggests that the TiO2(B)/anatase heterostructured TiO2 nanobelt can function as an ideal support and electron transfer mediator for plasmonic Au NPs.
It has been shown that the photocatalytic performance of Au-based plasmonic photocatalysts can be effectively improved by the incorporation of a second metal.43–48 Herein, we synthesized a series of bimetallic Au–Pd/TiO2-NB nanostructures by a similar method to that used in the synthesis of the Au/TiO2-NB samples. Fig. 5a,b show the representative TEM images of Au0.75Pd0.25/T-2. Just like the morphology of the Au/TiO2-NB nanostructures, small-sized metal NPs were highly dispersed on the surface of the TiO2 nanobelts. A narrow size distribution with an average diameter of 2.1 nm was obtained. As shown in Fig. S5,† the average diameters of the metal NPs in the Au0.5Pd0.5/T-2, Au0.25Pd0.75/T-2 and Pd1/T-2 nanostructures are 2.0, 1.9, and 1.7 nm, respectively. The particle sizes of the Au–Pd NPs decreased with the decrease of the Au/Pd molar ratio which is in agreement with the previous report.58 In addition, the HR-TEM image in Fig. 5d reveals that the metal NPs in Au0.75Pd0.25/T-2 are well-faceted and the measured lattice spacing of 2.32 Å is just between the values of Au(111) (2.35 Å in PDF no. 65-8601) and Pd(111) (2.25 Å in PDF no. 65-2867), indicative of Au–Pd alloy NPs.
 | ||
| Fig. 5 Representative (a, b) TEM and (d) HR-TEM images of Au0.75Pd0.25/T-2, and (c) its corresponding particle size distribution. | ||
It is known that the characteristic absorption peak of Pd NPs is in the UV region at a wavelength of 330 nm,59–61 which is overlapped by the absorption peak of TiO2. However, an increase of the baseline in the visible light region was observed for the Pd1/T-2 sample compared with the bare TiO2 nanobelts (T-2) in Fig. 6a, which can be attributed to light scattering or the interband electronic transitions of Pd NPs.62,63 For the bimetallic Au–Pd/TiO2-NB nanostructures, the Au alloyed with Pd led to the characteristic LSPR absorption peak of Au NPs at 570 nm broadening or even disappearing, corresponding to the color change of the sample from purple to gray (digital pictures inset in Fig. 6a). The Pd 3d and Au 4f XPS spectra of Pd1/T-2 and Au0.75Pd0.25/T-2 are shown in Fig. 6b–d. It can be seen that the Pd 3d3/2 peak in Pd1/T-2 could be fitted by two peaks at 340.5 and 342.5 eV which are attributed to metallic Pd and PdO species respectively.64,65 The existence of PdO is due to the easy oxidation of Pd NPs upon contact with O2 in air or water at room temperature. Compared to the values for Au 4f detected in Au1/T-2 (Fig. 2b), the corresponding binding energies in Au0.75Pd0.25/T-2 shifted to the higher values of 84.0 and 87.7 eV, while the peak of Pd 3d3/2 (339.1 eV) negatively shifted about 1.4 eV relative to the data of metallic Pd detected in Pd1/T-2. This result indicates that alloying Au with Pd gives Au a tendency to lose electrons. Given that the work function of metallic Pd (5.6 eV) is larger than that of metallic Au (5.3 eV), the migration of electrons from Au sites to Pd sites can occur.47,59–61 The positive shift of Au 4f and negative shift of Pd 3d indicate that a redistribution of electrons occurs in Au0.75Pd0.25/T-2, charging Au positively and Pd negatively when the equilibrium is reached.
 | ||
| Fig. 6 (a) Diffuse reflectance UV-vis spectra of Au1/T-2, Pd1/T-2 and Au0.75Pd0.25/T-2, and XPS spectra of (b) Pd 3d in Pd1/T-2, (c) Au 4f and (d) Pd 3d in Au0.75Pd0.25/T-2. | ||
The surface charge heterogeneity of Au–Pd alloy NPs will contribute to the improved catalytic performance of bimetallic nanostructures.59–61Fig. 7 shows that all bimetallic Au–Pd/T-2 nanostructures exhibited enhanced catalytic activity compared to their monometallic counterparts in the dark. The Pd/T-2 sample showed slightly enhanced catalytic activity compared to bare TiO2 nanobelts under visible light irradiation and no photocurrent response was observed (Fig. S6, ESI†), indicating the nonplasmonic metal nature of metallic Pd. The remarkable visible light-promoted catalytic performance was observed for all bimetallic Au–Pd/T-2 nanostructures, and the activity enhancement is strongly dependent on the Au/Pd ratio. Au0.75Pd0.25/T-2 (ICP-measured Au/Pd molar ratio is 2.4) showed the highest light-promoted activity; 35.2 μmol of benzaldehyde was produced under visible light irradiation, which is almost three times that obtained with Au1/T-2 (12.3 μmol). For the Au0.5Pd0.5/T-2 and Au0.25Pd0.75/T-2 samples, even though the Au–Pd NPs sizes become smaller, the photocatalytic efficiency is still lower than that of Au0.75Pd0.25/T-2. These data suggest a plasmonic Au mediated benzyl alcohol oxidation over bimetallic nanostructures under visible light irradiation. From Fig. 7, it can be seen that the light-promoted activity enhancement of Au0.75Pd0.25/T-2 is much higher than that of monometallic Au/T-2, indicative of a promoting effect from alloying Au with Pd for aerobic oxidation under visible light irradiation. This visible light-promoted synergistic effect of Au–Pd alloy NPs for benzyl alcohol oxidation can be ascribed to charge transfer between Au and Pd, and similar findings were found for the ZrO2 supported Au–Pd alloy NP photocatalysts reported by Zhu et al.59–61 The redistribution of hot electrons within alloy NPs might decrease the amount of electrons injected from alloy NPs into nanobelts to some extent. It is shown that a weaker photocurrent response of 0.83 μA cm−2 was measured for Au0.75Pd0.25/T-2 (Fig. S6, ESI†) compared to the value of 1.50 μA cm−2 obtained for Au1/T-2, though comparable light absorptions were measured for these two nanostructures. Accordingly, a less active O–O− species is expected to be produced on the TiO2 nanobelt surface in Au0.75Pd0.25/T-2 compared to the case of monometallic Au1/T-2, which would result in a decrease in benzaldehyde formation. Therefore, the remarkably improved visible light-promoted activity obtained for Au0.75Pd0.25/T-2 suggests that the synergistic effect resulted from charge transfer in the alloy NPs under visible light irradiation.
 | ||
| Fig. 7 Amount of benzaldehyde formed during the oxidation of benzyl alcohol over the Au–Pd/T-2 photocatalysts. | ||
It is generally accepted that alcohol oxidation over both Pd and Au NPs involves sequential cleavage of O–H and C–H bonds, followed by abstraction of the hydrogen atom from the intermediate metal-hydride species.8,66,67 The light-promoted catalytic activity of bimetallic Au–Pd/T-2 nanostructures can be attributed to efficient electron transfer between alloy NPs and the heterostructured nanobelt and the redistribution of hot electrons within alloy NPs. It has been proven that the electronic structure of Pd strongly impacts its oxygen activation efficiency, and electrons transferred into Pd tend to have substantial effects on its ability to activate oxygen molecules.68–70 Therefore, the hot electrons transfer from Au to both the TiO2 nanobelt and to Pd sites, which plays a key role in the activation of oxygen molecules, and facilitates the cleavage of the O–H bond of the alcohol. Simultaneously, the holes left in the Au sites can react with the alkoxide intermediate, accelerating the cleavage of C–H bonds.
On the basis of the results presented and discussed above, a possible mechanism for the visible light-promoted aerobic oxidation of benzyl alcohol over the Au–Pd/T-2 photocatalysts is proposed in Fig. 8. It illustrates that the oxidation of benzyl alcohol proceeds through cooperation between the bimetallic Au–Pd alloy NPs and the heterostructured nanobelt. In the first step, plasmon activation of Au sites transfers hot electrons to Pd sites and the heterostructured nanobelts. The hot electrons in the Pd sites and the heterostructured nanobelts can populate unoccupied orbitals of oxygen molecules yielding a transient anion O–O− species,27,71,72 which can cleave the O–H bond of the alcohol to form an alkoxide intermediate. In the second step, the intermediate undergoes a rapid hydride transfer from C–H to the positively charged Au to form benzaldehyde and a Au–H species.8 The third step will be the elimination of the Au–H species and the recovery of the active sites. The proposed mechanism gives a reasonable explanation for the light-promoted catalytic performance of bimetallic Au–Pd/T-2 nanostructures.
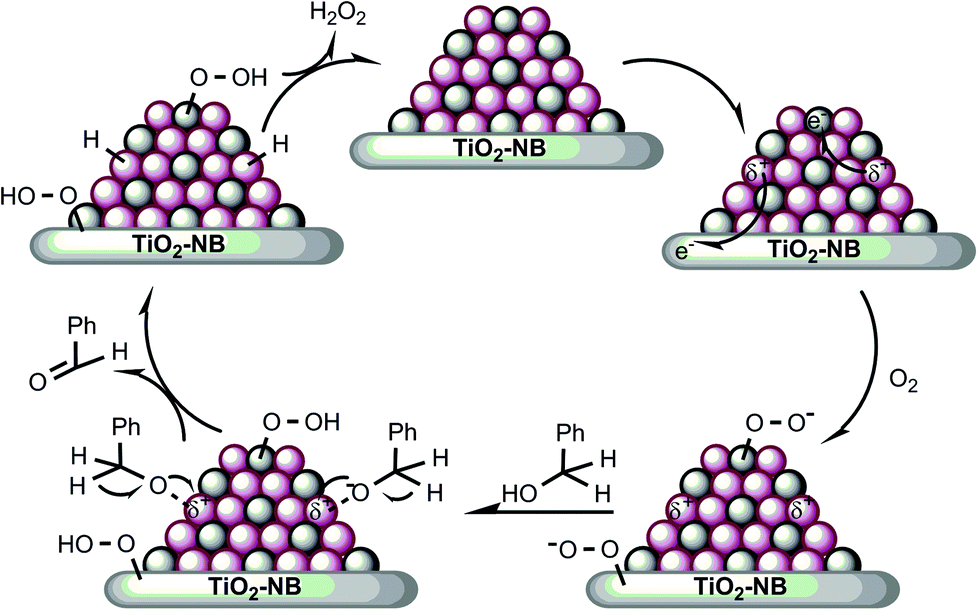 | ||
| Fig. 8 Proposed mechanism for the aerobic oxidation of benzyl alcohol over the Au–Pd/T-2 nanostructures driven by visible light irradiation. | ||
4. Conclusions
In conclusion, a deposition–precipitation method has been applied to synthesize highly dispersed small-sized Au and Au–Pd alloy NPs on TiO2 nanobelts. The one-dimensional nanobelt consisting of a TiO2(B)/anatase heterojunction enables efficient transfer of hot electrons from the Au NPs to the TiO2 nanobelts. This was confirmed by the superior visible light-promoted activity and much higher photocurrent response of the dual heterostructured Au/(TiO2(B)/anatase) nanostructure compared to the Au/TiO2(B) and Au/anatase counterparts. The bimetallic Au–Pd/TiO2-NB nanostructures showed a remarkably improved visible light driven activity compared to the monometallic Au/TiO2-NB sample, which can be attributed to the plasmon-mediated charge distribution from Au sites to Pd sites within alloy NPs. We anticipate that the findings obtained here are useful in the utilization of solar energy and contribute to the design and synthesis of more efficient plasmonic photocatalysts.Acknowledgements
We gratefully acknowledge the financial support of the National Natural Science Foundation of China (grant no. 51372142, 21176144, 21171106), National Science Fund for Distinguished Young Scholars (NSFDYS: 50925205), Innovation Research Group (IRG: 51321091), and the “100 Talents Program” of Chinese Academy of Sciences.Notes and references
- S. Arrii, F. Morfin, A. J. Renouprez and J. L. Rousset, J. Am. Chem. Soc., 2004, 126, 1199–1205 CrossRef CAS PubMed.
- H. Y. Fan, C. Shi, X. S. Li, S. Zhang, J. L. Liu and A. M. Zhu, Appl. Catal., B, 2012, 119–120, 49–55 CrossRef CAS PubMed.
- Y. Maeda, Y. Lizuka and M. Kohyama, J. Am. Chem. Soc., 2013, 135, 906–909 CrossRef CAS PubMed.
- E. Sacaliuc, A. M. Beale, B. M. Weckhuysen and T. A. Nijhuis, J. Catal., 2007, 248, 235–248 CrossRef CAS PubMed.
- J. Huang, T. Akita, J. Faye, T. Fujitani, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2009, 48, 7862–7866 CrossRef CAS PubMed.
- C. Qi, J. Huang, S. Bao, H. Su, T. Akita and M. Haruta, J. Catal., 2011, 281, 12–20 CrossRef CAS PubMed.
- J. Huang, W. L. Dai, H. Li and K. Fan, J. Catal., 2007, 252, 69–76 CrossRef CAS PubMed.
- A. Abad, C. Almela, A. Corma and H. García, Chem. Commun., 2006, 3178–3180 RSC.
- H. Liu, Y. Liu, Y. Li, Z. Tang and H. Jiang, J. Phys. Chem. C, 2010, 114, 13362–13369 CAS.
- G. M. Mullen, L. Zhang, E. J. Evans Jr., T. Yan, G. Henkelman and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 6489–6498 CrossRef CAS PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- M. Xiao, R. Jiang, F. Wang, C. Fang, J. Wang and J. C. Yu, J. Mater. Chem. A, 2013, 1, 5790–5805 CAS.
- A. Bumajdad and M. Madkour, Phys. Chem. Chem. Phys., 2014, 16, 7146–7158 RSC.
- R. Jiang, B. Li, C. Fang and J. Wang, Adv. Mater., 2014, 26, 5274–5309 CrossRef CAS PubMed.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- X. Wang, C. Liow, D. Qi, B. Zhu, W. R. Leow, H. Wang, C. Xue, X. Chen and S. Li, Adv. Mater., 2014, 26, 3506–3512 CrossRef CAS PubMed.
- Z. Bian, T. Tachikawa, P. Zhang, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 458–465 CrossRef CAS PubMed.
- F. Su, T. Wang, R. Lv, J. Zhang, P. Zhang, J. Lu and J. Gong, Nanoscale, 2013, 5, 9001–9009 RSC.
- S. Mubeen, G. Hernandez-Sosa, D. Moses, J. Lee and M. Moskovits, Nano Lett., 2011, 11, 5548–5552 CrossRef CAS PubMed.
- J. Long, H. Chang, Q. Gu, J. Xu, L. Fan, S. Wang, Y. Zhou, W. Wei, L. Huang, X. Wang, P. Liu and W. Huang, Energy Environ. Sci., 2014, 7, 973–977 CAS.
- A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852–14853 CrossRef CAS PubMed.
- D. C. Hurum, A. G. Agrios and K. A. Gray, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- T. Kawahara, Y. Konishi, H. Tada, N. Tohge, J. Nishii and S. Ito, Angew. Chem., Int. Ed., 2002, 41, 2811–2813 CrossRef CAS.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef CAS PubMed.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- D. Tsukamoto, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2012, 134, 6309–6315 CrossRef CAS PubMed.
- Y. Wen, B. Liu, W. Zeng and Y. Wang, Nanoscale, 2013, 5, 9739–9746 RSC.
- W. Zhou, L. Gai, P. Hu, J. Cui, X. Liu, D. Wang, G. Li, H. Jiang, D. Liu, H. Liu and J. Wang, CrystEngComm, 2011, 13, 6643–6649 RSC.
- W. Li, C. Liu, Y. Zhou, Y. Bai, X. Feng, Z. Yang, L. Lu, X. Lu and K. Y. Chan, J. Phys. Chem. C, 2008, 112, 20539–20545 CAS.
- D. Yang, H. Liu, Z. Zheng, Y. Yuan, J. C. Zhao, E. R. Waclawik, X. Ke and H. Zhu, J. Am. Chem. Soc., 2009, 131, 17885–17893 CrossRef CAS PubMed.
- B. Liu, A. Khare and E. S. Aydil, ACS Appl. Mater. Interfaces, 2011, 3, 4444–4450 CAS.
- D. Yang, J. Zhao, H. Liu, Z. Zheng, M. O. Adebajo, H. Wang, X. Liu, H. Zhang, J. C. Zhao, J. Bell and H. Zhu, Chem. – Eur. J., 2013, 19, 5113–5119 CrossRef CAS PubMed.
- H. H. Lo, N. O. Gopal, S. C. Sheu and S. C. Ke, J. Phys. Chem. C, 2014, 118, 2877–2884 CAS.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- M. Law, J. Goldberger and P. Yang, Annu. Rev. Mater. Res., 2004, 34, 83–122 CrossRef CAS.
- J. Tian, Z. Zhao, A. Kumar, R. I. Boughton and H. Liu, Chem. Soc. Rev., 2014, 43, 6920–6937 RSC.
- Y. Guan, N. Zhao, B. Tang, Q. Jia, X. Xu, H. Liu and R. I. Boughton, Chem. Commun., 2013, 49, 11524–11526 RSC.
- Q. Jia, D. Zhao, B. Tang, N. Zhao, H. Li, Y. Sang, N. Bao, X. Zhang, X. Xu and H. Liu, J. Mater. Chem. A, 2014, 2, 16292–16298 CAS.
- H. L. Jiang and Q. Xu, J. Mater. Chem., 2011, 21, 13705–13725 RSC.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- P. Montes-Navajas, M. Serra and H. Garcia, Catal. Sci. Technol., 2013, 3, 2252–2258 CAS.
- Y. Sugano, Y. Shiraishi, D. Tsukamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2013, 52, 5295–5299 CrossRef CAS PubMed.
- A. Zielińska-Jurek, E. Kowalska, J. W. Sobczak, W. Lisowski, B. Ohtani and A. Zaleska, Appl. Catal., B, 2011, 101, 504–514 CrossRef PubMed.
- N. Zhou, L. Polavarapu, N. Gao, Y. Pan, P. Yuan, Q. Wang and Q. H. Xu, Nanoscale, 2013, 5, 4236–4241 RSC.
- A. Tanaka, K. Fuku, T. Nishi, K. Hashimoto and H. Kominami, J. Phys. Chem. C, 2013, 117, 16983–16989 CAS.
- A. Cybula, J. B. Priebe, M. M. Pohl, J. W. Sobczak, M. Schneider, A. Zielińska-Jurek, A. Brückner and A. Zaleska, Appl. Catal., B, 2014, 152–153, 202–211 CrossRef CAS PubMed.
- P. Kittisakmontree, B. Pongthawornsakun, H. Yoshida, S. I. Fujita, M. Arai and J. Panpranot, J. Catal., 2013, 297, 155–164 CrossRef CAS PubMed.
- E. W. McFarland and J. Tang, Nature, 2003, 421, 616–618 CrossRef CAS PubMed.
- J. Y. Park, H. Lee, J. R. Renzas, Y. Zhang and G. A. Somorjai, Nano Lett., 2008, 8, 2388–2392 CrossRef CAS PubMed.
- S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279–285 CrossRef CAS PubMed.
- S. Higashimoto, N. Kitao, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2010, 274, 76–83 CrossRef CAS PubMed.
- C. J. Li, G. R. Xu, B. Zhang and J. R. Gong, Appl. Catal., B, 2012, 115–116, 201–208 CrossRef CAS PubMed.
- Y. Wang, K. Hang, N. A. Anderson and T. Lian, J. Phys. Chem. B, 2003, 107, 9434–9440 CrossRef CAS.
- T. Tachikawa, S. Tojo, M. Fujitsuka and T. Majima, Langmuir, 2004, 20, 2753–2759 CrossRef CAS.
- Y. Bai, W. Li, C. Liu, Z. Yang, X. Feng, X. Lu and K. Y. Chan, J. Mater. Chem., 2009, 19, 7055–7061 RSC.
- Y. Hong, X. Jing, J. Huang, D. Sun, T. Odoom-Wubah, F. Yang, M. Du and Q. Li, ACS Sustainable Chem. Eng., 2014, 2, 1752–1759 CrossRef CAS.
- S. Sarina, S. Bai, Y. Huang, C. Chen, J. Jia, E. Jaatinen, G. A. Ayoko, Z. Bao and H. Zhu, Green Chem., 2014, 16, 331–341 RSC.
- S. Sarina, H. Zhu, E. Jaatinen, Q. Xiao, H. Liu, J. Jia, C. Chen and J. Zhao, J. Am. Chem. Soc., 2013, 135, 5793–5801 CrossRef CAS PubMed.
- Q. Xiao, S. Sarina, A. Bo, J. Jia, H. Liu, D. P. Arnold, Y. Huang, H. Wu and H. Zhu, ACS Catal., 2014, 4, 1725–1734 CrossRef CAS.
- Y. Sugano, K. Fujiwara, Y. Shiraishi, S. Ichikawa and T. Hirai, Catal. Sci. Technol., 2013, 3, 1718–1724 CAS.
- S. Sarina, H. Y. Zhu, Q. Xiao, E. Jaatinen, J. Jia, Y. Huang, Z. Zheng and H. Wu, Angew. Chem., Int. Ed., 2014, 53, 2935–2940 CrossRef CAS PubMed.
- M. C. Militello and S. J. Simko, Surf. Sci. Spectra, 1997, 3, 387–394 CrossRef.
- M. C. Militello and S. J. Simko, Surf. Sci. Spectra, 1997, 3, 395–401 CrossRef.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS PubMed.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, J. Am. Chem. Soc., 2009, 131, 7189–7196 CrossRef CAS PubMed.
- W. E. Kaden, T. Wu, W. A. Kunkel and S. L. Anderson, Science, 2009, 326, 826–829 CrossRef CAS PubMed.
- W. E. Kaden, W. A. Kunkel, M. D. Kane, F. S. Roberts and S. L. Anderson, J. Am. Chem. Soc., 2010, 132, 13097–13099 CrossRef CAS PubMed.
- R. Long, K. Mao, M. Gong, S. Zhou, J. Hu, M. Zhi, Y. You, S. Bai, J. Jiang, Q. Zhang, X. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 3205–3209 CrossRef CAS PubMed.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CAS.
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044–1050 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr05905k |
| This journal is © The Royal Society of Chemistry 2015 |